

1 Introduction
Brain interactions with the endocrine system involve at least three different sequences of events [1]. (1) The first type of interaction consists of neuroendocrine effects of peripheral hormones such as thyroid hormones (TH) and sex steroids on the brain; before acting, those hormones can be transformed into other active hormones such as triiodothyronine derived from thyroxine through deiodinase or oestradiol derived from testosterone through aromatase; these transformations can occur in peripheral tissues or centrally in the brain. (2) The brain exerts also neuroendocrine effects on peripheral hormones through the hypothalamic pituitary system; here, a complex cascade of players is initiated by neuropeptides and neurotransmitters in the brain, such as kisspeptin or GABA. They regulate the synthesis and the neurosecretion of neurohormones such as Gonadotrophin Releasing Hormone (GnRH) and Corticotrophin Releasing Hormone (CRH) which, in turn, stimulate the secretion of pituitary hormones (LH, FSH and ACTH, respectively). These hormones will then stimulate the secretion by the gonads and the adrenals of other hormones, which will ultimately affect target tissues. (3) Finally, there are local brain neuroendocrine effects through neuro-hormones and neurosteroids produced and acting in the brain.
Among the public health issues raised following the exposure to Endocrine Disrupting Chemicals (EDCs), neurodevelopmental disorders as well as alterations of puberty timing and disturbances of energy balance involve prominently or partly neuroendocrine disruption. Considering the different neuroendocrine pathways, it makes sense that disruption of neuroendocrine effects results from interference at the precise step where those neuroendocrine effects (i.e. effects of brain hormones or hormone effects on the brain) occur. However, interference with any former step in the sequence of events will obviously also end up in neuroendocrine disruption. Understandably, the mode of action can be typically endocrine when a chemical interferes directly with hormone action while the mode of action can be different when a chemical interferes upstream to hormone action. Still, the net result is neuroendocrine disruption.
In this review, neuroendocrine disruptive effects of PCBs and BPA will be used as case studies to illustrate some mechanistic characteristics of neuroendocrine disruption.
2 Different pathways of neuroendocrine disruption
The pathway illustrated in Fig. 1 might provide a mechanistic paradigm for some neurodevelopmental disorders including impaired cognition. A study on the probability of disease causation based on epidemiological and toxicological evidence has established a strong probability (70–100%) of intellectual quotient (IQ) loss (13 million units each year) in Europe due to organophosphate pesticide exposure [2]. In the USA, exposure to polybrominated flame retardants appears to be the leading factor for a comparable effect [3]. Both compounds can interact with thyroid hormone action, which is an essential factor of brain development, thus accounting for a neuroendocrine disruptive mechanism involving some brain effects of peripheral hormones. Taking only the EDCs with high probability causation into consideration, the global cost of EDC exposure for the European Union has recently been estimated in the range of 163 billion euros per year [4] with neurodevelopmental disorders accounting for above 90% of the total. The period of exposure plays an important role. Reduction of IQ has also been associated with prenatal or early postnatal exposure to polychlorinated biphenyls (PCBs), a family of persistent EDCs used for decades for their isolating properties [5,6]. In rodents, deficits in spatial memory have also been reported in adult rats following gestational and lactational exposure to Aroclor 1254, a mixture of PCBs [7]. We have recently published that the same Aroclor 1254 exposure disrupts the developmental maturation of excitatory synapses in newborn neurons at weaning in mouse dentate gyrus. When compared to mature neurons, newborn neurons are preferentially recruited in hippocampal circuits and involved in learning and memory processes. Thus, an alteration of the maturation of their synapses is likely to alter memory and behaviour, but the link between those mechanistic findings and hippocampal function remains to be established [8]. Attention-deficit hyperactivity disorders and autism, two other neurodevelopmental disturbances with altered behaviour, are associated respectively with exposure to polybrominated/organophosphate compounds and phthalates though probabilities of causation (20–69%) are lower than for cognitive deficits [2,3]. Behavioural modifications such as internalization and externalization are difficult to evaluate in humans, as the effects are often moderate and can only be linked to the exposure by correlative studies, in contrast to animal models enabling substantiation of causal relationships with EDCs. Moreover, the underlying molecular, cellular and network mechanisms are only rarely fully elucidated.

The action of hormones on the brain provides a first possible pathway of neuroendocrine effects and neuroendocrine disruption. Here, peripheral glands synthesize and secrete hormones into the bloodstream. The levels of binding proteins that are produced in the liver can influence the amounts of free bioactive hormones. Also hormones can be transformed into other active hormones by enzymes present in various tissues, such as testosterone aromatized into oestradiol and T4 deiodenized into T3. Interference with any of those steps can ultimately affect the action of peripheral hormones on the brain and explain neuroendocrine disruption. Testost.: testosterone; T4: thyroxine; TBG: Thyroxin Binding Globulin; T3: triiodothyronine; SHBG: sex hormone binding globulin; TR: thyroid hormone receptor; ER: Estrogen receptor; AR: androgen receptor; GnRH: gonadotrophin releasing hormone; TRH: thyrotrophin releasing hormone; TSH: thyroid stimulating hormone; LH: luteinizing hormone. Masquer
The action of hormones on the brain provides a first possible pathway of neuroendocrine effects and neuroendocrine disruption. Here, peripheral glands synthesize and secrete hormones into the bloodstream. The levels of binding proteins that are produced in the liver can ... Lire la suite
As indicated in Fig. 1, crucial peripheral events could explain the disruptive effects of chemicals beside interference with hormone receptors in the brain. EDCs could affect the liver synthesis of carrier proteins influencing the free hormone fraction that is available for biological activity. They could metabolize hormones into more active or inactive compounds or into different hormones. Such enzymatic processes can also take place in the brain where aromatase transforms testosterone into oestradiol [9,10]. Like polybrominated and organophosphate compounds, PCBs are known to interfere with the thyroid hormone system through a reduction of serum thyroxin concentrations. In rats, foetal exposure to Aroclor 1254 increased cell cycle exit and delayed radial neuronal migration in the developing cortex [11]. Because those effects were temporally associated with a reduction of thyroxin levels in maternal serum, a mechanistic relation between the brain effects of Aroclor 1254 and insufficient thyroxin action could be hypothesized. Noteworthy, the classical concepts of endocrinology may not simply apply to this condition since primary hypothyroidism is expected to involve increased TSH levels that are not seen in this particular case of low thyroxin after PCB exposure [12]. Among possible explanations, an activation of the hypothalamic thyroid hormone receptors by PCBs could result in reduced TRH secretion and prevent subsequent TSH stimulation, despite low thyroxin levels.
The second possible pathway of neuroendocrine disruption (Fig. 2) shows a different sequence of mechanistic events leading to altered function of the endocrine system. The neuroendocrine effect that is disrupted by EDCs takes place among the initial steps, while it was the ultimate step in the pathway illustrated in Fig. 1. In other words, brain disorders can be consequential to disruption of hormone effects by EDCs (Fig. 1) and brain disorders following EDC exposure can be causal to disruption of the hormonal system (Fig. 2). For instance, several reproductive disorders originate from altered production and action of brain neuropeptides such as kisspeptin, a stimulator of GnRH secretion that does not belong to the endocrine system, but controls a cascade of downstream hormones i.e. hypothalamic GnRH, pituitary LH and FSH and, ultimately, gonadal hormones including sex steroids [13]. For instance, we showed that neonatal exposure to the EDC diethylstilbestrol resulted in reduced hypothalamic mRNA expression of Kiss1, the gene encoding kisspeptin [14]. After exposure to a high dose of BPA (> 0.5 mg/kg/d), Navarro et al. have shown a reduction of Kiss1 mRNA expression on postnatal day 30 in the hypothalamus [15]. Similarly, Patisaul et al. found a reduction in kisspeptin immunoreactivity in the arcuate nucleus at adulthood [16]. Kisspeptin being a major gatekeeper of puberty onset, such reduction in its expression is unexpected in face of the early vaginal opening reported after high doses of BPA exposure [17]. The reduction in kisspeptin expression could then be part of a mechanism reactive to BPA exposure.
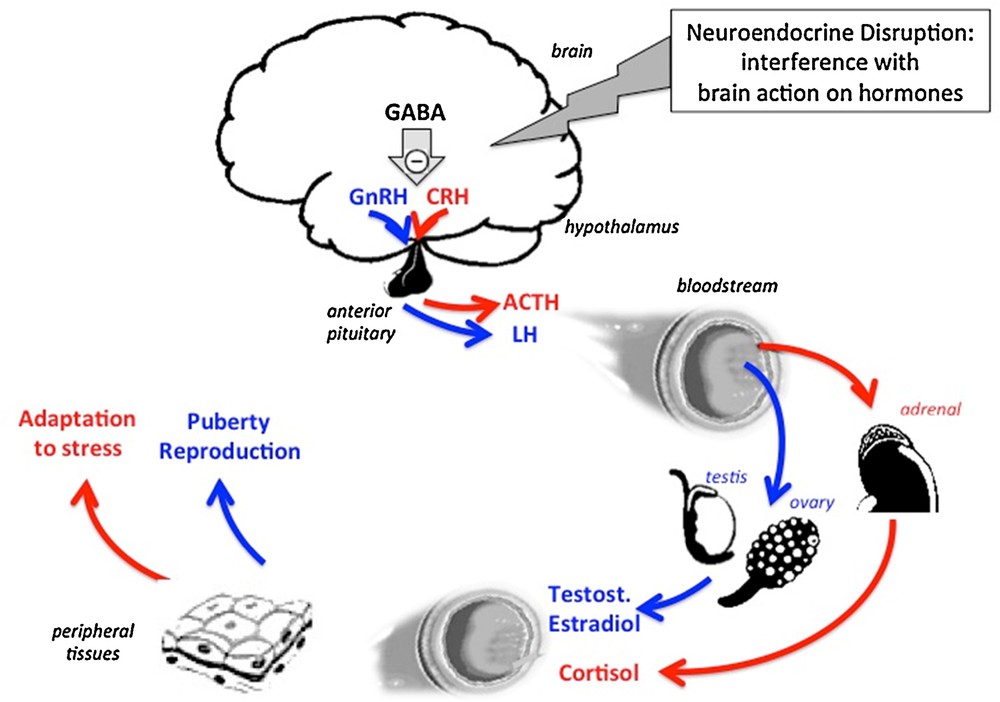
The action of the brain on peripheral hormones provides a second possible pathway of neuroendocrine effects and neuroendocrine disruption. Here, peptidergic neurons in the hypothalamus synthesize and secrete releasing (or inhibitory) hormones into the bloodstream of the portal hypothalamic-pituitary system. Consequently, the pituitary gland secretes hormones into the general circulation that will in turn cause steroid hormones to be released by peripheral glands such as the gonads and the adrenals and cause peripheral effects in target tissues. Interference with any of the steps (neuropeptides, neurotransmitters) upstream to peptidergic neurons in the brain can ultimately affect the action of the hormones released from peripheral glands as a result of neuroendocrine disruption. GABA: gamma-aminobutyric acid; GnRH: gonadotrophin releasing hormone; CRH: corticotrophin releasing hormone; ACTH: adreno-corticotrophic hormone; LH: luteinizing hormone; Testost.: testosterone. Masquer
The action of the brain on peripheral hormones provides a second possible pathway of neuroendocrine effects and neuroendocrine disruption. Here, peptidergic neurons in the hypothalamus synthesize and secrete releasing (or inhibitory) hormones into the bloodstream of the portal hypothalamic-pituitary system. ... Lire la suite
Other upstream non-endocrine factors such as transmitters can also account for neuroendocrine disruption. Recently, we have reported that an early postnatal exposure of female rats to an environmentally relevant dose of BPA (25 ng/kg/day) for two weeks is followed by delayed puberty. This effect involves a brain mechanism since the developmental increase in frequency of GnRH release is delayed as well. Interestingly enough, a higher BPA dose of 5 mg results in opposing effects. The mechanism involves respectively increased or reduced tone of GABAergic neurotransmission that plays an inhibitory control on GnRH secretion [17]. Here, neuroendocrine disruption might be initiated by a non-endocrine mechanism. Other examples of inhibitory GABA control in the brain are the endocrine components of stress, i.e. the cascade Corticotrophin releasing Hormone (CRH), ACTH and cortisol resulting in stress response such as hyperglycaemia [18,19]. Noteworthy, the mechanistic analysis of endocrine disruption is complex when involving the axes “hypothalamus–pituitary gland–peripheral gland–target tissues” due to possible effects of EDCs at their different levels.
The third type of neuroendocrine interaction (Fig. 3) involves both hormone production and action taking place in the brain. A classic example is the pineal gland, which produces melatonin acting in hypothalamic areas where circadian rhythms are controlled [20]. Such an action is exerted via melatonin in peripheral blood and in cerebrospinal fluid [21]. Melatonin also has receptors in different peripheral tissues, presumably contributing to the circadian rhythmicity of different functions [20]. Though endocrine disruption of this pathway has not been reported so far, it represents a potential target of neuroendocrine disruption.

The production and action of hormones in the brain provides a third possible pathway of neuroendocrine effects and neuroendocrine disruption. Here, the pineal gland synthesizes and secretes melatonin into the bloodstream as well as into cerebrospinal fluid. Consequently, the pineal hormone can have peripheral effects, while it has also central effects, particularly in the suprachiasmatic nucleus. Here, all the components of neuroendocrine function and potential disruption take place in the brain.
3 Non-specificity of region and receptor involved in neuroendocrine disruption
The regional specificity of physiological hormone action either in the brain or in any peripheral organ relies on the expression of specific hormone receptors in given tissues and places where hormones arrive from the bloodstream. Likewise, if an EDC would interfere with a particular hormone receptor only, endocrine disruption would be specific of that receptor and alter given functions in every place where those receptors are controlling or influencing some functions. For instance, oestrogens as well as estrogenic EDCs would act everywhere oestrogen receptors (ER) are expressed in the brain and in the rest of the body. The reality is more complex because receptors are not univocal due to the expression of different subtypes that may show different affinities for EDCs versus endogenous hormones. Such is the case for the oestrogen-sensitive receptors GPR30 and ERRγ that bind BPA preferentially [22–24]. The same applies for neurotransmitters like GABA and GABA receptors, which are expressed in the whole brain. There are several subunits of GABAA receptors and BPA seems to interfere with the delta subunit [17,25] that is expressed in the extrasynaptic space [26,27]. Beyond that mechanism, the general interference of EDCs with GABA neurotransmission through blood circulation is very different from the effect of GABA as an endogenous compound released locally by some GABAergic neurons. The specific endpoints that are affected by changes in GABA neurotransmission vary among brain regions depending on the role of the neurons that GABA impinges on. As opposed to that, endocrine disruptor effects are spread in the whole brain through circulation and can interfere with many different endpoints. Thus, when an EDC like BPA interferes with a specific neuroendocrine function through an upstream ubiquitous neurotransmitter like GABA [17], other disruptive effects can occur everywhere GABA plays a role. For instance, after exposure prenatally and early postnatally to the same low dose of BPA as we used (25 ng/kg/day), Cabaton et al. [28] and Zalko et al. [29] have shown that whole brain GABA estimated via nuclear magnetic resonance spectroscopy shows increased concentrations as opposed to a reduction after exposure to a 1000 times higher dose of BPA. This experiment demonstrates that the impact of BPA is not restricted to the hypothalamic neuroendocrine system. Other studies have reported that GABA neurotransmission could be affected in different brain regions after early-life exposure to BPA. Ogi et al. have recently shown that prenatal and lactational exposure to BPA leads to increased levels of GABA and glutamate in almost all brain regions and of norepinephrine in the cortex, hypothalamus and thalamus. These modifications were also associated with behavioural alterations in female rats [30]. It has been shown that BPA could delay the perinatal chloride shift in cortical neurons because of a reduced expression of potassium chloride cotransporter 2 (Kcc2) mRNA associated with epigenetic effects and modify the neuronal ability to migrate in vitro [31]. In the basolateral amygdala, Zhou et al. have shown that perinatal exposure to BPA led to an increase of postsynaptic neuronal excitability due to GABAergic disinhibition [32]. The mRNA level of glutamic acid decarboxylase 67 was reduced, while DNA methyltransferase 1 mRNA was overexpressed, suggesting again an epigenetic mechanism. These effects in the basolateral amygdala were also associated with long-term anxiety-like behaviours. Taken together, those data show that BPA can affect neurotransmission in several brain regions with behavioural alterations. Early exposure to BPA could therefore play a role in the pathogenesis of human neurodevelopmental disorders. Such a hypothesis is supported by some studies reporting correlations between the level of BPA in the urines of young children or of their mother and some atypical behaviours [33,34] or increase of anxious and depressive experiences [35]. BPA is commonly viewed as an estrogenic substance that can act trough oestrogenic receptors and disrupt the physiological action of oestrogens. However, BPA action is much more complex. Unlike hormones that have high affinity for only one kind of receptor, an EDC-like BPA can bind to many kinds of receptors. In addition to classical and non-classical oestrogen receptors, BPA can bind and have an antagonist effect on androgen receptors [36–38] and thyroid hormone receptors [39,40]. BPA can also bind the glucocorticoid receptor [41,42] and the aryl hydrocarbon receptor [43,44]. Thus, BPA targets are multiple, including neurotransmitters, epigenetic regulation and hormone receptors belonging to different systems.
PCBs provide another interesting case study in the sense that their causal involvement in neurodevelopmental disturbances is quite clear, though no direct endocrine mode of action or interference with hormone receptors is firmly established so far to account for those effects. Prenatal and/or early postnatal exposure to PCBs, which causes alteration of cognitive functions in rodents as well as in humans [6,7], is associated with various cellular effects. We reported a moderate but significant reduction in the number of newborn neurons in adult mice hippocampus as well as a disruption of the synaptic maturation [8]. However, since cognitive functions are not depending on the hippocampus only and PCBs can cause alterations in other brain areas; the combination of those effects could all together explain the behavioural phenotype. Notably, A1254 delays radial neuronal migration in the developing cortex [11]. So, several alterations happening at the cellular and circuitry levels are leading to the alteration of the cognitive functions as the final outcome. The cellular effects are themselves probably due to alterations at the molecular levels. Even if some molecular modifications have been identified [45,46], a full mechanistic picture is still missing and further investigations are needed in order to identify the precise mode of action of PCBs in the brain.
4 Concluding remarks
The mechanism of endocrine disruption is complex because the physiology of the endocrine system is complex. The hormonal receptors are inserted into cascades of events that ultimately determine the physiological effects of hormones and well as their alterations by EDCs. Neuroendocrine disruption provides an illustration of those concepts. These realities may not be reflected neither in in vitro paradigms focusing exclusively on interactions at the receptor level nor in in vivo conditions arbitrarily restricted to a given type of hormone receptor (e.g., receptors to oestrogens, androgens or thyroid hormones) at the price of ignoring interactions between different parts of the endocrine systems and the role of non-strictly endocrine components such as neurotransmitters and neuropeptides. Therefore, an adverse neuroendocrine effect of EDCs does not necessarily involve a direct endocrine mode of action. Moreover, disturbed endocrine function can be mechanistically a consequence rather than a cause of neuroendocrine disruption.