

1 Introduction
Endocrine-disrupting chemicals (EDCs) are compounds that interfere with hormonal systems and induce developmental, reproductive, neurological, immune, or metabolic diseases in humans and wildlife [1–3]. Many EDCs are man-made chemicals released into the environment like plasticizers, pesticides, flame retardants, preservatives, pharmaceuticals, and cosmetics [4,5]. Some naturally occurring EDCs can also be found in plants or fungi. The sources of exposure to EDCs are diverse and vary widely around the world. Single exposure can occur for people working with high quantities of pesticides, fungicides, and industrial chemicals. However, the more common exposures are due to a broad mix of chemicals and contaminants present at low-concentrations. These complex mixtures enter the food chain and accumulate in animals and humans. Exposure occurs through drinking water, breathing polluted air, ingestion of food or, contacting contaminated soils.
EDCs can affect the endocrine systems of an organism in a wide variety of ways. These include mimicking natural hormones, antagonizing their action or modifying their synthesis, metabolism, and transport. Moreover, these substances can act via multiple pathways, including membrane receptors, the aryl hydrocarbon receptor, or the enzymatic machineries involved in hormone metabolism. However, most of the reported harmful effects of EDCs are attributed to their interference with hormonal signaling mediated by nuclear receptors (NRs) [6]. Human NRs are a family of 48 ligand-regulated transcription factors that control a plethora of biological processes such as development, organ homeostasis, metabolism, immune function, or reproduction. They harbor transcription regulation as well as DNA- and ligand-binding properties embedded in three distinct structural domains, and respond to a large variety of small endogenous ligands such as hormones, vitamins, fatty acids or metabolites. Upon ligand-binding, conformational changes in the C-terminal region of the ligand-binding domain (LBD) allow the receptors to recruit transcriptional coactivators. Originally, most of the studies have focused on NRs involved in reproductive processes, in particular the estrogen (ERα and ERβ) and the androgen (AR) receptors. More recently, studies have shown that the activity of the pregnane X receptor (PXR), the estrogen-related receptor γ (ERRγ), the thyroid hormone receptors (TRs), the retinoid X receptors (RXRs), or the peroxisome proliferator-activated receptors α and γ (PPARα and γ) is also affected by EDCs.
The group of molecules acting as NR environmental ligands is highly heterogeneous and comprises compounds that are often distantly related to endogenous ligands in terms of size or chemical structure. This group contains substances as chemically different as bisphenols, phthalates, parabens, dioxins, alkylphenols, organotins, polychlorinated biphenyls, perfluoroalkyls, or benzophenones, as well as natural compounds such as the phytoestrogen genistein, or the mycoestrogen α-zearalanol (Fig. 1). This large structural diversity renders the interaction of EDCs with their biological targets poorly understood and hardly predictable. Our recent studies combining structural, biophysical, and cell-based assays have revealed that chemicals bind to NRs with affinities ranging from sub-nanomolar to high micromolar values via diverse binding mechanisms [5,7,8]. Since humans and wildlife are simultaneously and chronically exposed to low doses of multiple contaminants, understanding the molecular mechanisms underlying the physiological consequences of exposure to environmentally-relevant concentrations of EDCs is of prime importance. In this review, we present the molecular details of three different mechanisms used by EDCs to bind to NRs with high-affinity and discuss some current hypotheses that may account for the low dose action of compounds with medium-to-weak affinities for NRs.
Chemical structures of the representative endocrine disruptors mentioned in this review.
2 α-Zearalanol is a high-affinity ligand of estrogen receptors
ERα and ERβ (NR3A1-2) are receptors for the sex hormone, 17β-estradiol (E2), which play important roles in the growth and maintenance of a diverse range of tissues such as the mammary gland, uterus, bone, or the cardiovascular system. Both ERs are widely distributed throughout the body, displaying distinct but overlapping expression patterns in a variety of tissues [9]. Although ERα and ERβ share similar mechanisms of action, several differences in the transcriptional abilities of each receptor as well as distinct phenotypes between gene null animals have been identified, suggesting that these receptors may regulate distinct cellular pathways [9].
Endogenous estrogens (E2, estriol, estrone) are high-affinity ligands of ERs with dissociation constants (Kd) comprised between 10 pM and 1 nM. On the contrary, xenoestrogens such as the phytoestrogens genistein and ferutinin, the pesticides DDT, HPTE (a methoxychlore metabolite), vinclozoline or chlordecone, the plasticizers bisphenols and phthalates, and the benzophenones and parabens used as UV-filters and preservatives, respectively, bind to ERs with a wide array of affinities ranging from 10 nM to 10 μM [4,10–12]. All of them bind to the hormone-binding site of ERs and engage in different sets of ligand–protein interactions according to their size and chemical structures. The smallest compounds making fewer contacts with the receptor cavity are generally associated with lower binding affinities, whereas bigger EDCs adopting a binding mode reminiscent of that used by the endogenous ligands are characterized by higher interaction capacities. This is, for example, the case of the mycoestrogen α-zearalanol (α-ZA), which acts as a full ERα agonist (Fig. 2A) and binds to ERs with high-affinity (Kd of 0.29 nM and 1.88 nM for ERα and ERβ, respectively, to be compared to 0.017 nM and 0.068 nM for E2) [4]. As observed in the ERα crystal structures, α-ZA recapitulates most of the interaction network observed with E2 (Fig. 2B, C), including the hydrogen bonds linking the phenol moieties of the two ligands and the polar residues H524 and E353 located at the two ends of the ligand-binding pocket (LBP). The remaining contacts involve essentially van der Waals interactions, the number and strength of which account for the different binding affinities of α-ZA and E2 towards both ER subtypes. This is particularly evident in the case of ERβ, for which we have previously shown that the replacement of ERα L384 and M421 by M336 and I373, respectively, imposes more space constraints in its LBP and renders this ER isotype more sensitive to variations in the size of the bound ligand [4]. Accordingly, α-ZA, which occupies a slightly bigger volume (Fig. 2C), displays a 7-fold lower affinity than E2 towards ERβ, whereas α-ZA and E2 bind equally well to ERα.
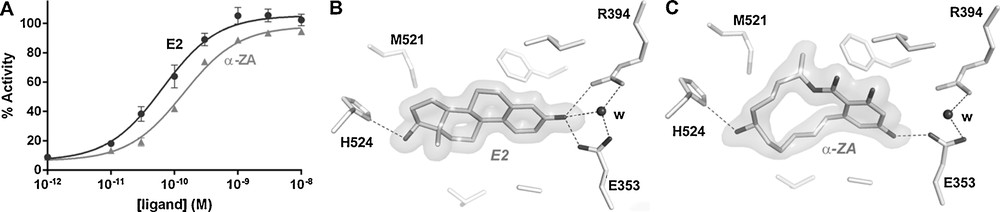
The mycoestrogen α-zearalanol is a full estrogen (ERα) agonist. Relative transcriptional activity of ERα in HGELN-ERα cells in the presence of estradiol or α-zearalanol (100% as 10 nM E2) (A). Interaction networks of estradiol (B) and α-zearalanol (C) with residues of the ERα–ligand-binding domain (LBD). The volume around the ligands represents their electron density.
3 Organotins are covalent ligands of many nuclear receptors
Organotin compounds are ubiquitously present throughout the environment due to their widespread use since the 1960s in many industrial and agricultural processes. Since the 1980s, they were found to be responsible for a wide variety of deleterious effects in the endocrine systems of humans and wildlife at nanomolar concentrations [13]. Organotins form a collection of more than 200 tin compounds containing a variety of mono-, di-, tri- or tetra-substituted organic groups. They do neither structurally nor chemically resemble known NR ligands, so the mechanism by which organotins act as endocrine disruptors has remained enigmatic until the structures of RXRα and PPARγ in complex with tribultyltin (TBT) and tripropyltin (TProT), respectively, were solved.
RXRα, β and γ (NR2B1-3) occupy a particular position in the NR superfamily as they are the common heterodimerization partners for one-third of the 48 family members. As such, RXRs play key roles in the control of many NR-dependent signaling pathways. RXRs heterodimers can be partitioned into two classes according to the nature of the partner receptor. The so-called permissive heterodimers (e.g., with PPARs, LXRs or PXR) can be activated upon ligand-binding to RXR, even in the absence of the partner receptor ligand. In contrast, in non-permissive RXRs heterodimers (e.g., with RARs, VDR, or TRs), the activation of RXR by its own ligand is subordinated to the presence of a ligand in the partner receptor. However, in both cases, RXR ligands and ligands of the partner receptors can act synergistically to activate heterodimers [14]. This regulatory control of nuclear signaling pathways by multiple RXR heterodimers allows environmental RXR ligands to potentially trigger a multitude of adverse effects on human health. The identity of the physiological RXR ligands is still debated, and there is particular uncertainty about the status of the historical “endogenous RXR agonist” 9-cis-retinoic acid (9-cisRA), because many groups have been unable to detect 9-cisRA in vivo. Recently, other vitamin A metabolites and some unsaturated fatty acids have been demonstrated to act as endogenous RXR ligands in several tissues [15]. Thus, the current trend is to assign an intracellular sensor function to RXRs that could bind a variety of fatty acids and metabolites with moderate affinities (μM range). In its heterodimeric form with RXRs, PPARγ (NR1C3) plays key roles in regulating adipogenesis, lipid metabolism, and glucose homeostasis through improving insulin sensitivity [16]. PPARγ ligands are diverse and include unsaturated fatty acids or prostaglandins with which they associate with rather weak affinities [17].
Few environmental ligands of RXRs and PPARγ have been reported up to now. They include the pesticide metabolite methoprene acid, some bisphenols, the 4-tert-octylphenol, and organotins for the first one, and halogenated bisphenols, phthalates, perfluorinated compounds, phytoestrogens and organotins for the second one [18–22]. Among them, organotins are, by far, those showing the strongest affinity for both receptors (Kd in the nanomolar range) [7,19,23,24]. The crystal structure of RXRα in complex with TBT [19] shows that as compared with 9-cisRA (Fig. 3A), the organotin occupies only a small part of the LBP (Fig. 3B). However, it also reveals that the high-affinity of TBT for RXRs derives from the formation of a covalent bond between the tin atom of the organotin and the sulfur atom of the conserved cysteine C432. Although TBT interacts with only a subset of LBP residues, it is engaged in enough essential contacts to efficiently stabilize RXRα in its active conformation. The particular position of C432 in helix H11 allows TBT to stabilize the C-terminal activation helix H12 in a position that is crucial for the recruitment of transcriptional coactivators. Hence, in addition to bind to RXR at very low-concentrations, TBT acts as a full agonist activating the receptor as efficiently as the reference synthetic CD3254 (Fig. 3C). In contrast, the crystal structure of PPARγ in complex with TProT [7] shows that the anchoring cysteine (C285) resides in helix H3, placing the organotin in a region of the LBP that does not allow the efficient stabilization of the active receptor conformation (Fig. 3D). This is in line with the weak PPARγ agonistic activity of the compound [19] (Fig. 3C). Thus, the efficient activation of the RXR-PPARγ by organotins results from the combined action of the compounds on the two heterodimer subunits. The discovery of this binding mode suggests that other NRs presenting a cysteine residue should be considered as potential targets of organotins, the functional outcome of this interaction being dictated by the position of the anchoring cysteine in the LBP. Accordingly, it has been reported that dibutyltin acts as a potent antagonist of the glucocorticoid receptor (GR), which contains two cysteine residues in its hormone-binding site [25].
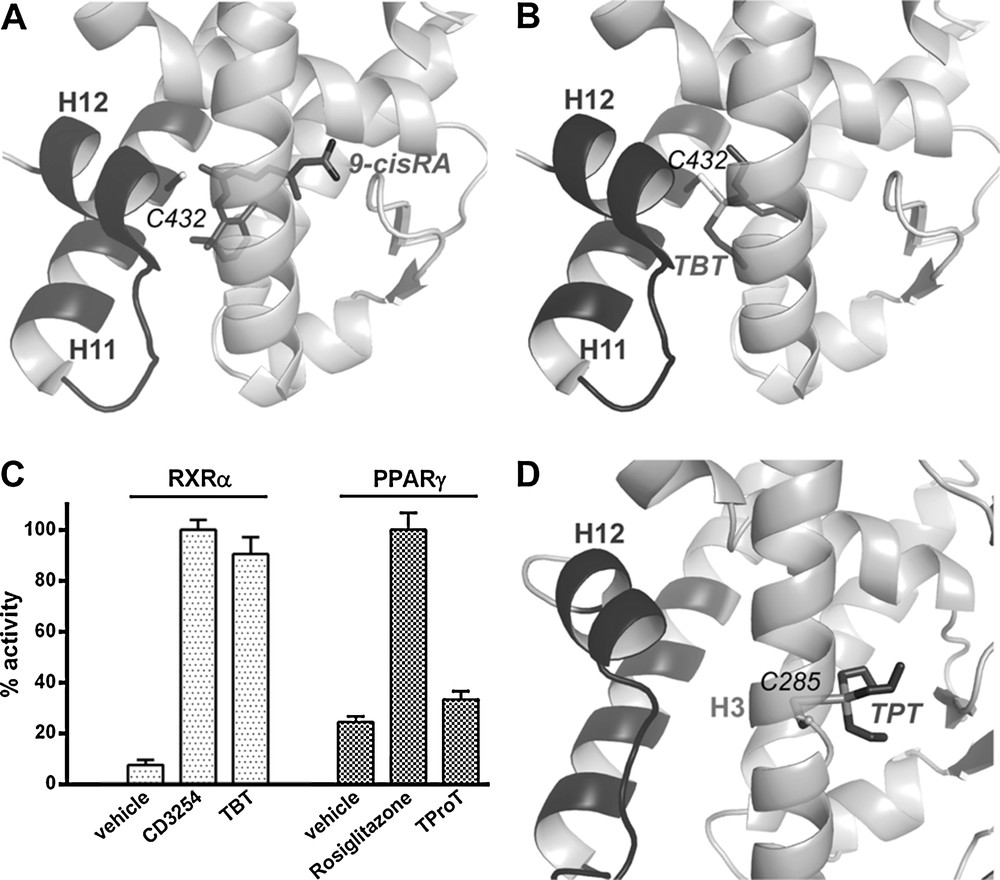
Organotins bind covalently to RXRα and peroxisome proliferator-activated receptor γ (PPARγ). Close-up view on the ligand-binding site of RXRα ligand-binding domain (LBD) structure bound to the 9-cis retinoic acid (A) and the tributyltin (B). Relative transcriptional activity of RXRα and PPARγ ligands (100% as CD3254 100 nM for RXRα and rosiglitazone 1 μM for PPARγ); concentrations: CD3254 10 nM, TBT 10 nM, rosiglitazone 1 μM, TProT 100 nM (C). Close-up view on the ligand-binding site of PPARγ LBD structure bound to the tripropyltin (D).
4 The pregnane X receptor and the cocktail effect
PXR (NR1I2) is involved in the biosynthesis, distribution, and metabolism of steroids, bile acids, and xenobiotics [26]. It plays a prominent role as protector of the endocrine system from chemical perturbation by sensing increases in the concentration of a multitude of external substances. Upon activation by xenobiotics, PXR forms heterodimers with RXR and binds to PXR response elements in the regulatory regions of target genes where it up-regulates the transcription of major detoxification genes such as the phase-I (CYP), phase-II (conjugating) and phase-III (ABC transporters) enzymes, and efflux proteins. By doing so, PXR induces the xenobiotic clearance and prevents other NRs from interactions with these chemicals. On the other hand, PXR activation has been linked to drug–drug interactions, deregulation of steroid homeostasis, chemoresistance, growth and aggressiveness of colon and hepatic cancers [27–30]. Unlike many NRs that tend to be specialized to bind few ligands with structural homologies, PXR is able to interact with a large number of structurally diverse compounds with medium affinities (Kd between 0.1 and 100 μM). The known ligands of PXR include pesticides, phenols, cosmetics, phytoestrogens, pharmaceuticals, etc. [8,31,32]. Crystallographic studies have revealed the unique characteristics of PXR that account for its promiscuous ligand-binding properties. Firstly, PXR possesses a large LBP that can accommodate compounds with larger volumes than that of classical NR ligands, and secondly, several loops of the LBD confer a high plasticity allowing the receptor to adopt different shapes according to the bound ligands. We recently demonstrated that a pharmaceutical estrogen (the contraceptive 17α-ethinylestradiol [EE2]) and a persistent organochlorine pesticide (trans-nonachlor [TNC]), both exhibiting low efficacy when studied separately, cooperatively bind to PXR, leading to synergistic activation [8]. Both biophysical and cell-based analyses showed that each ligand enhances the binding affinity of the other one, so the binary mixture binds 100-fold more avidly to PXR than TNC and EE2 alone, and induces a substantial biological response at doses at which each chemical individually is inactive (Fig. 4A, B). High-resolution crystal structures showed that, individually, EE2 and TNC are too small to make all the necessary interactions ensuring high binding affinity and effective stabilization of the active conformation of the receptor. In contrast, when associated in a binary mixture, EE2 and TNC fill a larger fraction of the PXR LBP. Moreover, eight van der Waals contacts could be measured between EE2 and TNC (Fig. 4C). These inter-ligand contacts generate a mutual stabilization of the compounds in the LBP and account for the enhanced binding affinity of the binary mixture. We therefore proposed the concept of “supramolecular ligand” that defines a molecular assembly consisting of two or more compounds that interact with each other inside the LBP of a receptor, resulting in the creation of a new entity with improved functional characteristics in regard to those of its individual components. This study provided the first detailed mechanistic explanation and a proof of concept for the synergistic action of a mixture (cocktail) of compounds via their simultaneous interaction with a NR, as well as new insight as to how low doses of EDCs or drugs may affect physiology and homeostasis.
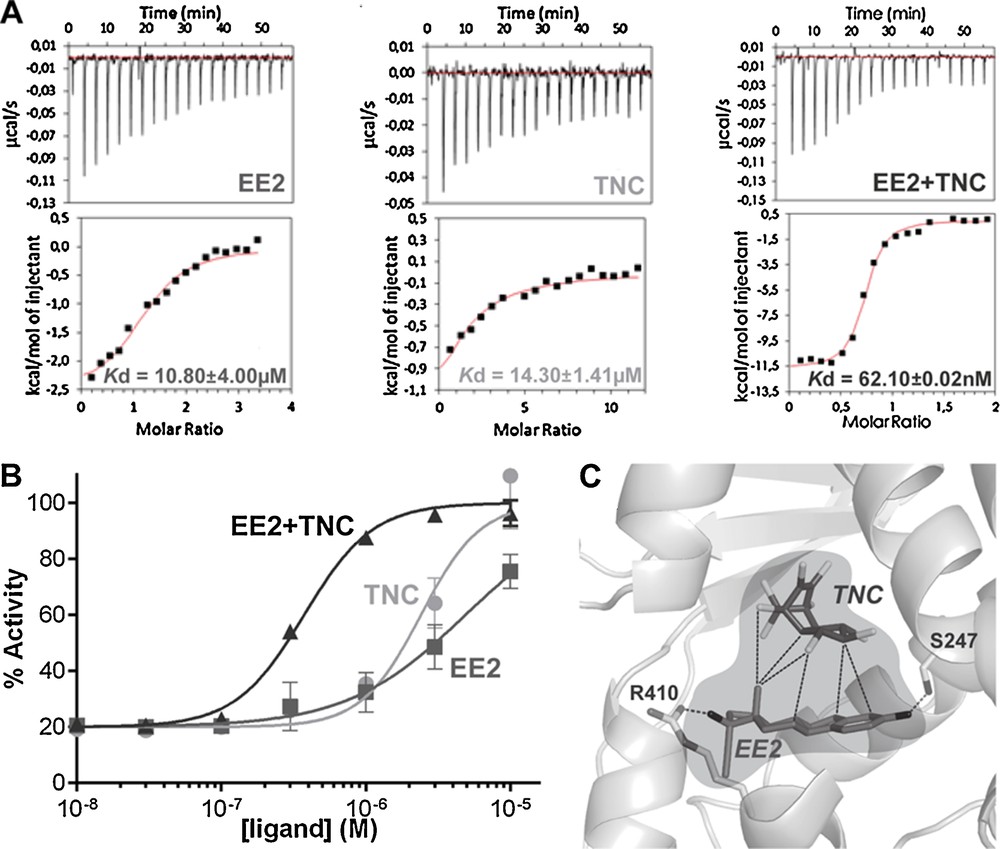
The cocktail effect highlighted in pregnane X receptor (PXR). Isothermal titration calorimetry characterization of PXR interaction with contraceptive 17α-ethinylestradiol (EE2) and trans-nonachlor (TNC), alone or in combination (A). Relative transcriptional activity of PXR in HG5LN GAL4–PXR–ligand-binding domain (LBD) cells in the presence of EE2 and TNC alone or in combination (100% as SR12813 3 μM) (B). The supramolecular ligand formed by EE2 and TNC inside the ligand pocket of PXR is surrounded in light grey; dashed lines represent the interactions between the two ligands and the hydrogen bonds between EE2 and the residues of PXR (C). Masquer
The cocktail effect highlighted in pregnane X receptor (PXR). Isothermal titration calorimetry characterization of PXR interaction with contraceptive 17α-ethinylestradiol (EE2) and trans-nonachlor (TNC), alone or in combination (A). Relative transcriptional activity of PXR in HG5LN GAL4–PXR–ligand-binding domain (LBD) cells ... Lire la suite
5 Concluding remarks
The deregulation of NR-mediated transcription accounts for the deleterious effects of many EDCs. Thus, characterization of the interaction between receptors and environmental compounds, both at the structural and functional levels, are important for the assessment of the global hormonal activity of a large number of chemicals as well as the development of robust in vivo, in vitro and in silico screening methods. Because EDCs are generally present at low-concentrations in the environment, the molecular basis for the health impact of chemically unrelated compounds at low doses has remained largely elusive. In this review, we have described three different mechanisms accounting for the high-affinity interaction between EDCs and NRs.
In spite of its structural differences with E2, the interaction of α-ZA (a mycoestrogen with non-estrogenic chemical structure) resembles that of E2. Indeed, the key contacts observed between the endogenous hormone and ER were found to be conserved with the mycoestrogen. As a consequence, α-ZA is one of the most active xenoestrogen that can modulate ER activity at concentrations as low as 0.1 nM. In contrast, we have shown that organotins such as TBT do not recapitulate any of the specific interactions made by the classical ligands of RXR and PPAR with their corresponding receptors. Instead, tin compounds use a Sn–S covalent interaction to bind to and modulate the transcriptional activity of the RXR–PPAR heterodimer at nanomolar concentrations. Covalent coupling between PPARs and pharmaceutical or natural compounds has also been reported [33,34]. It is thus not excluded that the low dose effects of some environmental compounds can also be explained by their covalent interaction with the dozen NRs containing a cysteine residue in their LBP. In the last reported mechanism, a pesticide and a pharmaceutical compound were found to interact with each other in the PXR LBP, forming a ‘supramolecular ligand’ that is a more potent activator than either of the two chemicals alone. It has been observed that the two compounds not only bind concomitantly to the LBP of the receptor, but they do so cooperatively, i.e. the binding of one molecule promotes the high-affinity binding of the second one. Structural studies revealed that the ligands mutually stabilize each other via strong inter-ligand contacts and that a large number of interactions with LBP amino acids allow the supramolecular ligand to tightly hold the receptor in the transcriptionally-competent conformation.
Considering the conservation of structural and functional NR features and the huge chemical and size diversity of xenobiotics, one can predict that the three mechanisms described in this review in the context of a given receptor/EDC couple are very likely to apply to many other NR and EDC family members. A synergistic/additive effect resulting from the simultaneous activation or inhibition of different signaling pathways could also account for the low dose action of certain chemicals. BPA, which is a moderate environmental agonist of ERs, ERRγ, PXR, and AR antagonist [10], is a good example of an EDC whose adverse effects could result from its combined estrogenic and anti-androgenic properties. Additionally, the health impact of EDCs at environmentally-relevant concentrations could be also due to the interaction of chemicals with other bona-fide targets that remain to be identified, such as membrane-associated receptors, hormones transporters or enzymes. Several research groups have proposed that the non-classical membrane G-protein coupled estrogen receptor (GPER/GPR30) or the membrane-associated estrogen receptors (mbERs) could mediate the effects of xenoestrogens through rapid non-genomic activation of signal transduction pathways [35,36]. It has been postulated that activation of the extranuclear-initiated signaling pathway might depend to a lesser extent on the affinity of ligand for the receptor than the nuclear pathway [36], but this hypothesis is still controversial and needs further validations.
Acknowledgments
This work was supported by funds from the INSERM, CNRS, ANR (TOXSYN ANR-13-CESA-0017-03), “Environnement cancer” project (SYNERPXR 1 A12156FS and SYNERPXR 2 C15010FS), ANSES (EVALPE EST-13-031), FBI (ANR-10-INBS-04-01), and FRISBI (ANR-10-INBS-05).
Vous devez vous connecter pour continuer.
S'authentifier