

1 Introduction
L’association d’un ligand à son récepteur membranaire déclenche en quelques secondes l’activation d’un réseau de signalisation complexe qui, en modulant l’expression des gènes, dicte le phénotype cellulaire. Les gènes immédiats/précoces sont transcrits en moins d’une heure, tandis que la transcription des gènes dont la régulation nécessite la synthèse de nouvelles protéines et/ou des remodelages notables de la chromatine se déroule sur plusieurs heures. À la suite de ces événements, les ARNm nouvellement transcrits subissent un processus de maturation, puis ils sont exportés dans le cytoplasme à travers les nucléopores pour être transportés dans différents compartiments subcellulaires. Jusqu’à présent, la plupart des études ont échoué à établir une stricte corrélation entre la transcription des ARNm par analyse transcriptomique à l’échelle du génome, et le répertoire cellulaire en protéines [1–5]. Ceci s’explique en partie par le fait que l’expression des gènes dépend aussi de la traduction de populations d’ARNm déjà transcrits, au moment de la perception d’un signal extracellulaire, comme cela a été initialement démontré au cours de l’embryogenèse précoce [6]. Or, l’une des caractéristiques importantes de la traduction des ARNm est la rapidité avec laquelle elle procède, suite à l’action d’un stimulus extracellulaire, puisque la machinerie de traduction est branchée de façon dynamique sur le réseau de signalisation induit par l’activation d’un récepteur. C’est pourquoi des variations subtiles de l’environnement telles que des modifications de la concentration en différents facteurs de croissance ou hormones, en régulant l’expression des gènes au niveau traductionnel, aboutissent à une modification très précoce du contenu cellulaire en protéines [7,8].
Des signaux extracellulaires de nature chimique très diversifiée, telles que des hormones, des neurotransmetteurs, des lipides, des aminoacides, des ions, ciblent les récepteurs à sept domaines transmembranaires couplés aux protéines G (RCPG). À ce jour, plus de 800 RCPG ont été identifiés dans le génome humain [9]. Ces récepteurs ont une structure tridimensionnelle flexible, qui oscille d’une façon dynamique entre des conformations inactives et actives, stabilisées par le ligand spécifique et par le couplage aux protéines de transduction du signal [10]. Les modifications conformationnelles du récepteur sont directement perçues par les protéines G et par les ß-arrestines. Ce mode d’activation est une caractéristique unique aux RCPG qui assure la transmission du signal au réseau de signalisation ad hoc en quelques secondes. Au sein de ce réseau, les protéines G et les ß-arrestines transduisent un signal qui peut contrôler la traduction des ARNm, soit en ciblant directement le complexe d’initiation de la traduction, et/ou en stimulant les voies de signalisation qui régulent l’assemblage de la machinerie traductionnelle [11,12].
L’impact de la signalisation des RCPG s’exerce principalement dans des cellules hautement spécialisées, telles que des neurones, des bâtonnets rétiniens, des cellules de Sertoli des gonades, des cellules immunocompétentes, des cellules endothéliales, dans lesquelles ces récepteurs peuvent jouer un rôle trophique. Par exemple, le récepteur de la FSH (RFSH) induit la néosynthèse de facteurs paracrines essentiels aux différents stades de la spermatogenèse dans la cellule de Sertoli, cellule nourricière de la lignée germinale dans le testicule [13]. De la même façon, le récepteur de l’endothéline 1 ET1R stimule l’hypertrophie des cardiomyocytes lors d’une accommodation à l’effort ou en conditions pathologiques via la régulation de la traduction des ARNm [14]. D’autres RCPG exercent un rôle trophique sur leurs cellules-cibles : c’est le cas des récepteurs de l’adénosine et des récepteurs purinergiques P2Y dans les cellules endothéliales, au cours de l’angiogenèse [15], ou des récepteurs de neurotransmetteurs de type muscarinique ou GABAB pendant la maturation du cerveau [16,17]. Dans les cellules différenciées, l’activation d’un RCPG par son ligand peut induire la traduction sélective de certains ARNm, avec un impact mineur mais néanmoins détectable sur les néosynthèses protéiques globales. Par exemple, l’activation du RFSH par la FSH induit la traduction sélective des ARNm vegfa et c-fos en quelques minutes, en absence d’effet sensible sur la transcription [18]. Bien que depuis le début des années 2000, plus de 160 publications de transcriptomes induits par un RPCG sont apparues, les traductomes de RCPG publiés n’excèdent pas la dizaine. Cette revue présentera brièvement ces traductomes, ainsi que les approches méthodologiques qui ont permis de les identifier.
2 Régulation du traductome par les RCPG
2.1 Récepteur de l’endothéline 1
Le premier traductome induit par un RCPG publié est celui du récepteur de l’endothéline (ET-1) dans les cardiomyocytes [19]. Ce récepteur est impliqué dans la prolifération des cellules en division et dans l’hypertrophie des cardiomyocytes. Les voies de signalisation intracellulaires impliquées dans l’hypertrophie des cardiomyocytes sont bien connues, cependant leur implication dans l’expression de ce phénotype spécifique demande à être précisée. Pour déterminer si les réponses transcriptionnelles précoces des cardiomyopathies à l’ET-1 sont sujettes à une régulation traductionnelle, Cullingford et al. ont comparé les profils d’expression des ARN totaux à ceux qui sont associés aux polysomes issus de cardiomyocytes exposés à l’ET-1 et de cellules non stimulées, par profilage polysomique et identification sur microarray Affymetrix. Ils ont démontré que plus de 80 % des ARNm induits précocement par ET-1 étaient aussi recrutés aux polysomes.
2.2 Récepteur de la GnRH
Plus récemment, la réponse au mis-repliement des protéines (UPR pour unfolded protein response), un mécanisme de contrôle qualité des protéines dans les cellules sécrétoires, a été explorée dans la lignée gonadotrope LßT2 dérivée de l’anté-hypophyse. Par profilage polysomique et identification sur microarray Affymetrix, cette étude a révélé que la GnRH induit une augmentation de la traduction de certains ARNm, tel que Dusp1, qui code une phosphatase participant au décodage de la pulsatilité du signal GnRH au niveau hypophysaire [20]. Simultanément, la GnRH induit une pause de la traduction de certains ARNm impliqués dans la fonction de reproduction comme ceux qui codent les chaînes a et ß de la LH [21]. De cette façon, la stimulation GnRH régule finement à la fois la qualité et la quantité des protéines sécrétées par les cellules gonadotropes de l’hypophyse.
2.3 Récepteurs D1 et D2 de la dopamine
La purification par affinité de la grande sous-unité ribosomale 60S marquée par un épitope permet de récupérer les ARNm associés à des polysomes par immunoprécipitation, à partir de tissus. Heiman et ses collègues [22] ont baptisé cette méthodologie « traduction par purification d’affinité des ribosomes », ou TRAP (voir Section 3.3). Cette approche nécessite des souris qui expriment une protéine rpL10a de la grande sous-unité ribosomale fusionnée, par exemple, à la eGFP, dans une population cellulaire spécifique du système nerveux central (CNS). La protéine de fusion L10a-eGFP est sous le contrôle des séquences régulatrices des récepteurs de la dopamine Drd1a ou Drd2, ce qui a permis d’isoler les ARNm associées aux polysomes de neurones striato-nigraux (Drd1) ou striato-pallidaux (Drd2), du striatum de souris et de les identifier par microarray Affymetrix. De cette façon a été mise en évidence une traduction sélective des ARNm associés aux polysomes dans les cellules striato-pallidales Drd2-positive (Adk, Plxdc1, BC004044, et Hist1h2bc) et dans les cellules striato-nigrales Drd1-positive (Slc35d3, Zfp521, Ebf1, Stmn2, Gnb4, et Nrxn1), qui sont des populations neuronales morphologiquement indiscernables.
2.4 Récepteurs métabotropiques du glutamate
Au niveau des synapses hippocampiques, l’activation des récepteurs mGluR induit une dépression à long terme (LTD, long-term depression), dont l’un des effecteurs majeurs est le facteur d’initiation de la traduction eIF2a. Au niveau des dendrites, l’activation des mGluR déclenche la synthèse rapide de protéines dont la fonction est de provoquer une dépression en augmentant l’endocytose des récepteurs AMPA (AMPAR) impliqués dans la dépolarisation de la membrane des neurones cibles du glutamate. Alors que l’augmentation de l’expression de certaines protéines en réponse à l’activation du mGluR est connue (Arc, Step, et Map1b) [23], les travaux de Di Prisco et al. [24] ont permis d’identifier certains ARNm dont la traduction est sélectivement régulée au cours de cette LTD, par profilage polysomal et identification par séquençage Illumina. Des neurones de souris en culture ont été traités par l’agoniste 3,5-dihydroxyphenylglycine (DHPG) sélectif des mGlur1 et 5, afin d’induire la phosphorylation d’eIF2a et, par conséquent, de stimuler la LTD. L’approche combinée du profilage polysomal et du séquençage à haut débit a montré que l’augmentation de la phosphorylation de eIF2a est associée à une augmentation significative de la traduction des ARNm du récepteur cholinergique Chrna6, du récepteur Gpr22 et de l’oligophrénine-1 (OPHN1). Cette protéine est décrite pour être impliquée dans la médiation des signaux intracellulaires au cours de la migration cellulaire et de la morphogenèse. La perte de fonctionnalité d’OPHN1 est corrélée à des déficiences intellectuelles liées au chromosome X. L’injection de shRNA Ophn1 dans l’hippocampe de souris montre une réduction de l’exploration des souris vers un objet préalablement exposé. Tout comme le contrôle de la traduction médié par la phosphorylation de eIF2a, OPHN1 est nécessaire à l’accomplissement des tâches d’apprentissage médiées par mGluR-LTD dans l’hippocampe. OPHN1 a également été récemment décrite pour être recrutée au cours de l’endocytose de AMPAR [25]. Grâce au traductome, l’identification des cibles d’eIF2a phosphorylé permettrait de trouver de nouvelles cibles pour les thérapies des troubles cognitifs liés à la LTD, y compris la maladie d’Alzheimer, la maladie de Parkinson, certaines déficiences intellectuelles et comportements addictifs.
2.5 Récepteur de la gonadotrophine LH
L’impact des signaux hormonaux sur la régulation de la spermatogenèse par les cellules somatiques de la gonade a été étudié grâce à l’approche RiboTag (voir Section 3.3) [26]. Des souris AMH-cre et Cyp17i-cre ont été croisées avec des souris RiboTag pour marquer avec un tag HA, les ribosomes des cellules de Sertoli et de Leydig de souris adultes, respectivement. Avant sacrifice, les animaux ont été traités par un antagoniste du GnRH pour amener la concentration d’hormones circulantes (FSH ou LH/testostérone) à un niveau de base. Après immunoprécipitation, les ARNm associés aux polysomes ont été identifiés par puce Affymetrix, en normalisant par rapport aux ARN totaux du testicule entier. Aucune augmentation de la régulation de la traduction n’est observée dans les cellules de Sertoli après 4 h de traitement par la FSH, et c’est principalement l’effet de la stimulation LH sur la régulation de la traduction des ARNm dans les cellules de Leydig qui a été décrite. Dans ces cellules, après une heure de stimulation, la LH augmente la traduction des ARNm codant les facteurs de transcription Nr4a1 et Egr1, ou encore celle de Rgs2, un régulateur du cycle cellulaire. Le nombre d’ARNm traduits augmente encore après 4 h de stimulation. Ces résultats démontrent que, chez le mâle adulte, la LH exerce un effet sur la régulation de la traduction dans la cellule de Leydig.
3 Les méthodes d’analyse du traductome
Bien que la concentration en ARNm soit couramment assimilée à l’abondance protéique, la comparaison de l’expression des ARNm et des protéines à l’échelle du génome global montre qu’il existe une faible corrélation entre les deux mesures [1,3,4,27]. Seulement 20 à 40 % des protéines totales auraient un niveau d’expression protéique directement proportionnel à leur niveau d’expression transcriptionnelle [1,2,27,28]. Ceci souligne l’importance des mécanismes de régulation post-transcriptionnelle dans l’homéostasie et la croissance cellulaire, mécanismes qui ne peuvent être appréhendés par la seule analyse comparative des quantités d’ARNm et de protéines. C’est pourquoi il est nécessaire d’associer à cette dernière une analyse systémique de la traduction des ARNm, c’est-à-dire de définir le traductome, qui vise à identifier l’ensemble des ARNm associés aux ribosomes au cours de la synthèse protéique. Il peut être obtenu à l’aide de plusieurs méthodes : profilage ribosomal, profilage polysomique et purification des ribosomes par affinité (Fig. 1).

Approches biochimiques du traductome. A. Le profilage ribosomique (ribosome profiling) sert à définir l’occupation des ARNm par les ribosomes. Des lysats de cellules traitées par la cycloheximide sont digérés par des nucléases pour éliminer les séquences d’ARNm qui ne sont pas protégées par les ribosomes (à gauche). Les complexes de monosomes qui en résultent sont purifiés par ultracentrifugation sur un gradient de saccharose. Les fragments protégés par les ribosomes sont récupérés et séquencés. En parallèle, l’ARNm total provenant d’une préparation similaire est fragmenté (à droite) et sert de contrôle de normalisation. B. Profilage polysomique (polysome profiling) : des lysats cytoplasmiques sont séparés sur un gradient de sucrose par ultracentrifugation pour séparer les petits ARN et ARNm libres, les sous-unités 40S, 60S, 80S et les polysomes. Les ARNm issus des fractions polysomales sont isolés et identifiés par micro-array ou RNA-seq. C. Les souris BacTRAP expriment une protéine ribosomale rpL10a fusionnée à EGFP (ribosomes verts) sous le contrôle de promoteurs activés dans des cellules spécifiques. Les complexes ARNm/ribosomes-rpL10a-EGFP sont immunopurifiés à partir de tissus de souris bacTRAP, et les ARNm associés sont identifiés. D. Dans les souris RiboTag, le gène rpL22 comprend un exon 4 de type sauvage floxé suivi du même exon 4 fusionné à HA. Lorsque ces souris sont croisées avec des souris exprimant la recombinase Cre sélectivement dans un type cellulaire donné, cette dernière, en excisant l’exon 4 sauvage, permet l’expression de rpl22 fusionné à HA, qui s’incorpore aux polysomes (ribosomes bleus). Les ARNm associés aux ribosomes rpL22-HA uniquement dans le type cellulaire d’intérêt sont récupérés par immunoprécipitation à l’aide d’un anticorps anti-HA et identifiés.
3.1 Profilage ribosomique (ribosome footprint ou ribosome profiling)
Le profilage ribosomique a révolutionné l’étude de la traduction des ARNm. Cette technique a été adaptée à différents systèmes cellulaires et organismes pour étudier le contrôle de la traduction face à différentes conditions de stress, action médicamenteuse ou dans le cadre de processus biologiques fondamentaux [29–31].
3.1.1 Principe
Depuis longtemps, les empreintes laissées par les nucléases sont reconnues comme un moyen de déterminer la position des ribosomes sur les ARNm en cours de traduction [32]. Dans le profilage ribosomique, il est possible de déterminer avec précision la position des ribosomes sur les ARNm, car seuls les fragments d’ARNm protégés par des ribosomes seront séquencés (Fig. 1A). Le traitement par des antibiotiques fige les ribosomes en phase d’initiation ou d’élongation de la traduction, ce qui masque physiquement 28 à 30 nucléotides du transcrit et les protège de la digestion par des nucléases. Cette trentaine de nucléotides forme une empreinte ou un motif d’occupation sur le transcrit.
Le profilage ribosomique permet de définir précisément les sites de l’initiation de la traduction. Cependant, la présence de plusieurs cadres de lecture chevauchant ou l’initiation sur des codons non-AUG en complique l’analyse. Pour contourner cette difficulté, le traitement par la harringtonine ou la lactimidomycine n’affecte pas l’élongation, mais immobilise les ribosomes spécifiquement présents sur les sites d’initiation de la traduction, révélant ainsi les cadres de lecture. L’utilisation de fortes doses de puromycine est utilisée comme contrôle, car elle provoque une terminaison rapide et prématurée de la traduction, et une perte des empreintes des ribosomes avant et après les sites d’initiation. L’utilisation combinée de ces trois drogues permet, non seulement de séquencer 28 à 30 nucléotides des transcrits liés aux ribosomes, mais aussi de déterminer l’abondance des ARNm. Des approches bio-informatiques comme le fragment length organization similarity score (FLOSS) [30] permettent de filtrer les séquences en fonction de la taille des fragments protégés par les ribosomes et de vérifier que les empreintes faites par les ribosomes soient bien celles de ribosomes fonctionnels 80S.
La technique de profilage ribosomique a été adaptée pour étudier l’impact de facteurs trans, agissant spécifiquement sur la traduction comme les microARN et les ribonucléoprotéines [33,34]. Sur un plan plus intégré, elle a permis d’évaluer la réponse traductionnelle de la levure face au stress oxydatif, l’action des inhibiteurs de la voie mTOR sur la multiplication les fibroblastes embryonnaires de souris [35], l’impact d’un choc thermique ou d’un stress protéotoxique sur la méiose des cellules de mammifères [36] ou bien encore la progression dans le cycle cellulaire de cellules humaines [37].
3.1.2 Limites de la méthode
Le profilage ribosomique comprend de nombreuses étapes (traitement des lysats par antibiotiques, digestion des lysats par des nucléases, élimination des ARNm non associés aux ribosomes, purification des ARNm liés), il n’est donc pas adapté au traitement de nombreux échantillons simultanément. Le choix de la nucléase peut induire des biais significatifs dans le profil des séquences nucléotidiques protégées par les ribosomes [38] (Tableau 1). Par exemple, la RNAse A ne clive qu’en 3' des nucléotides C et U.
Avantages et limites des méthodes d’analyse des ARNm en cours de traduction.
| Ribosome Profiling | Polysome Profiling | TRAP | |
| Advantages | Méthode très résolutive qui permet de déterminer le taux d’occupation des ribosomes par analyse RSS, la position des sites d’initiation, la vitesse de traduction d’un ARNm, l’identification des ORFs, l’identification des sites de pause L’approches bio-informatique par « fragment length organization similarity score » (FLOSS) limite les faux-positifs |
Le niveau d’expression des ARN peut être quantifié par séquençage haut débit. Permet l’identification de transcrits traduits, au niveau du génome entier. Se prête aussi à une approche gène-candidat | Permet l’étude de la traduction en condition physiologique et pathologique |
| Limites | |||
| Méthode | Nécessite une grande quantité de matériel de départ Le choix de la RNAse peut induire des biais significatifs dans le profil des séquences protégées par les ribosomes. Risque de faux positifs : contamination des fractions du gradient par des ARN ribosomaux |
Nécessite de grandes quantités d’échantillons Nécessite des équipements spécifiques et coûteux pour l’ultracentrifugation et un système de fractionnement reproductible Risque de faux-positifs : contamination des fractions du gradient par des ARN ribosomaux |
Risques de faux-positifs par fixation aspécifique à l’anticorps d’immunoprécipitation |
| Analyse | Ne permet pas de distinguer les monosomes et les polysomes L’identification des fragments d’ARN protégés dépend de l’annotation du génome d’intérêt Amplification des fragments protégés, très courts, particulièrement délicate En analysant les ribosomes individuels et leurs empreintes plutôt que des transcrits entiers, il y a une perte d’informations par rapport au profilage des polysomes Ne permet pas d’évaluer l’efficacité de traduction des ARNm mais uniquement de mesurer la densité de ribosomes sur un transcrit donné |
La présence de cadres de lecture (ORF) des régions 5'UTR sur lesquels se fixent des ribosomes peut fausser l’analyse | Cette approche ne permet pas de distinguer les monosomes et les polysomes |
L’identification des fragments d’ARN qui sont protégés par le ribosome dépend de la qualité de l’assemblage et de l’annotation du génome. Les fragments d’ARN protégés par les ribosomes sont très courts, par conséquent les méthodes de séquençage en paired-end ne peuvent pas être utilisées. Une caractéristique inhérente à la méthode de profilage ribosomique est que la digestion par la nucléase dégrade les extrémités 5’ et 3’UTR des transcrits, qui peuvent contenir des informations régulatrices importantes.
Il est également possible d’utiliser, dans les échantillons contrôles, des inhibiteurs de l’initiation de la traduction comme la patéamine A qui affecte la forme libre du facteur d’initiation de la traduction eIF4A, ou des inhibiteurs non spécifiques comme l’EDTA (acide éthylène diamine tétra-acétique) qui bloquent la fixation des ribosomes aux ARNm. La comparaison avec ces conditions contrôles permet alors de vérifier que les séquences identifiées dans les échantillons d’intérêt proviennent réellement d’empreintes ribosomales et non d’artéfacts. Cependant, ces inhibiteurs peuvent provoquer un décalage du cadre de lecture en modifiant la distribution des ribosomes sur les régions codantes et de ce fait induire un biais. Ainsi, l’utilisation de la cycloheximide aurait tendance à enrichir les empreintes de ribosomes situées à proximité des sites d’initiation de la traduction.
Le ribosome release score (RSS) et le disengagement score (DS) ont été développés pour discriminer, parmi les ARNm associés à des ribosomes, ceux qui sont codants de ceux qui ne le sont pas [39,40]. Le RSS se base sur l’hypothèse que sur les transcrits codants, le nombre de ribosomes associés en aval du codon stop décroît drastiquement, tandis que l’association des ribosomes avec des ARN non codants reste homogène tout au long de la séquence.
3.2 Profilage polysomique (polysome profiling)
3.2.1 Principe
L’exploration du traductome peut être faite par sédimentation d’ARNm à haute vitesse sur un gradient de sucrose. Cette approche consiste à séparer les ARNm associés aux ribosomes des ARN libres non associés (Fig. 1A). Ainsi, les ARNm sont séparés en fonction du nombre de ribosomes qui leur sont associés.
Des études à l’échelle du génome entier ont utilisé le profilage polysomique pour comparer l’état de traduction des ARNm dans différents types de cellules (saines par rapport à des cellules cancéreuses), ou des cellules soumises à des conditions spécifiques qui mettent en évidence des ARNm dont la traduction est régulée. En effet, à travers cette technique, tout changement d’abondance observé pour un ARNm du gradient est indicatif d’une régulation au niveau traductionnel. Dans la pratique, des profils à basse résolution sont obtenus en comparant la teneur en ARNm issus des fractions de densité élevée du gradient, qui contiennent les polysomes, à des fractions du gradient de densité plus faible, qui contiennent des ARNm libres, non traduits et les petits ARNs codants ou non codants.
Ensuite, les niveaux d’expression des ARNm liés aux ribosomes sont quantifiés de manière globale ou par approche gène-candidat par qRT-PCR [41]. L’inconvénient de l’analyse quantitative par qRT-PCR est qu’elle est limitée à quelques gènes à la fois, mais c’est la moins onéreuse. Dans le cas de l’identification par microarrays, les sondes oligonucléotidiques doivent être conçues individuellement pour chaque espèce et l’identification se limite aux produits d’épissage couverts par les oligonucléotides qui se trouvent sur la puce. Depuis plus récemment, les ARN sont identifiés et quantifiés par séquençage ARN à haut débit (RNAseq). Grâce au séquençage haut débit, il est possible d’analyser jusqu’à des dizaines de millions de séquences en parallèle. Cette profondeur de séquençage permet de mesurer l’abondance d’ARNm spécifique à partir d’un échantillon biologique complexe.
Les premiers exemples de profilage polysomique concernent les changements globaux du traductome dans différentes conditions physiologiques chez la levure [8]. L’approche a également été adaptée aux cellules de mammifères pour étudier, par exemple, la réponse à divers agents qui induisent l’apoptose [42], le stress du réticulum endoplasmique (RE) [43], l’hypoxie [44], l’infection virale [45], les rayonnements UV [46]. Ces agents stimulent le recrutement sélectif d’ARNm dans les fractions polysomales comme par exemple des ARNm codant les enzymes de réparation de l’ADN [46]. L’analyse comparative de lignées cellulaires leucémiques [47] ou lors de la reprogrammation de la traduction au cours de la différenciation des adipocytes [48] démontre le recrutement sélectif d’ARNm qui sont traduits en protéines, offrant ainsi des cibles potentielles au développement de nouveaux médicaments et traitement médicaux.
3.2.2 Limites de la méthode
Bien que le profilage polysomique soit utilisé depuis les années 1960, cette technique présente certaines limitions, dont les majeures sont présentées dans le Tableau 1. En effet, elle nécessite des équipements spécifiques tel qu’un système de fractionnement performant et reproductible. De plus, le profilage polysomique nécessite une grande quantité de cellules de départ, afin de récupérer une quantité raisonnable d’ARN à l’issue du fractionnement sur le gradient de sucrose. Une alternative à l’utilisation d’un gradient de sucrose consiste à purifier les ribosomes par chromatographie d’affinité (TRAP) en utilisant des anticorps dirigés contre une protéine de la sous-unité 60S des ribosomes, fusionnée à une étiquette moléculaire telle que HA (hémagglutinine), FLAG (Asp-Tyr-Lys-Asp-Asp- Asp-Asp-pLys) ou GFP (Green Fluorescent Protein).
3.3 TRAP (Translating Ribosome Affinity Purification) : purification des ribosomes par affinité à partir de tissus
Dans les tissus, il est impossible d’isoler les ARNm liés aux polysomes dans un type cellulaire donné, sans risque de contamination par les cellules environnantes. Pour y remédier, Heiman et al. ont développé une technique de purification des ribosomes par chromatographie d’affinité, appelée Translating Ribosome Affinity Purification (TRAP) [49].
3.3.1 Principe
Cette technique permet d’identifier in situ les ARNm en cours de traduction en comparant les ARNm traduits dans un type cellulaire spécifique avec ceux qui sont traduits dans l’ensemble du tissu, ou bien dans des conditions physiopathologiques distinctes. Dans cette approche, l’organisme modèle est génétiquement modifié, afin qu’une protéine de la sous-unité 60S du ribosome soit porteuse d’une étiquette (Fig. 1C). En effet, la sous-unité ribosomale 60S est recrutée à l’ARNm une fois que la petite sous-unité 40S a reconnu le codon AUG initiateur. La plus petite entité reconnue dans la méthode TRAP est donc le monosome. Une fois le tissu d’intérêt isolé, les cellules sont lysées et les ribosomes sont isolés sur colonne d’affinité. Il est alors possible d’identifier et de quantifier les ARNm associés par microarray ou par analyse RNAseq. C’est ainsi qu’à partir de souris transgéniques dont la protéine ribosomale L10a (Rpl10a) de la sous-unité ribosomale 60S est couplée à la protéine fluorescente enhanced green fluorescent protein (eGFP) dans le striatum, une centaine d’ARNm permettant de discriminer quatre types de neurones a été identifiée [49]. Grâce à cette technique, l’équipe de Halbeisen a recherché le niveau de corrélation entre le transcriptome et le traductome dans différentes conditions de stress. Ainsi, cette équipe a démontré, chez la levure Saccharomyces cerevisiae, que dans les cellules soumises à un stress intense, il y a une forte coordination entre transcriptome et traductome, alors que les cellules soumises à un moindre stress montrent des variations principalement au niveau du traductome [50]. Ces expériences indiquent que la régulation de la traduction est un élément-clé pour une adaptation rapide à des changements subtils de l’environnement.
Cette méthode s’est ensuite considérablement améliorée grâce au développement de modèles d’expression conditionnelle qui permettent un contrôle spatio-temporel du marquage des ribosomes, telles que les souris Ribo-Tag (Fig. 1D). Ces souris ont notamment permis d’explorer le traductome des cellules de Leydig stimulées in vivo par la LH [26]. Plus récemment, l’équipe de Hupe a créé deux lignées de souris exprimant la recombinase Cre : une lignée l’exprimant sélectivement dans la cellule endothéliale et une autre lignée l’exprimant dans le cerveau et les reins [51] afin d’étudier les mécanismes qui régulent la différenciation des cellules endothéliales au cours du développement embryonnaire. De plus, Nectow et al. ont développé deux stratégies complémentaires [52]. Dans la première approche, dénommée Retro-TRAP (Fig. 2A, à gauche), une lignée de souris transgénique exprime la protéine de fusion NBL10 (Rpl10a couplée en N-terminal à un nanobody anti-GFP) sous le contrôle du promoteur de la synapsine humaine (SYN-NBL10), entraînant l’expression de NBL10 dans tous les neurones. Après l’injection d’un virus traceur rétrograde exprimant la GFP tel que le virus de l’adénovirus canin (CAV)-GFP dans une région cérébrale définie, il est possible de disséquer les loci présynaptiques et d’identifier les neurones qui ont envoyé leurs projections dans la zone d’injection, sur la base des ARNm traduits, après une immunoprécipitation anti-GFP. En effet, le CAV-GFP transduit spécifiquement les neurones. La deuxième approche s’appuie sur cette stratégie pour définir les profils de traduction de sous-ensembles de neurones (Fig. 2A, à droite). Dans une lignée de souris exprimant la recombinase Cre-dans un type de neurone spécifique est injecté un virus AAV-FLEX-NBL10 conçu pour exprimer de manière conditionnelle la construction NBL10 dans la région souhaitée du cerveau. La protéine de fusion NBL10 peut alors s’exprimer et s’intégrer dans les ribosomes. Les neurones l’exprimant sélectivement sont révélés là encore par injection d’un CAV-GFP. Ainsi, seuls les neurones doubles positifs (exprimant à la fois la GFP et la protéine de fusion NBL10) peuvent être récupérés par immunoprécipitation avec un anticorps anti-GFP. À partir de ces deux approches, une analyse d’expression génique par RNAseq est possible en comparant les ARNm marqueurs spécifiques de la région du cerveau considérée (Rétro-TRAP) et les gènes des neurones spécifiques de cette région (Rétro-TRAP cellule-spécifique).
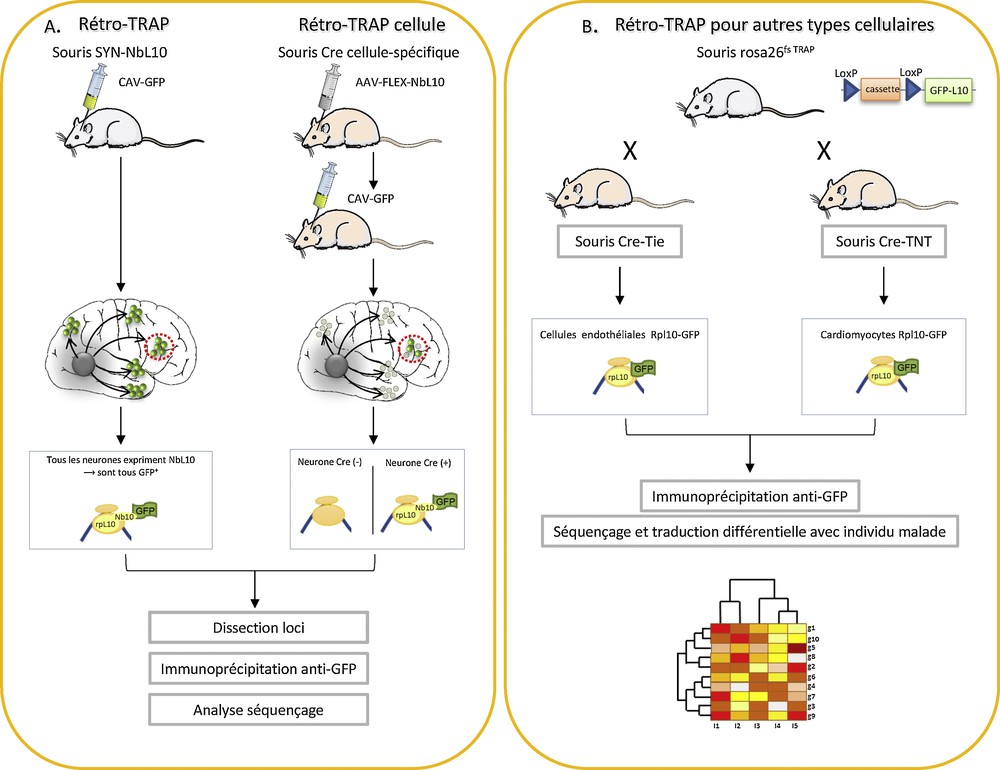
Représentation schématique des stratégies expérimentales de la méthode TRAP. A. Le Rétro-TRAP consiste à utiliser des souris transgéniques exprimant de manière constitutive la protéine ribosomale NBL10 dans tous les neurones, grâce au promoteur de la synapsine. Un adénovirus canin CAV-GFP injecté dans une région cérébrale spécifique (cercle gris), circule de manière rétrograde du site d’injection jusqu’à différentes structures du cerveau, en transduisant les neurones spécifiquement. La dissection d’un locus (zone pointillée) permet d’identifier les neurones NBL10-GFP qui se sont projetés dans la zone d’injection, par immunoprécipitation des ribosomes sur des billes anti-GFP. Le Rétro-TRAP cellule-spécifique consiste à croiser les souris SYN-NbL10 avec des souris cre-dépendante conditionnelle dans une sous-population de neurones. L’injection d’un adénovirus AAV-FLEX-NBL10 dans une région spécifique du cerveau permet de révéler les ribosomes cre-double-positifs, qui possèdent à la fois la protéine de fusion NBL10 et le marquage GFP dans un locus spécifique du cerveau (zone pointillée), par immunoprécipitation sur billes anti-GFP. B. Retro-TRAP dans les cellules non-neuronales. Cette approche consiste à marquer in vivo les ribosomes d’un type cellulaire spécifique dans un tissu. Les recombinants conditionnels sont obtenus par croisement de souris Rosa26fs TRAP, qui contient un transgène GFP-Rpl10 dont l’expression est entravée par l’ajout d’une cassette Néo loxée, avec des souris Cre-Tie ou Cre-TNT pour révéler l’expression de la protéine ribosomale GFP-Rpl10, après excision de la cassette Néo, respectivement dans les cellules endothéliales ou dans les cardiomyocytes.
De même, Zhou et al. ont généré des souris Rosa26fsTRAP, chez qui la protéine ribosomale Rpl10a est couplée à la GFP pour marquer les cellules souches embryonnaires (Fig. 2B) [53]. Puis ils ont analysé l’expression spécifique de la GFP-Rpl10 dans les cellules endothéliales Cre-Tie et dans les cardiomyocytes Cre-troponine (TNT). Les données de séquençage suggèrent une expression différentielle de plus de 1500 gènes entre l’analyse Tie-TRAP des cellules endothéliales et TNT-TRAP des cardiomyocytes. Les auteurs observent également un différentiel de traduction entre ces données et celles de cardiomyocytes issus de souris ayant subi un pontage aortique expérimental. Ce différentiel de traduction de certains ARNm a été confirmé par séquençage et qRT-PCR, montrant que l’analyse TRAP est appropriée pour l’identification de gènes différentiellement traduits dans un modèle pathologique (Fig. 2B).
La technique TRAP présente l’avantage incomparable de visualiser la traduction dans des conditions physiologiques ou pathologiques, in vivo. Contrairement aux approches traditionnelles, elle combine la détection simultanée de tous les ARNm traduits avec la spécificité du type cellulaire. L’information obtenue est spécifique d’un type cellulaire, tout en minimisant les risques de dégradation des ARNm qui peut survenir au cours des longues procédures de séparation exposées dans les autres approches ci-dessus.
3.3.2 Limites de la méthode
Cette approche isole à la fois les monosomes et les polysomes. Elle n’est donc pas aussi résolutive que le profilage polysomal (Table I). La principale source de faux-positifs est due à la présence de certains ARNm très abondants adhérant de façon non spécifique à la colonne de purification par affinité. En effet, Doyle et al. ont observé qu’un petit nombre d’ARNm était constamment enrichi dans tous les ensembles de données analysées, même chez les souris témoins non transgéniques [54]. Les auteurs indiquent que certains ARNm pourraient interagir avec des anticorps monoclonaux ou avec les billes des colonnes de purification en l’absence de la protéine de fusion eGFP. De plus, certaines lignées de souris transgéniques présentent une expression de la protéine Cre qui n’est pas suffisamment spécifique du type cellulaire ciblé.
En dépit de ces limitations, la méthode TRAP est un outil qui ouvre de nombreuses perspectives pour l’étude de la traduction en conditions physiologiques et pathologiques.
4 Perspectives pour l’analyse du traductome
L’ensemble des technologies exposées dans cette revue permet de répondre à des questions fondamentales concernant la dynamique de traduction des ARNm. Par exemple, la spectroscopie par résonance magnétique nucléaire (RMN) couplée à la spectrométrie de masse a permis récemment d’étudier le repliement co-traductionnel chez E. coli [55]. Cette approche permet une compréhension de la dynamique conformationnelle d’une chaîne naissante liée à un ribosome et ainsi de comprendre à l’échelle atomique les contraintes moléculaires appliquées à ce processus. Par ailleurs, on peut également s’interroger sur le nombre de protéines qui peuvent être synthétisées par un seul ARNm. En effet, certaines classes de protéines, tels que les récepteurs ou les protéines régulatrices, pourraient être traduites beaucoup plus efficacement que d’autres, bien que faiblement exprimées au niveau transcriptionnel. En théorie, la néosynthèse de quelques nouvelles molécules de récepteurs pourrait suffire à modifier les caractéristiques de signalisation dans une cellule donnée. Ainsi, des transcrits de faible abondance pourraient avoir un effet physiologique important, relayé par l’efficacité de leur traduction.
Classiquement, les méthodes d’analyse du traductome négligent les ARNm associés aux monosomes. Cependant, des données récentes suggèrent que l’importance du monosome dans l’efficacité de traduction est maintenant à reconsidérer [56].
L’avènement de la technologie TRAP offre maintenant la possibilité passionnante de pouvoir découvrir des variations dans le traductome entre différentes cellules d’un même tissu, et entre cellule saine et malade, entre statuts physiologiques différents, et même entre compartiments subcellulaires distincts [57,58]. À l’avenir, le perfectionnement de l’analyse du traductome nécessitera de miniaturiser les méthodes analytiques, afin d’isoler les ribosomes plus facilement, plus rapidement et de façon plus fiable, à partir d’un petit nombre de cellules ou même à l’échelle d’une cellule unique. Enfin, l’approfondissement des méthodes biostatistiques et bio-informatiques sera décisif pour générer des modèles de régulation et de signalisation réalistes. Cette standardisation des méthodes d’analyse du traductome popularisera alors son utilisation, aussi largement que l’est aujourd’hui celle du transcriptome.
Remerciements
Les auteurs remercient le département PHASE de l’Institut national de la recherche agronomique, et la Région Centre qui financent la thèse d’A.T. Nous remercions également le Centre national de la recherche scientifique.