


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Disharmonious overgrowth syndromes are characterized by asymmetrical overgrowth. Diagnosis can be made early during embryonic development, at birth or later [1, 2]. These syndromes are mostly due to somatic mutations [3]. Mechanistically, a cell acquires a mutation during embryogenesis that affects the regulation of tissue growth. Cells (with or without the mutation) will continue to divide during embryonic development to form organs and tissues. This will result in an individual with two genetically distinct cell populations. Consequently, only one part of the body will be affected by the overgrowth.
The severity of the symptoms depends on the affected gene and its role in signaling pathways involved in the regulation of cell growth, survival and/or proliferation. Clinical manifestations will also dependent on the location and timing of the post-zygotic mutation. Moreover, the later the mutation appears during embryogenesis, the more localized the damage will be, but of course this does not always correlate with the severity of the phenotype.
In the last decade, new sequencing methods have allowed the identification of several genes involved in segmental overgrowth syndromes. One of the pathways frequently found with mutations in these syndromes is the PI3-Kinase pathway.
2. The PI3-Kinase signaling pathway
PI3-Kinases are a large family of enzymes that phosphorylate membrane phospholipids (phosphoinositides) at the 3-position of the inositol group. Phosphatidylinositol (3,4,5)-triphosphate (PIP3) levels are also regulated by phosphatases that hydrolyze them to phosphatidylinositol (3,4)-bisphosphate (PIP2). One of the main negative regulators of this pathway is the phosphatase PTEN (phosphatase and TENsin homolog).
The human PI3-Kinase family is composed of eight members divided into four classes according to their structure, regulation, and substrate (class Ia, Ib, II and III). Phosphoinositides allow the recruitment at the membrane of several proteins that possess a particular motif: the PH domain (Pleckstrin homology). Thus, the serine/threonine kinase AKT is recruited and phosphorylated by another protein kinase, phosphatidylinositol 3-dependent kinase 1 (PDK1) on threonine at position 308. It is then phosphorylated by mTORC2, on serine at position 473. AKT activation affects numerous effectors regulating cell proliferation, cell size and survival (including mTOR) (Figure 1).
PI3K-AKT—mTOR signaling pathway and inhibitors undergoing (or discontinued) clinical trials in PROS syndrome. This pathway is downstream of the growth factor receptor. The receptor activates PI3-Kinase, which phosphorylates phosphatidylinositol biphosphate (PIP2) to phosphatidylinositol triphosphate (PIP3). This allows the recruitment of AKT, which is phosphorylated (by PDK1 and mTORC2) and thus activated. AKT in turn activates many substrates including mTOR. PTEN is a phosphatase that acts as an inhibitor of PI3K, countering the action of PI3K by transforming PIP3 into PIP2. Numerous molecules are being tested in the treatment of PROS syndrome, which are inhibitors of the various players in the PI3K-AKT mTOR pathway: inhibitors of PI3-Kinases (Taselisib, Alpesilib), AKT inhibitors (Miransertib) and mTOR inhibitors (Sirolimus).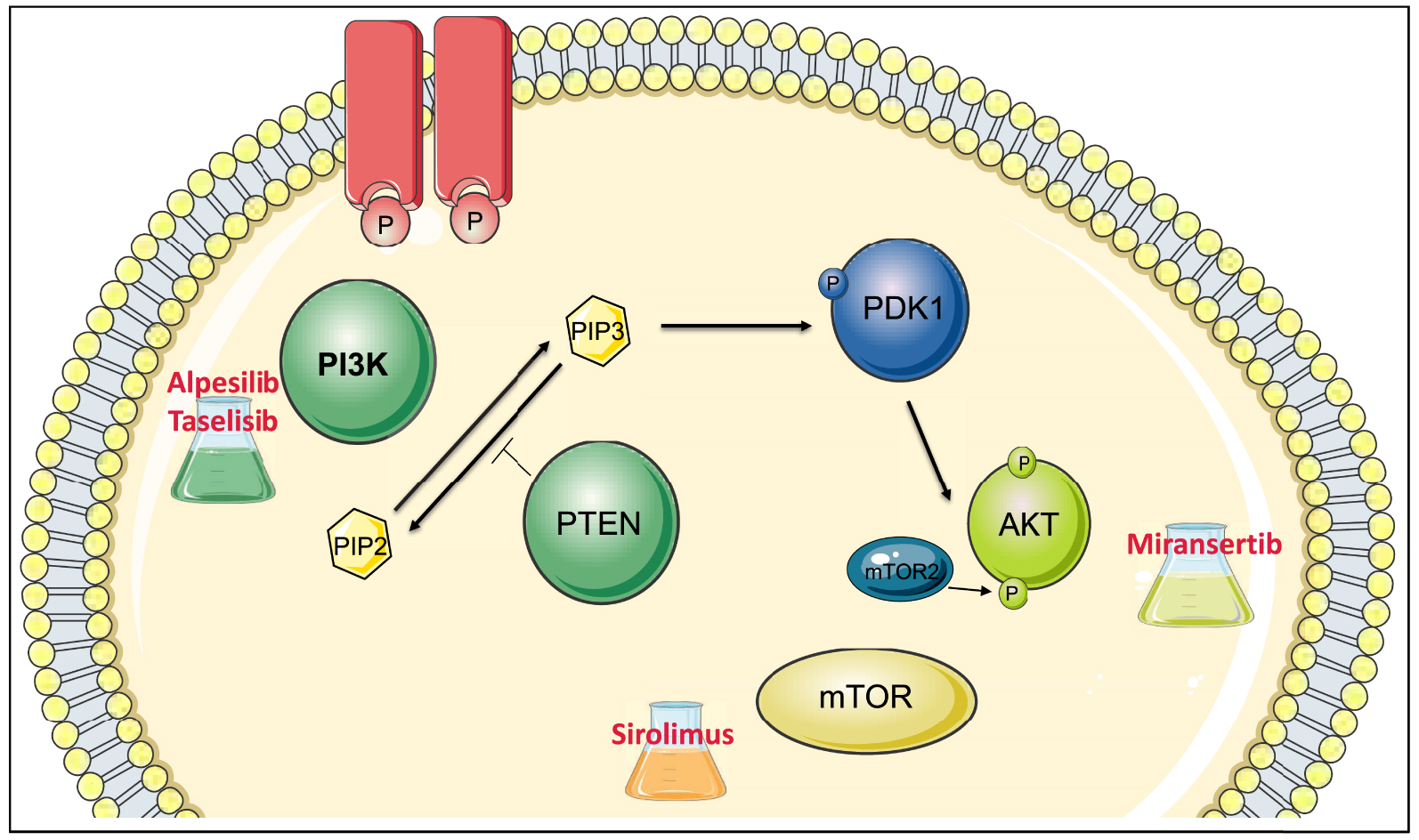
The various mouse models of PROS syndrome. Different mouse models have been created, using transgenes induced during embryonic or postnatal life.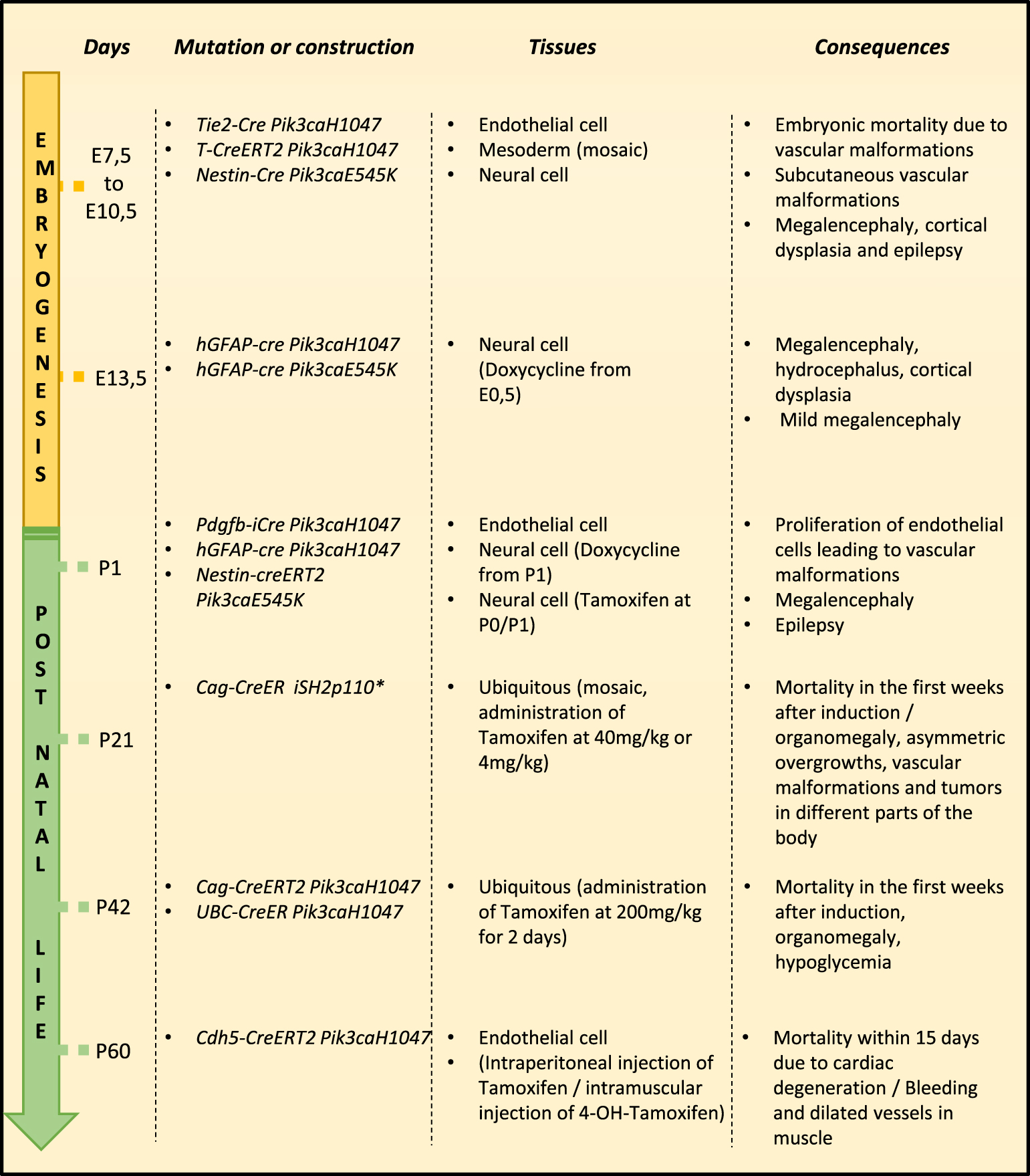
Class Ia PI3K is composed of two subunits, a catalytic subunit, p110 (α, β, or δ) and a regulatory subunit (p85α or β, p50α or p55β or γ). The PIK3CA gene encodes for the p110α catalytic subunit. Somatic activating mutations in this gene cause overgrowth syndromes and cancers. Highly activating germline mutations in this gene are likely to be predominantly lethal, as there are only very few cases reported [4]. PIK3CA mutations were identified in cases of disharmonious overgrowth for the first time in 2012 [5].
3. PIK3CA-related overgrowth spectrum
Overgrowth syndromes linked to PIK3CA mutations were grouped into an entity called PROS (PIK3CA-Related Overgrowth Spectrum) at a consensus conference organized by the National Institute of Health (NIH) in 2014 [6]. Several pathologies with very heterogenic phenotypes were then grouped under the name PROS syndrome: FAO (fibroadipose overgrowth), HHML (hemihyperplasia-multiple lipomatosis), DMEG (dysplatic megalencephaly), MCAP (megalencephaly-capillary malformation) and CLOVES (congenital lipomatous asymmetric overgrowth of the trunk with lymphatic, capillary, venous, and combined-type vascular malformations, epidermal naevi, scoliosis/skeletal and spinal anomalies, OMIM #613089) and K-T syndrome (or Klippel-Trenaunay OMIM #149000) which was included in 2016.
PROS syndrome is therefore an entity that gather patients with very diverse clinical conditions. The clinical spectrum can range from isolated macrodactyly to more disorders such as observed in CLOVES patients: scoliosis, vascular malformations, lipomatous malformations… All tissues derived either from ectoderm (nervous system, epidermis…) or mesoderm (muscle, dermis, adipose tissue…) may be affected. PROS can also affect the central nervous system with megalencephalia or focal cortical dysplasia.
More than thirty activating mutations in the PIK3CA gene have been identified in PROS [7]. They are scattered along the gene. Mutations in histidine 1047 (His1047) and glutamate 545 (Glu545) are the most common mutation, as in cancers [7].
4. Animal models
The majority of animal models involving PIK3CA mutations are tumorigenesis models. However, several approaches have been used to obtain a mouse model of PROS and to understand its pathophysiology precisely (Figure 1). Various strategies have been implemented with different transgenes and different timing.
4.1. During embryogenesis
In patients with PROS syndrome, the mutation appears during embryogenesis. A first approach has been to create mouse models allowing overactivation of the PI3K-AKT-mTOR pathway from embryogenesis.
The ubiquitous induction (using CMV-Cre transgenic mice) of the H1047R mutation or a MYR-p110α construct (allowing p110α to be addressed to the membrane and thus activated) from the zygote stage results in death in utero. This death is due to abnormalities in vessels and hemorrhage [8, 9]. These data confirm that germline mutations in the PIK3CA gene are very likely to be lethal.
To overcome this limitation, some teams induced the mutation specifically in certain tissues or leaflets during embryogenesis (Figure 2). Castillo et al. used an inducible model. They used mice expressing a Cre-recombinase under the control of the transcription factor T promoter (Brachyury) to obtain mosaic transcription of PIK3CAH1047R in the mesoderm. By administration of a low dose of tamoxifen to pregnant mice, they obtain an expression from embryonic day 7.5 [10]. These mice were born with vascular malformations and localized bleeding in various parts of the body such as PROS patients but without visible hypergrowth. The authors aged these mice and treated them with rapamycin. Rapamycin is an inhibitor of a player in the PI3-Kinase signaling pathway, mTOR. They were able to observe a reduction in the volume of vascular malformations (by about 25%) as well as an arrest of bleeding.
Castel et al. as well as Di Blasio et al. adopted a similar murine model by inducing PIK3CAH1047R transcription in endothelial cells leading to embryonic death due to vascular malformations [11, 12].
As mentioned above, PROS can also affect the central nervous system. In order to study the mechanisms leading to brain malformations, Roy et al. created several mouse models. One of these models allows the expression of the E545K mutant in neural progenitor cells at embryonic day E8.5 (through the Nestin-Cre promoter) [13]. In the mutant mice the authors observed severe megalencephaly, increased neural cell size, cortical dysplasia and epilepsy. The authors then created transgenic mice expressing the H1047R and E545K mutations of PIK3CA later in embryogenesis, at embryonic day 13.5 (hGFAP-Cre promoter). These two mutations result in distinct phenotypes. Mice expressing the H1047R mutant present severe hydrocephalus, severe megalencephaly, cortical dysplasia and die before weaning. Mice expressing the E545K mutant survive to adulthood and do not show hydrocephalus but mild megalencephaly.
4.2. In post-embryonic life
Another strategy to obtain an animal model of PROS was to induce PIK3CA mutations at different stages of post-embryonic life.
Roy et al. created a third mouse model, mice expressing the mutant H1047R and E545K early in postnatal life, at P1 (using the inducible hGFAP-Cre and Nestin-CreER promoters). Phenotypes are less severe (or absent) than when mutants are expressed from embryogenesis. Indeed, the mice expressing the H1047R mutant show no difference from the control mice. In mice expressing the E545K mutant, the authors did not observe any increase in brain size, but the mice did experience epileptic seizures. The authors used the two mouse models with epileptic seizures (the mice expressing the E545K mutant at E8.5 and P1) to test a class I pan-PI3Kinase inhibitor, BKM120. This inhibitor drastically reduces the number and duration of epileptic seizures.
Similarly, Castillo et al. induced the PIK3CA H1047R mutation postnatally in P1 mice (by administration of tamoxifen) in endothelial cells using the inducible Pdgfb-iCreER promoter [10]. Mice develop retinal endothelial cells, proliferation, loss of arteriovenous markers, and loss of pericytes. The authors used these mice as a preclinical model to test the impact of rapamycin on vascular malformations. In this model, they observe a reduction of the number of endothelial cells as well as their proliferation.
Castel et al. created a mouse model in which the H1047R mutant is ubiquitously expressed (with the CAG, CMV early enhancer/chicken β actin promoter) and tamoxifen dependent (CAGG-CreER). These mice develop vascular malformations and die early [11].
Likewise, overexpression of the H1047R mutation of PIK3CA in a ubiquitous (via the Ubiquitin C promoter) and tamoxifen dependent (UcreERT2) manner leads to increased weight in mice with organomegaly as well as hypoglycemia, increased systemic vascularization and early mortality [14].
These models were able to reproduce some of the lesions found in patients, but none of these models were able to summarize all the phenotypes of PROS syndrome.
Our team used a constitutively active PIK3CA construct (iSH2p110*) [15]. This construct results in a more pronounced activation of the AKT/mTOR pathway in mice than models using mutations found in patients. Breeding these mice with mice carrying CAGG-creER recombinase led us to obtain an ubiquitous activation of the mutant following tamoxifen administration. A high dose of tamoxifen (40 mg⋅kg−1) results in early and sudden death of the mice within the first week of life as observed in previous studies [14]. However, by decreasing the dose of tamoxifen (4 mg⋅kg−1) we were able to obtain mice with a lower rate of mosaicism, which survive longer (about two months) and thus recapitulate most of the phenotypes observed in the patients. Indeed, these animals develop numerous organ anomalies, vascular and lymphatic malformations, localized tumors and scoliosis. This mouse model therefore presents very similar manifestations to those observed in patients with PROS. We used this mouse model to test a specific inhibitor of p110α, BYL719 (or Alpesilib). This treatment was administered after the onset of the different malformations. We observed a halt in the progression of the various malformations, but also their regression, from the first days of treatment. We also observed an increase in the life span of the animals and an improvement in the structure of the organs. As the mTOR inhibitor rapamycin had already shown efficacy in vascular malformations, we compared its efficacy to BYL719. Rapamycin improved the survival of the mice, but we did not see significantly changes organ malformations or the various vascular malformations. These data suggest that BYL719 may be a good therapeutic option for patients with a PIK3CA activating mutation.
5. Clinical trial
Treatments currently available for patients with PROS are not very effective, consisting mainly of surgery, amputations, sclerotherapy and endovascular procedures. Genetic alterations in the PI3K/AKT/mTOR signaling pathway have frequently been identified in cancers and this pathway represents a major therapeutic target for the pharmaceutical industry. Recently developed mouse models now allow these molecules to be tested. Several molecules have been tested or are currently being studied in clinical trials to investigate their efficacy in the treatment of PROS.
5.1. mTOR inhibitor
One of the first molecules tested in PROS syndrome was an mTOR inhibitor, rapamycin, already used in clinic as an immunosuppressant. A clinical trial was conducted jointly in France, the United Kingdom and the United States (NCT02443818, EudraCT: 2014-000484-41 and NCT02428296), comprising 39 participants (22 adolescents over 16 years old and 17 adults). A modest effect was observed on malformations affecting lean tissue (muscles, connective tissues…) and those with an adipose component (with a decrease of about 7%). This treatment caused side effects quite disabling for almost 40% of participants [16].
5.2. AKT inhibitor
Another approach currently used is to block the activity of a kinase downstream p110α and upstream mTOR: AKT. Several AKT inhibitors are currently in clinical trials in oncology, including the pan-AKT inhibitor, miransertib (MK-7075 formerly ARQ092). This inhibitor was recently used in two patients with PROS [17]. It was clinically effective in one patient, with a 15% reduction in malformations, and improved the quality of life in the second patient, including a reduction in seizures. The treatment was well tolerated in both patients. This treatment is currently in a phase I/II clinical trial for patients with Proteus syndrome (due to a mosaic mutation in AKT1) as well as for patients with PROS syndrome (NCT03094832).
5.3. P110α inhibitors
Several p110α inhibitors initially developed in oncology have been or are currently being tested in patients with PROS syndrome.
5.3.1. Taselisib
Taselisib is a strong inhibitor of p110α but also of P110δ and p110γ. It has been tested in a clique trial (NCT03290092) in several centers in France. This phase Ib/IIa trial has consisted of evaluating the safety and efficacy of Taselisib in 30 patients with segmental mosaic overgrowth. It was stopped prematurely due to serious adverse events caused by the inhibition of p110γ and p110δ.
5.3.2. Alpesilib
One of the most promising perspective for treating PROS syndrome appears to be Alpelsib, which has a much higher specificity for p110α.
Our team has treated now more than 60 patients with PROS syndrome with Alpesilib (BYL719) at the Necker-Enfants Malades hospital. The first 19 patients (including children) have been published recently [15]. Some of these patients (8 of them) had previously received rapamycin without significant improvement of their condition. The results were very encouraging, with all patients showing an improvement in their quality of life, including a decrease in tiredness. All patients showed marked improvements with cessation of bleeding, improved organ function, better pain control (both patients taking opioids were able to discontinue them) and a reduction in the volume of malformations (by almost 40% after 6 months of treatment). All patients are currently receiving treatment. Patients had no or few side effects and the treatment had no impact on growth. The treatment was given at lower doses than those given in oncology (250 mg for adults and 50 mg for children), which may explain the few side effects observed.
Another team has recently published the results of the first North American patient treated with BYL719 [18]. This patient experienced an improvement in pain within the first few days and after two months of treatment the deformities had decreased by about 50%. A 17-year-old female patient with severe genital damage was first treated with rapamycin for one year without significant improvement and then treated with BYL719. Her condition then improved markedly allowing surgical reconstruction [19].
An international clinical trial (NCT04589650) will be launched soon to compare the efficacy of Alpelisib in patients with PROS. The study will include 150 participants divided into three groups: an adult group, a group of children aged 6–17 years and later a group of young children aged 2–5 years. The first two groups will be randomized with half of the participants receiving the placebo and the other half receiving the Alpesilib.
6. Conclusions and perspectives
In conclusion, PROS are serious genetic conditions that may alter the quality and quantity of life. But, thanks to the recent progress made in the understanding of disease progression, the development of powerful genetic tools to identify mutation and the discovery of potential targeted therapies open new avenues and hope for patients.
French version
1. Introduction
Les syndromes d’hypercroissance dysharmonieuse sont caractérisés par une croissance excessive et localisée d’un ou plusieurs tissus. Ils peuvent être observables dès le développement embryonnaire, à la naissance ou encore n’être visibles qu’au cours des premières années de vie [1, 2]. Ces syndromes sont majoritairement dus à des mutations somatiques [3]. Une cellule acquiert durant l’embryogenèse une mutation qui va affecter la régulation de la croissance tissulaire. Les cellules, contenant ou non la mutation, vont continuer à se diviser durant le développement de l’embryon afin de former les organes et tissus. Cela va aboutir à un individu possédant deux populations cellulaires génétiquement distinctes, avec pour conséquence seulement une partie du corps affectée par la pathologie liée à la mutation.
La gravité des symptômes dépend en partie du gène muté et de son importance dans les voies de signalisation impliquées dans la régulation de la croissance cellulaire, de la survie et/ou de la prolifération. Les atteintes cliniques sont aussi dépendantes de la localisation et du moment de l’apparition post-zygotique de la mutation. En outre, plus la mutation apparait tardivement au cours de l’embryogenèse plus l’atteinte sera localisée mais bien entendu cela ne corrèle pas toujours avec la gravité du phénotype.
Depuis une dizaine d’années, de nouvelles techniques de séquençage ont permis d’identifier un certain nombre des gènes et des voies de signalisation impliquées dans les syndromes de surcroissance segmentaire. Une des voies fréquemment retrouvées avec des mutations dans ces syndromes est la voie de la PI3-Kinase.
2. La voie de signalisation de la PI3-Kinase
Les PI3-Kinases constituent une vaste famille d’enzymes qui phosphorylent des phospholipides membranaires, les phosphoinositides, sur la position 3 du groupement inositol. Les niveaux des phosphatidylinisotol (3,4,5)-triphosphate (PIP3) sont aussi régulés par des phosphatases qui les hydrolysent en phosphatidylinisotol (3,4)-bisphosphate (PIP2). L’un des principaux régulateurs négatifs de cette voie est la phosphatase PTEN (phosphatase and TENsin homolog).
La famille des PI3-Kinases est composée de huit membres chez l’homme divisés en quatre classes d’après leur structure, leur régulation et leur substrat (classe Ia, Ib, II et III). Les phosphoinositides permettent le recrutement à la membrane d’un certain nombre de protéines qui possèdent un motif particulier : le domaine PH (Pleckstrin homology). Ainsi la sérine/thréonine kinase AKT est recrutée et phosphorylée par une autre protéine kinase, la phosphatidylinositol 3-dependent kinase 1 (PDK1) sur la thréonine en position 308. Elle est ensuite phosphorylée par mTORC2, sur la sérine en position 473. L’activation d’AKT va affecter de nombreux effecteurs régulant la prolifération cellulaire, la taille cellulaire et la survie (notamment la protéine mTOR) (Figure 1).
La voie de signalisation PI3K-AKT — mTOR et les inhibiteurs en cours (ou arrêtés) d’essai clinique dans le cadre du syndrome de PROS. Cette voie se situe en aval des récepteurs aux facteurs de croissance. Le récepteur active la PI3-Kinase, qui phosphoryle le phosphatidylinositol biphosphate (PIP2) en phosphatidylinositol triphosphate (PIP3). Cela permet le recrutement d’AKT qui va être phosphorylé (par PDK1 et mTORC2) et ainsi activé. AKT va à son tour activer de nombreux substrats dont mTOR. PTEN est une phosphatase jouant un rôle d’inhibiteur d’AKT, en contrant l’action de la PI3K par la transformation du PIP3 en PIP2. De nombreuses molécules sont à l’essai dans le traitement du syndrome de PROS, ce sont des inhibiteurs des différents acteurs de la voie PI3K-AKT mTOR : des inhibiteurs des PI3-Kinases (Taselisib, Alpesilib), des inhibiteurs d’AKT (Miransertib) et des inhibiteurs de mTOR (Sirolimus).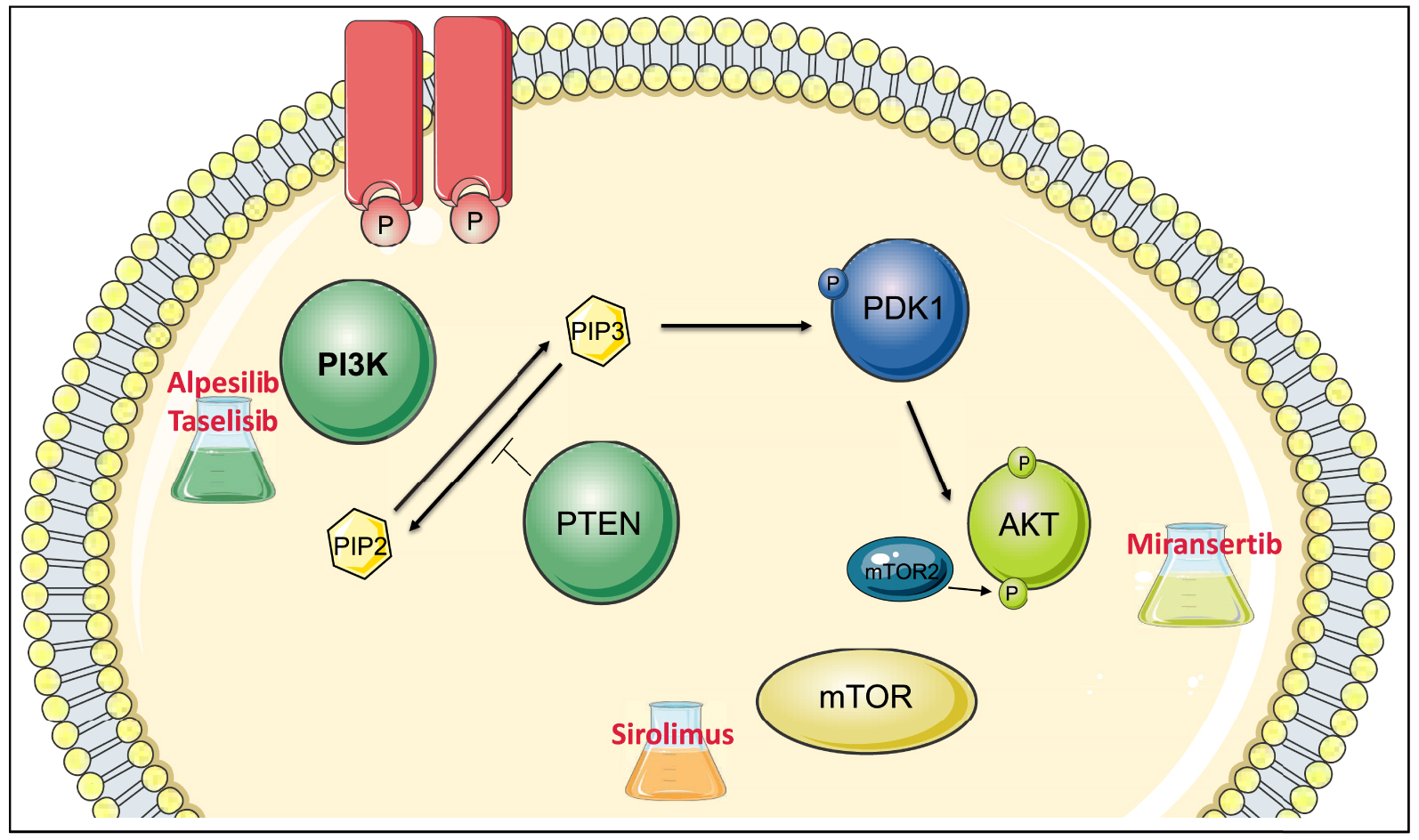
La PI3K de classe Ia est composée de deux sous-unités, une sous unité catalytique, p110 (α, β, ou δ) et une sous-unité régulatrice, p85 (p85α ou β, p50α ou p55β ou γ). Le gène PIK3CA code pour la sous-unité catalytique p110α. Des mutations activatrices somatiques de ce gène sont à l’origine de nombreuses pathologies humaines, notamment de nombreux cancers. Il est probable que des mutations germinales fortement activatrices de ce gène soient majoritairement létales ; il n’existe en effet que de très rares cas répertoriés de mutations activatrices germinales touchant le gène PIK3CA [4]. Des mutations activatrices de ce gène ont été identifiées dans des cas d’hypercroissance dysharmonieuse pour la première fois en 2012 [5].
3. Les syndromes d’hypercroissance liés à une mutation du gène PIK3CA
Les syndromes d’hypercroissance liés à une mutation du gène PIK3CA ont été regroupés dans une entité appelée PROS (PIK3CA — Relates Overgrowth Spectrum) lors d’une conférence de consensus organisé par le National Institute of Health (NIH) en 2014 [6]. Sous la dénomination syndrome de PROS ont alors été rassemblées plusieurs pathologies avec des phénotypes très différents : le FAO (Fibroadipiose overgrowth), HHML (hemihyperplasia-multiple lipomatosis), DMEG (dysplatic megalencephaly), le MCAP (megalencephaly-capillary malformation) et le CLOVES (congenital lipomatous asymetric overgrowth of the trunk with lymphatic, capillary, venous, and combined-type Vascular malformations, epidermal naevi, scoliosis/skeletal and spinal anomalies, OMIM #613089) et le syndrome K-T (ou Klippel-Trenaunay OMIM #149000) qui a été inclus en 2016.
Le syndrome de PROS est donc une entité regroupant des patients ayant des présentations cliniques très diversifiées et de gravité extrêmement variable. Ces présentations peuvent aller d’une simple macrodactylie isolée aux symptômes plus sévères retrouvés chez les patients CLOVES : scoliose, malformations vasculaires, malformations lipomateuses… Les patients atteints du syndrome de PROS sont particulièrement affectés dans les tissus issus de l’ectoderme (système nerveux, épiderme…) et le mésoderme (muscle, derme, tissus adipeux…). Le syndrome de PROS peut aussi affecter le système nerveux central, avec notamment des mégalencéphalies, des malformations du cortex cérébral et des dysplasies corticales focales.
Plus d’une trentaine de mutations activatrices du gène PIK3CA ont été identifiées dans le syndrome de PROS [7]. Elles sont disséminées sur toute la longueur du gène. On retrouve en particulier des mutations de l’histidine 1047 (His1047) et du glutamate 545 (Glu545) hélicoïdal qui sont aussi des sites ≪ hotspots ≫ de mutations dans de nombreux cancers [7].
4. Modèles animaux
La majorité des modèles animaux impliquant des mutations de PIK3CA sont des modèles de tumorigenèse. Cependant, plusieurs approches ont été utilisées afin d’obtenir un modèle murin du syndrome de PROS et comprendre précisément sa physiopathologie (Figure 1), incluant des modèles transgéniques avec des mutations constitutives ou conditionnelles. Plusieurs stratégies ont été mises en œuvre avec des différences dans le transgène utilisé et dans la temporalité.
4.1. Lors de l’embryogenèse
Chez les patients atteints du syndrome de PROS la mutation apparait au cours de l’embryogenèse, des équipes ont donc créé des modèles murins permettant l’activation de la voie PI3K-AKT-mTOR dès l’embryogenèse.
L’induction de manière ubiquitaire (par l’utilisation de souris transgénique CMV-Cre) de la mutation H1047R ou d’une construction MYR-p110α (permettant d’adresser p110α à la membrane et ainsi de l’activer) dès le stade zygote entraine la mort in utero due à des anomalies du développement des vaisseaux et des hémorragies… [8, 9]. Ces données confirment que des mutations germinales du gène PIK3CA sont très probablement létales.
Pour surmonter cette limitation, d’autres équipes ont induit la mutation spécifiquement dans certains tissus ou feuillets au cours de l’embryogenèse (Figure 2). Castillo et al. ont utilisé un modèle inductible. Ils ont utilisé des souris qui expriment une Cre-recombinase sous le contrôle du prometteur du facteur de transcription T (Brachyury) afin d’obtenir une transcription en mosaïque de PIK3CAH1047R dans le mésoderme à partir du jour embryonnaire 7.5 après une administration de tamoxifène à faible dose chez la mère [10]. Ces souris présentent à la naissance des malformations vasculaires et des saignements localisés dans différentes parties du corps rappelant ainsi le phénotype du syndrome de PROS mais sans hypercroissance visible. Les auteurs ont laissé vieillir ces souris et les ont traitées avec de la rapamycine. La rapamycine est un inhibiteur d’un acteur de la voie de signalisation de la PI3-Kinase, mTOR. Ils ont pu observer une diminution du volume des malformations vasculaires (d’environ 25%) ainsi qu’un arrêt des saignements.
Les différents modèles murins du syndrome de PROS. Différents modèles murins ont été créés, utilisant des transgènes induits au cours de la vie embryonnaire ou en post natal.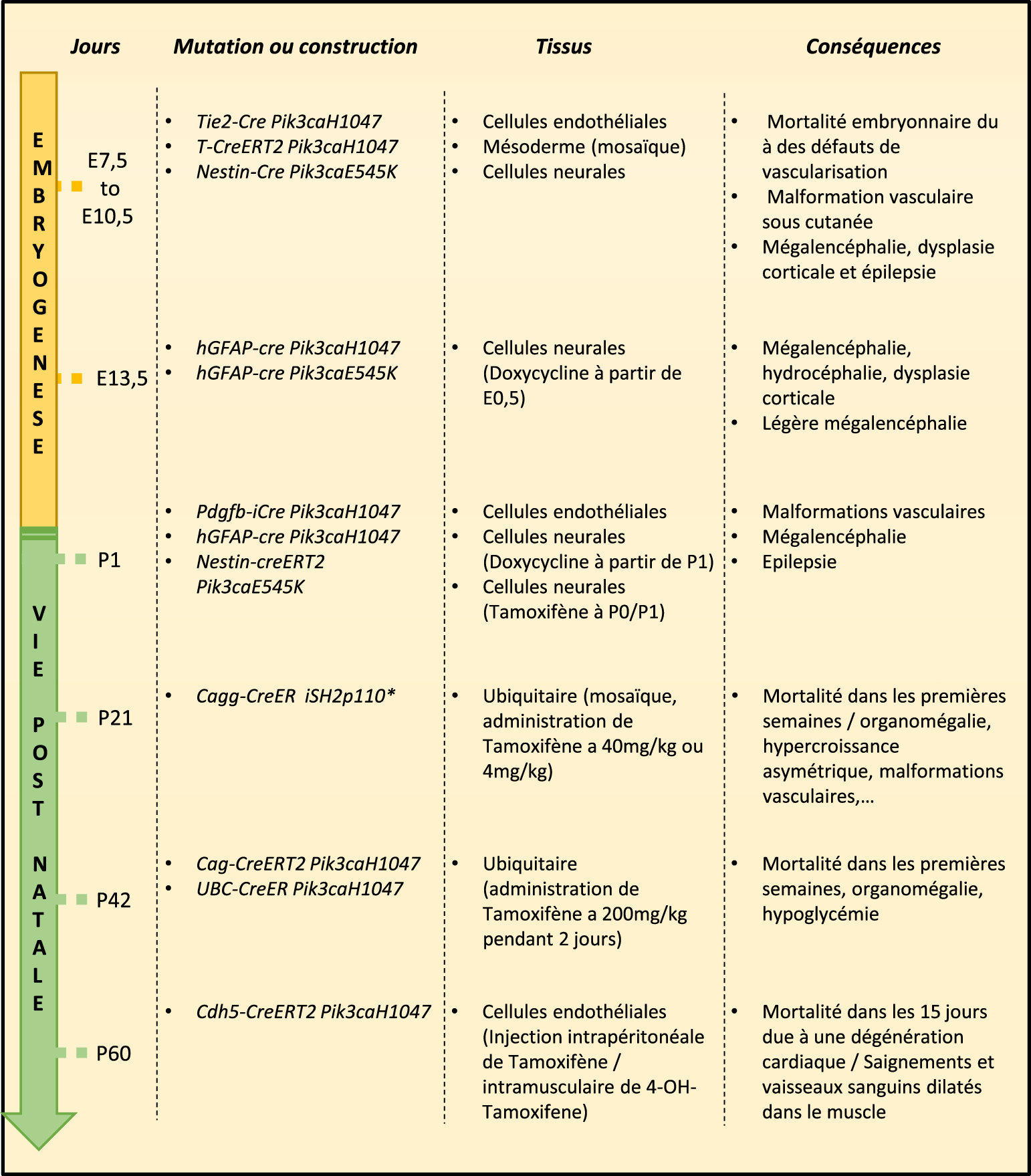
Castel et al. ainsi que Di Blasio et al. ont adopté un modèle murin assez proche en induisant la transcription PIK3CAH1047R dans les cellules endothéliales entrainant une létalité embryonnaire due à des défauts de vascularisation [11, 12].
Le syndrome de PROS peut aussi toucher le système nerveux central. Afin d’étudier les mécanismes menant aux malformations du cerveau dans le syndrome de PROS, Roy et al. ont créé plusieurs modèles murins. Un de ces modèles permet l’expression du mutant E545K dans les cellules progénitrices neurales dès le jour embryonnaire E8.5 (par le promoteur Nestin-Cre) [13]. Chez les souris mutantes, les auteurs ont pu observer une forte mégalencéphalie, une augmentation de la taille des cellules neurales, une dysplasie corticale et de l’épilepsie. Les auteurs ont ensuite créé des souris transgéniques exprimant les mutations H1047R et E545K de PIK3CA plus tardivement au cours de l’embryogénèse, au jour embryonnaire 13.5 (promoteur hGFAP-Cre). Ces deux mutations entrainent des phénotypes distincts. Les souris exprimant le mutant H1047R présentent une hydrocéphalie sévère, une forte mégalencéphalie, une dysplasie corticale et meurent avant le sevrage. Les souris exprimant le mutant E545K survivent jusqu’à l’âge adulte et ne présentent pas d’hydrocéphalie mais une légère mégalencéphalie.
4.2. Lors de la vie post embryonnaire
Une autre stratégie afin d’obtenir un modèle animal du syndrome de PROS a consisté à induire des mutations de PIK3CA à différents stades de la vie post-embryonnaire.
Roy et al. ont créé un troisième modèle murin, des souris exprimant les mutants H1047R et E545K au début de la vie post natale, à P1 (en utilisant les promoteurs inductibles hGFAP-Cre et Nestin-CreER). Les phénotypes sont moins sévères (ou absents) que lorsque les mutants sont exprimés dès l’embryogenèse. En effet, les souris exprimant le mutant H1047R ne présentent aucune différence avec les souris contrôles. Chez les souris exprimant le mutant E545K, les auteurs n’ont pas observé d’augmentation de la taille du cerveau mais les souris présentent des crises d’épilepsie. Les auteurs ont utilisé les deux modèles de souris avec des crises d’épilepsie (les souris exprimant le mutant E545K des E8.5 et à P1) pour tester un pan-inhibiteur des PI3Kinases de classe I, le BKM120. Cet inhibiteur permet de faire diminuer drastiquement le nombre et la durée des crises d’épilepsie.
De la même manière, Castillo et al. ont induit la mutation PIK3CAH1047R en post natal chez des souris à P1 (par administration de tamoxifène) dans les cellules endothéliales en utilisant le promoteur inductible Pdgfb-iCreER [10]. Les souris développent une prolifération des cellules endothéliales de la rétine, une perte des marqueurs artérioveineux et une perte des péricytes. Les auteurs ont utilisé ces souris comme modèle pré-clinique pour tester l’impact de la rapamycine sur les malformations vasculaires. Elle a permis dans ce modèle de diminuer le nombre de cellules endothéliales ainsi que leur prolifération.
Castel et al. ont créé un modèle murin permettant d’exprimer le mutant H1047R de manière ubiquitaire (avec le promoteur CAG, CMV early enhancer/chicken β actin) et tamoxifène dépendant (CAGG-CreER). Ces souris développent des malformations vasculaires et meurent précocement [11].
De même, la surexpression de la mutation H1047R de PIK3CA de manière ubiquitaire (via le promoteur Ubiquitine C) et tamoxifène dépendante (UcreERT2) entraine chez la souris une augmentation du poids avec une organomégalie ainsi qu’une hypoglycémie, une augmentation de la vascularisation générale ainsi qu’une mortalité précoce [14]. Ces modèles ont permis de reproduire une partie des lésions retrouvées chez les patients, mais aucun de ces modèles n’a permis de récapituler l’ensemble des phénotypes observés.
Notre équipe a utilisé un construit de PIK3CA constitutivement actif (iSH2p110∗) [15]. Ce construit entraine une activation de la voie AKT/mTOR plus marquée chez la souris que les modèles utilisant les mutations retrouvées chez les patients. Ce modèle permet une activation ubiquitaire du mutant par administration de tamoxifène (en utilisant le promoteur CAGG-creER). Une dose élevée de tamoxifène (40 mg⋅kg−1) entraine une mort précoce et soudaine des souris dès la première semaine de vie, comme observé dans des études précédentes [14]. Cependant, en diminuant la dose de tamoxifène (4 mg⋅kg−1) nous avons pu obtenir des souris qui présentent un plus faible taux de mosaïcisme, qui survivent plus longtemps (environ deux mois) et ainsi développent la majorité des phénotypes observés chez les patients. En effet, ces animaux développent de nombreuses anomalies des organes, des malformations vasculaires et lymphatiques, des tumeurs localisées ainsi que des scolioses. Ce modèle murin présente donc des manifestations très similaires à celles observées chez les patients atteints du syndrome de PROS. Nous avons utilisé ce modèle murin pour tester une molécule inhibitrice spécifique de p110α, le BYL719. Ce traitement a été administré après l’apparition des différentes malformations. Nous avons observé un arrêt de la progression des différentes malformations, mais aussi leur régression, dès les premiers jours de traitement. Nous avons aussi constaté un allongement de la durée de vie des animaux et une amélioration de la structure des organes. Comme l’inhibiteur de mTOR, la rapamycine avait déjà montré une efficacité dans les malformations vasculaires nous l’avons administré à notre modèle murin. La rapamycine a amélioré la survie des souris mais nous n’avons pas noté d’amélioration des malformations au sein des organes ni des différentes malformations vasculaires. Ces données suggèrent que le BYL719 pourrait être une bonne option thérapeutique pour les patients porteurs d’une mutation activatrice de PIK3CA.
5. Essai clinique
Les traitements actuellement disponibles pour traiter les patients atteints du syndrome de PROS sont peu efficaces, ils consistent principalement à de la chirurgie, des amputations, des sclérothérapies et des procédures endovasculaires. Les altérations génétiques de la voie de signalisation PI3K/AKT/mTOR ont été identifiées comme fréquemment affectées dans des cancers, cette voie représente donc une cible thérapeutique majeure pour les industriels de la pharmacie. Certains de ces composés pourraient être logiquement repositionnés dans le syndrome de PROS (Figure 2). Les modèles murins développés récemment permettent maintenant de tester ces molécules. Plusieurs molécules ont été testées ou sont en cours d’étude dans des essais cliniques afin d’étudier leur efficacité dans le traitement du syndrome de PROS.
5.1. Inhibiteur de mTOR
L’une des premières molécules testées dans le syndrome de PROS a été un inhibiteur de mTOR, la rapamycine, déjà utilisée en clinique comme immunosuppresseur. Un essai clinique a été mené conjointement en France, au Royaume-Uni et aux Etats-Unis (NCT02443818, EudraCT: 2014-000484-41 et NCT02428296). Un effet modeste a pu être observé sur les malformations touchant les tissus maigres (muscles, tissus connectifs…) et ceux à composante adipeuse (avec une diminution d’environ 7%) mais au prix d’effets secondaires assez invalidants pour près de 40% des participants [16].
5.2. Inhibiteur d’AKT
Une autre approche utilisée actuellement est celle de bloquer l’activité d’une kinase en en aval de p110α et amont de mTOR : AKT. Plusieurs inhibiteurs d’AKT sont actuellement en essai clinique en oncologie, notamment l’inhibiteur pan-AKT, le miransertib (MK-7075 anciennement ARQ092). Cet inhibiteur a été utilisé récemment chez deux patients atteints du syndrome de PROS [17]. Il s’est révélé efficace cliniquement chez un des patients, avec une diminution de 15% des malformations et a amélioré la qualité de vie du second patient, avec notamment une diminution des crises d’épilepsie. Le traitement a été bien toléré chez les deux patients. Ce traitement est actuellement en essai clinique de phase I/II pour les patients atteints du syndrome de Protée (du a une mutation mosaïque de AKT1) ainsi que pour les patients présentant un syndrome de PROS (NCT03094832).
5.3. Inhibiteurs de p110α
Plusieurs inhibiteurs de p110α développés initialement en oncologie, ont été ou sont actuellement testés chez les patients atteints du syndrome de PROS.
5.4. Taselisib
Le Taselisib est un puissant inhibiteur des PI3-Kinase de classe I notamment de p110α mais également avec une forte activité inhibitrice de p110δ et p110γ. Il a été testé dans un essai clique (NCT03290092) dans plusieurs centres en France. Cet essai de phase Ib/IIa a consisté à évaluer la tolérance et l’efficacité du Taselisib chez 30 patients présentant une hypertrophie segmentaire en mosaïque. Il a été arrêté prématurément du a des effets secondaires sévères liés à l’inhibition de p110δ et p110γ.
5.5. Alpesilib
L’une des pistes les plus prometteuses pour traiter le syndrome de PROS semble être l’Alpelsib dont la spécificité pour p110α est bien plus importante.
Notre équipe a traité plus de 60 patients atteints du syndrome de PROS avec l’Alpesilib (BYL719) à l’hôpital Necker-Enfants Malades. Les 19 premiers patients (comprenant des enfants) ont été publiés récemment [15]. Certains de ces patients (8 d’entre eux) avaient reçu de la rapamycine auparavant sans amélioration notable de leur pathologie. Les résultats ont été très encourageants, tous les patients ont une amélioration de leur condition de vie avec notamment une diminution de la fatigue. Les patients ont tous présenté des améliorations nettes avec un arrêt des saignements, une amélioration du fonctionnement des organes atteint, un meilleur contrôle de la douleur (les deux patients prenant des opioïdes ont pu les arrêter) une réduction du volume des malformations (de près de 40% après 6 mois de traitement). Tous les patients continuent actuellement à recevoir le traitement. Les patients n’ont pas (ou peu) présenté d’effets secondaires et le traitement n’a pas eu d’impact sur la croissance. Le traitement a été administré à des doses plus faibles que celles données en oncologie (250 mg pour les adultes et 50 mg pour les enfants), cela pourrait expliquer le peu d’effets secondaires observés.
Une autre équipe a récemment publié les résultats sur le premier patient d’Amérique du Nord traité au BYL719 [18]. Ce patient a ressenti dès les premiers jours une amélioration de la douleur et après deux mois de traitement les malformations ont diminué d’environ 50%. Une patiente de 17 ans avec de sévères atteintes aux parties génitales a été d’abord traitée avec de la rapamycine pendant un an sans amélioration notable puis traitée au BYL719. Son état s’est alors nettement amélioré permettant une reconstruction chirurgicale [19].
Un essai clinique (NCT04589650) va être lancé sur plusieurs sites aux Etats-Unis, en France, en Australie, en Espagne et en Irlande. L’étude va comprendre 150 participants répartis en trois groupes : un groupe d’adultes, un groupe d’enfants de 6 à 17 ans puis plus tardivement un groupe de jeunes enfants de 2 à 5 ans. Les deux premiers groupes seront randomisés avec la moitié des participants recevant le placebo et l’autre moitié recevant l’Alpesilib.
6. Conclusions et perspectives
Le syndrome de PROS est une pathologie dont la physiopathologie commence à être de mieux en mieux comprise. Sa prévalence n’est pas connue mais très certainement sous-estimée étant donné l’extrême diversité des phénotypes associés à ces syndromes et la difficulté d’établir un diagnostic génétique pour les pathologies présentant des mutations mosaïques. Les symptômes pouvant être extrêmement invalidants, il est urgent de pouvoir proposer un traitement efficace pour les patients. L’établissement de modèles murins a permis de mettre en évidence l’efficacité potentielle de certaines molécules inhibitrices de la voie PI3K-AKT-mTOR. Certaines de ces molécules ont été ou sont en cours d’évaluation par des essais cliniques ouvrant des perspectives thérapeutiques encourageantes pour les patients atteints du syndrome de PROS.