

1 Introduction
Catalytic phases (CP) – solids with a well-defined structure – constitute an important class of catalytic materials [1,2], which includes transition metals and their oxides and sulfides, heteropolyacids, perovskites, zeolites, pillared clays, etc. The chemical functionality of these materials is determined by their surface crystallography. Atoms or groups of atoms in specific coordination at the surface of CPs display unique chemical properties enabling chemisorption of reagents/intermediates/products in a way that facilitates the formation of catalytic cycles. Ensembles of Co–Mo–S atoms at the edge planes of MoS2 crystals with a layered structure [3] or ensembles of SO4–Zr–O atoms at the surface of ZrO2 with a tetragonal structure [4] are typical examples. In many cases, CPs create catalytic cycles with higher efficiency per active site than those obtained with molecular catalysts. The combination of the unique properties of CPs with their thermal and chemical stability has led to widespread application of CP-based catalysts in industrial processes.
The development of CP-based catalysts for specific organic reactions, such as hydrogenation (HYD), oxidation, isomerization and condensation, began some decades ago [1,2], and today preparation strategies for CP-based catalytic materials have been optimized to yield efficient compositions and composite structures. Further improvements in catalyst performance may be achieved by adding the ‘novel dimension’ provided by dispersing the CPs in ordered mesoporous silicas (OMS) [5–8]. As matrixes for CPs, MCM-41 and SBA-15 OMS with a hexagonal structure or MCM-48 and SBA-16 OMS with a cubic array of nanotubular mesopores have a number of advantages over conventional catalyst supports:
- • The surface area of OMS is two to three times higher than that of traditional mesoporous supports, like silica gels and activated alumina. This greater surface area gives a higher concentration of active sites on a weight basis, while facilitating maintenance of the optimal surface concentration of CPs and of their precursors, which controls the CP sintering-dispersion [9] at proportionally higher CP loadings.
- • Limitations on particle growth inside the OMS mesopore channels with controlled diameters maintain a high dispersion of the CP at loadings beyond those dictated by the requirements for a surface diffusion mechanism for CP sintering.
- • It is possible to control the locations of the CPs in the OMS matrix or to provide a combination of different CP locations in a single nanocomposite, as shown schematically in Fig. 1: The CPs may be located inside the mesopores (as nanoparticles or as a thin layer), inside the micropores that arise within the pore walls after removal of single polymer chains belonging to the corona of co-polymer surfactant micelles, or at the external surface of the OMS microcrystals; in this way, the distance between the active sites and the order of their accessibility to reacting molecules may be controlled.
- • The stabilization, inside the OMS matrix, of the nanoparticles or clusters of CPs with unusual surface crystallography and the high thermostability of the embedded CP structures provides efficient catalytic cycles.
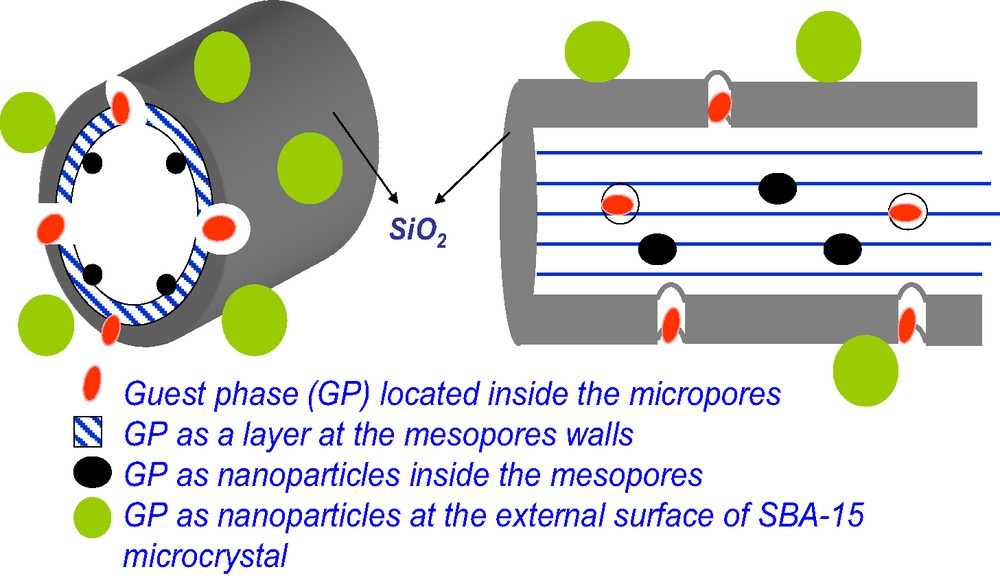
Possible locations of CPs in the OMS matrix.
2 Conditions for the preparation of high-performance host/guest catalytic composites on mesostructured silica
The catalytic reaction rate (r) of a CP is determined by the specific external surface area and surface chemistry of its crystals and the reaction conditions:(1)
Modern methods for the preparation of thermostable porous solids, i.e. pure metals or transition-metal oxides, yield materials with a particle size ≥10 nm [10]. Smaller particles of <10 nm should be stabilized inside porous matrixes, like OMS, that restrict particle growth and provide high accessibility to reacting molecules. In this case, the condition for the preparation of a host/guest composite catalytic material with activity (CP surface area) higher than that in the bulk reference CP is:(5)
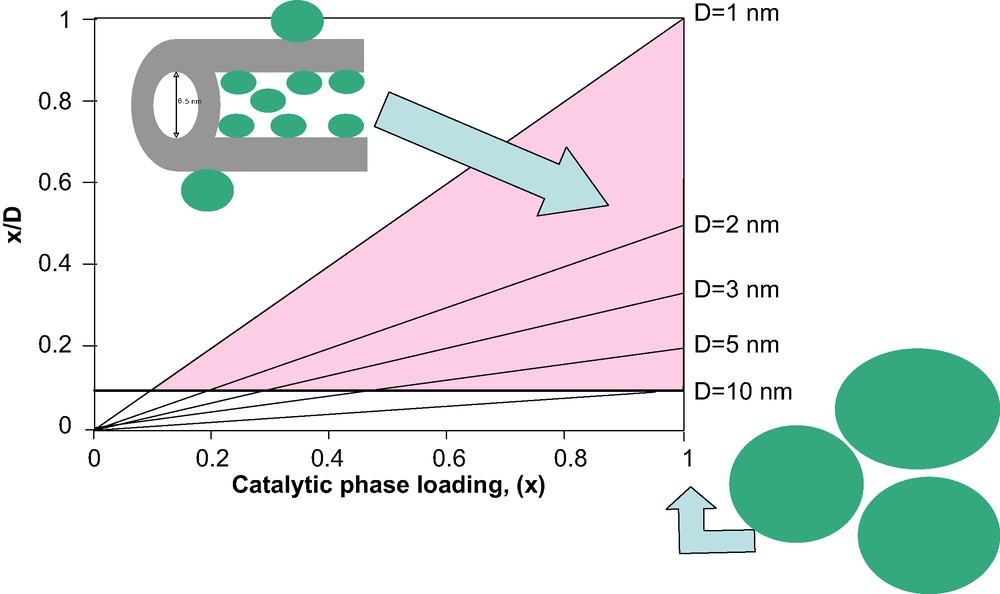
Relationship between the CP loading (x) and the x/D ratio at different values of CP particle size.
Thus, the requirements for an efficient host/guest catalytic composite are nanoparticle (nanocrystal) assembly mode of the CP (and not an epitaxial layer), a particle size < 10 nm and > 50 wt.% of CP located such that the CP is accessible to the reacting molecules, without complete filling of the mesopore volume. Estimation of the loadings of different CPs (i.e. WS2, ZrO2, TiO2) corresponding to 50% and 100% filling of the mesopore volume in hexagonal SBA-15 OMS with a pore volume of 1 cm3 g–1 is shown in Fig. 3. Calculations based on the theoretical densities of silica and of sulfide and oxide CPs [11] show that embedding of the CP at 65–80 wt.% loadings leaves 50% of OMS mesopore volume empty and thus available for transporting reacting molecules and reaction products.

Effect of mesopore filling in OMS on the CP loading in CP/OMS composites.
In the preparation of high-loaded CP/OMS composite materials, it is necessary to maintain uniform distribution of the CP inside the matrix without plugging of the mesopores, especially for OMS with a 1D hexagonal structure. It has been demonstrated that the normalized surface area (NSA) of the composite may be calculated as [12,13]:(6)
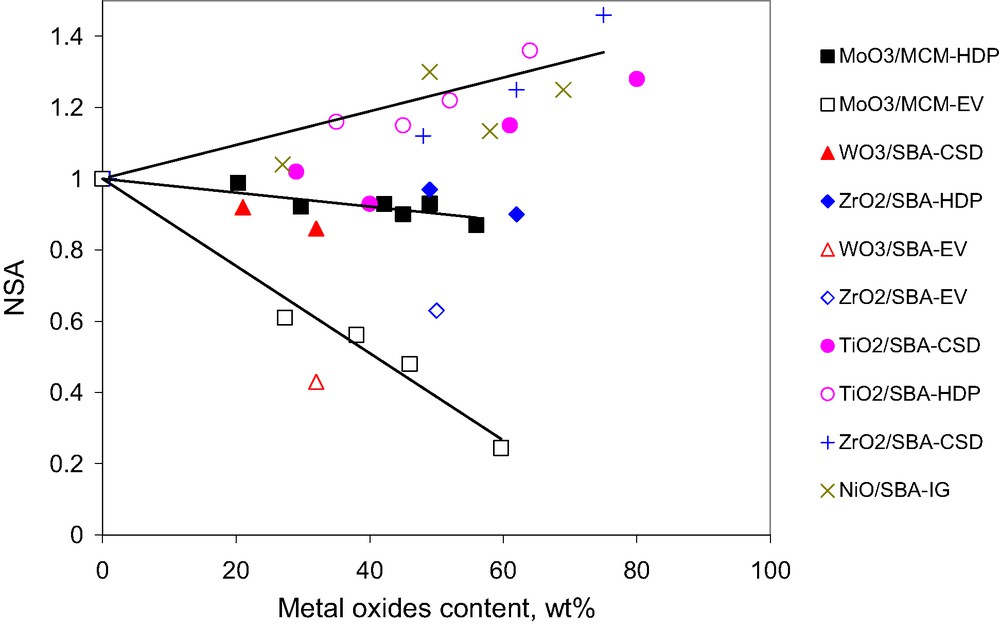
NSA measured for different metal oxide–OMS composites as a function of guest-phase loadings obtained with different insertion methods: HDP, homogeneous deposition precipitation; EV, impregnation-evacuation; CSD, chemical solution decomposition; IG, internal gelation [13].
Since one of the main advantages of host/guest CP/OMS composites is the smaller particle size of the CP vis-à-vis that of known (reference) catalytic materials, it is expected that increasing the CP dispersion will improve the catalyst performance. This is, however, not always the case, as has been found, for example, for the photocatalytic oxidation of different substrates with (anatase) TiO2. Here, the competing effects of effective particle size on charge-carrier dynamics (electron-hole recombination rate) and the amount of available surface active sites leads to a decrease of the quantum yield in photocatalytic reactions for a TiO2 with crystal size of < 10–25 nm [14,15]. However, in the aerobic oxidation of volatile organic compounds (VOCs), which does not include photo-effects, a TiO2/OMS catalytic material exhibiting a ‘pure chemical catalytic cycle’, displayed superior performance to that of the bulk nanostructured reference TiO2 [16]. In another example, insertion of layered WS2 crystals inside the nanotubular channels of SBA-15 decreased the length of the WS2 slabs – which determines the exposure of the active side crystal plane – by a factor of 2 compared with that in WS2/Si-gel and WS2/Al2O3 [17]. This effect – and not the change of the WS2 stacking number of crystal layers – caused significant enhancement of HYD and hydrodesulfurization (HDS) activity of the WS2 CP after promotion with nickel [17].
Retaining the crystal structure of the CP after insertion in the OMS matrix is critical to its catalytic performance. For example, formation of layered nanoslabs of MoS2 [12,18] and WS2 [17,18], tetragonal ZrO2 [11,16,19] and TiO2 anatase [11,16] or CsxH3–xPW12O40 with Keggin structure of the polyanion [20] inside OMS mesoporous channels takes full advantage of increasing CP dispersion at high loadings in hydrotreating, oxidation or acid-catalyzed reactions. A strong interaction of the CP with OMS will hinder the formation of the desired CP structure and will yield a composite with low catalytic activity. For example, insertion of ZrO2 in an SBA-15 matrix by precipitation of zirconium hydroxide inside the channels yielded amorphous zirconia, with high thermal stability, which could not be converted to the tetragonal phase and displayed low acidity/activity performance after sulfidation [21].
Information on the effect of the location of the CP inside the OMS matrix on catalytic performance is very limited. Generally, the CP is inserted inside the nanotubular channels of OMS to form ensembles, as described above. For the SBA-type OMS, some of the CP probably enters the micropores inside the pore walls: In the study of Yuranov et al. [22], for example, a metallic palladium or PdO CP could be preferentially located inside the micropores of SBA-15, but neither the portion of CP located in micropores nor the contribution of this portion of the CP to the catalytic reaction rate was estimated in that study. It is likely that additional amounts of CP (whether the same type or another type) inside the micropores or at the outer surface of OMS microcrystals can improve the performance of CP/OMS composite catalytic materials. As was shown by Rao et al. [20], the simultaneous deposition of a Cs2.5H0.5PW12O40 salt at the external surface of SBA-15 microcrystals and inside the nanotubular mesopores substantially increased the acidity and catalytic activity of the composite in acid-catalyzed reactions compared with the Cs2.5H0.5PW12O40/SBA-15 composites where the CP was located only at the external surface or only inside the mesopores.
3 Preparation and characterization of high-loaded host/guest CP/OMS composites
Insertion of a CP into OMS during the synthesis of the matrix is a complex process for high-loaded (> 50 wt.% CP) composites, because the requirements for the optimal conditions for OMS crystallization and the formation of the desired CP phase by different chemical reactions are incompatible in most cases. The post-synthetic inclusion of CPs necessitates fixation of their precursors inside the nanotubular channels to facilitate good dispersion and to prevent plugging of the pores during subsequent preparation steps (drying, calcination, reduction, etc.). This requires the proper balance between the strength of CP-OMS interaction and the driving force for CP formation to prevent pore plugging and to obtain a CP with the required structure. For example, the adsorption of transition-metal-containing precursors on the surface of silanol-rich OMS leads to blocking of the mesopores with well-packed guest metal oxides at loadings > 40 wt.%, yielding a composite surface area < 50 m2 g–1 [23]. It has been shown that plugging of the mesopores at high CP loadings can be minimized by using calcined OMS with low concentrations of silanols, where fixation of large amounts of precursors can be achieved by various methods, depending on the chemical properties of the guest material, i.e. by homogeneous deposition precipitation (HDP), by chemical solution decomposition (CSD) or by multistep grafting (MG).
3.1 Homogeneous deposition precipitation
HDP comprises two stages: a highly soluble CP-precursor (designated A) is converted in solution to a precursor with low solubility (designated BL, stage 1) that will precipitate at stage 2 exclusively on the surface of the silica support as the CP, due to the nonspecific interaction of the precursor with silica [24]. The conversion of A → BL requires an additional controlling component that usually generates a metal hydroxide of low solubility after slow basification of the suspension of the support in the CP-precursor solution. Since basification causes degradation of the OMS structure, a different HDP chemistry should be used here. For example, for deposition of Mo-oxide [12] or W-, or Mo-sulfides [17,18] inside the nanotubular channels of MCM-41 or SBA-15, ultrasonication of the corresponding Mo- and W-carbonyls is used instead of insertion of a controlling component. In the presence of oxygen or elemental sulfur, respectively, the ultrasonication converts the carbonyls directly to the corresponding oxides or sulfides. Such treatment has been used to produce Co–MoS2/MCM-41, Co–MoS2/SBA-15 and Ni–WS2/SBA-15 CP/OMS composites with 63–67 wt.% loadings, a high surface area of > 200 m2 g–1, a crystal slab length of 3.2–3.5 nm, and low plugging of the mesopores of Δ = 0.07 – 0.27 (Table 1). HRTEM, combined with selected area EDX, showed the nanocrystals of WS2 and MoS2 to be located inside the nanotubular pores of OMS (Fig. 5, upper left), with uniform distribution along the channels.
Characteristics of host/guest CP/OMS composite catalytic materials
| Catalytic material (and preparation method) | CP loading (wt.%) | CP structure | CP particle size (nm) | Surface area (m2 g–1) | Pore volume (cm3 g–1) | Average pore diameter (nm) | NSA | References |
| Co–MoS2/MCM-41(HDP) | 63.3 | Layered slabs | 3.5 × 5.0 | 275 | 0.43 | 6.2 | 0.93 | [12] |
| Co–MoS2/SBA-15 (HDP) | 64.2 | Layered slabs | 3.4 × 4.5 | 256 | 0.32 | 5.0 | 0.90 | [18] |
| Ni–WS2/SBA-15 (HDP) | 67.4 | Layered slabs | 3.2 × 6.5 | 210 | 0.25 | 4.7 | 0.73 | [17,18] |
| ZrO2/SBA-15 (CSD) | 62.0 | Tetragonal | 4.5 | 380 | 0.34 | 3.7 | 1.22 | [11,16,19] |
| TiO2/SBA-15 (CSD) | 40.0 | Anatase | 8.5 | 445 | 0.45 | 4.0 | 0.93 | [11,16] |
| TiO2/SBA-15 (HDP) | 61.0 | Anatase | 5.5 | 400 | 0.40 | 3.7 | 1.36 | [11,16] |
| Al2O3/MCM-41 (MG) | 38.0 | Amorphous | – | 542 | 0.89 | 6.1 | 1.01 | [26,27] |
| Cs2.5H0.5PW12O40/SBA-15 (MG) | 60.0 | Condensed Keggin units | 7.5 | 338 | 0.36 | 3.8 | 0.78 | [20] |
| WO3/SBA-15 (CSD) | 32.0 | Amorphous | <1.5 | 468 | 0.62 | 6.0 | 0.87 | [25] |
| H3PW12O40–Al2O3/SBA-15 (MG) | 25 (Al2O3), 35 (HPW) | Amorphous, | – | 300 | 0.43 | 5.7 | 0.82 | – |
| Isolated Keggin units |

Nanocrystals of CP stabilized in the nanotubular pores of mesostructured silica at high loadings: a – 62% WS2/SBA-15(HDP)[16]; b– 61% TiO2/SBA-15(HDP) [19]; c – 62% ZrO2/SBA-15 (CSD) [18]; d – 60% Cs2.5H0.5PW12O40/SBA-15 (MG) [20].
Another nondestructive HDP route that may be used is based on the hydrolysis of a metal alkoxide within the OMS mesopores, where water plays the role of the controlling component. Immersion in water of SBA-15 OMS whose pores were filled with a solution of Ti(n-BuO)4 in n-decane led to the deposition of titanium hydroxide exclusively inside the nanotubular pores [11,16]. Calcination of this material yielded a TiO2/SBA-15 composite with 61 wt.% loading of the anatase phase of 5.5 nm crystal size and no plugging of OMS mesopores; Δ = – 0.36 (Table 1). The uniform distribution of the TiO2 nanocrystals in nanotubular channels of SBA-15 OMS and their anatase structure was confirmed by XRD, HRTEM and Fourier-transform of the periodicity of the nanocrystal images into electron diffraction patterns (Fig. 5, upper right).
3.2 Chemical solution decomposition
Embedded tetragonal zirconia nanocrystals cannot be produced by HDP, since zirconium hydroxide interacts strongly with OMS pore walls. Therefore, a different method is needed to produce this CP/OMS, and the CSD insertion method has been selected [11,16,19]. Heating to 493 K in an autoclave of SBA-15 OMS, whose pores had been filled with Zr(n-PrO)4 in presence of an excess of n-PrOH not contacting with the wet sample, caused thermal decomposition of the alkoxide. Maintaining the material at elevated temperatures in an atmosphere of saturated vapor of the solvent retained the liquid solution inside the pores until thermal decomposition of the alkoxide took place. After calcination, a ZrO2/SBA-15 composite with 62 wt.% loading of tetragonal 4.5-nm crystals and no plugging of OMS mesopores (Δ = – 0.22) was obtained (Table 1). Uniform distribution of ZrO2 nanocrystals in nanotubular channels of SBA-15 OMS and the tetragonal structure of the ZrO2 crystals were confirmed by XRD, HRTEM and Fourier-transform of the periodicity of the nanocrystal images into electron diffraction patterns (Fig. 5, lower left). Implementation of the CSD method for insertion of 40 wt.% TiO2 in an SBA-15 matrix yielded an anatase nanocrystal ensemble with a crystal size of 8.5 nm and Δ = 0.07 [11,16]. The very strong interaction of W-species formed during CSD of W-carbonyl with the silica walls inside the mesopores of SBA-15 OMS prevented the formation of a WO3 nanocrystalline phase, even with the CSD method [25] (Table 1). Instead, a thin layer of WO3 with no pore plugging (Δ = 0.13) was formed on the surface of nanotubular OMS mesopores.
3.3 Multistep grafting
Grafting alumina or cesia species on the internal surface of OMS mesopores and external surface of their microcrystals forms multiple anchoring sites for fixation of a metal oxide guest CP as a second layer. The four-step grafting of the MCM-41 OMS surface with Al(sec-BuO)3 – including intermediate hydrolysis of the adsorbed alkoxide species – yielded 38 wt.% of alumina CP as amorphous clusters without mesopore plugging; Δ = 0 (Table 1) [26,27]. SBA-15 grafted with 38 wt.% Al2O3 immobilized up to 35 wt.% of H3PW12O40 heteropolyacid (by adsorption from aqueous solution) as an isolated molecular species without plugging the mesopores, Δ = –0.2 (Table 1). SBA-15 grafted with CsPrO adsorbed 60 wt.% H3PW12O40 heteropolyacid to form 7.5-nm nanocrystals of Cs2.5H0.5PW12O40 by the reaction 2.5 Si–O–CsPrO + H3PW12O40 → Si–OH + Cs2.5H0.5PW12O40 (CP) + 2.5PrOH, with a small extent of mesopore blocking; Δ ~ 0.2 (Table 1). HRTEM combined with selected area EDX showed the nanocrystals of the Cs2.5H0.5PW12O40 salt to be located mostly inside the nanotubular pores of OMS (Fig. 5, lower right), with uniform distribution along the channels [20].
The current state of the art thus provides the means for insertion of various CPs at > 50 wt.% loadings in OMS matrices without substantial mesopore plugging. The optimal insertion strategy – HDP, CSD or MG – is dictated by the chemical nature of the CP and its precursors.
4 Examples of high-performance host/guest CP/OMS catalytic materials
Some examples of the hexagonal OMS, MCM-41 and SBA-15, as matrixes for improvement the catalytic materials based on different CPs are presented below:
4.1 Co–Mo/OMS and Ni–W/OMS
The rate constants for the HDS of dibenzothiophene (DBT) and the HYD of toluene catalyzed by high-loaded MoS2- and WS2-based materials inserted in OMS matrices are compared in Table 2 with those for conventional Co–Mo/Al2O3 and Ni–W/Al2O3 catalysts. Slabs of MoS2 and WS2 phases promoted with Co or Ni were inserted into OMS nanotubes [12,17] at about 60 wt.% loadings, so that their length did not exceed 3.5 nm. The reference Co–Mo/Al2O3 catalyst supported on alumina, with ~20 wt.% of (Co + Mo) CP, had a slab length similar to that of the Co–Mo–S/SBA-15 composite and an activity comparable to that of the commercial sample. Increasing the Co–Mo loading in alumina to about 40 wt.% caused a substantial (more than threefold) decrease of the surface area and a drop (more than twofold) of HDS activity (Table 2). The surface area of ~300 m2 g–1 exhibited by the MCM-41 or SBA-15 matrices loaded with ~40 wt.% of Co–Mo facilitated good CP accessibility. This improved accessibility at high CP loading is the reason for the ~1.7 times higher HDS activity of Co–Mo/OMS composites compared with that of the conventional Co–Mo/Al2O3 catalyst. For the Ni–W/SBA-15 composite, insertion of the Ni–W–S CP inside the nanotubular channels of SBA-15 decreased the WS2 slab length by a factor of 2 compared with that in Ni–W/Al2O3 (Table 1). This decrease, together with the twofold higher CP loading and higher surface area of Ni–W/SBA-15 composite, yielded an approximately fivefold higher value of the rate constant for the HYD of toluene catalyzed by Ni–W/SBA-15 than that for the reaction catalyzed by the conventional Ni–W/Al2O3 material. The high-loaded Ni–W/SBA-15 host/guest composite exhibited high HDS activity similar to that of Co–Mo/Al2O3 (Table 2), which makes it an excellent potential catalyst for deep HDS of middle petroleum distillates, where a combination of high HDS and HYD activities is essential for removal of hindered S-compounds [28].
Rate constants for HDS of DBT and hydrogenation of toluene with MoS2- and WS2-based catalysts [12,17,18]
| Catalyst | W (Mo) (wt.%) | Co (Ni) (wt.%) | Surface area (m2 g–1) | Slab length (nm) | KHYD (628 K) (cm3 g–1 h–1) | KHDS (593 K) (cm3 g–1 h–1) |
| Co–Mo/Al2O3 (commercial) | 17.6 | 4.5 | 220 | – | 0.9 | 57 |
| Co–Mo/Al2O3 | 14.7 | 4.4 | 200 | 3.5 | – | 63 |
| Co–Mo/Al2O3 | 29.7 | 9.4 | 60 | – | – | 24 |
| Co–Mo/MCM-41 | 28.6 | 10.1 | 320 | – | – | 98 |
| Co–Mo/SBA-15 | 30.0 | 9.2 | 297 | 3.4 | – | 102 |
| Ni–W/Al2O3 | 26.4 | 16.4 | 158 | 7.3 | 0.9 | 28 |
| Ni–W/SBA-15 | 44.5 | 5.7 | 212 | 3.6 | 4.4 | 54 |
Since in high-loaded CP/OMS host/guest catalytic materials the tubular mesopores of OMS matrix are partially filled with CP, the question of unrestricted accessibility of the existing active sites to reacting molecules is of great practical interest. For the HDS of DBT conducted in the liquid phase, calculation of the Thiele modulus φ was performed using the molecular diffusion coefficient of DBT in a decane–octadecane reaction mixture, calculated according the Wilke-Chang correlation [29]. At the reaction temperature (593 K), the diffusion coefficient was equal to 2 × 10–4 cm2 s–1. The effective diffusion coefficient inside the pores is normally lower by an order of magnitude than the molecular diffusion coefficient due to the porosity and pore tortuosity of the catalyst pellet [30]. A Thiele modulus of 0.4 was calculated by taking an effective diffusion coefficient of 2 × 10–5 cm2 s–1, the diameter of catalyst pellet as 1.3 mm, and a high value for the zero-order HDS rate constant (measured by Vradman et al. [18] for a Co–M-/SBA-15 catalyst) of 102 cm3 g–1 h–1 normalized to the initial DBT concentration KHDS = K/Co (Co, inlet DBT concentration; K, pseudo-zero-order HDS rate constant). This value of the Thiele modulus yields an effectiveness factor of > 0.95 and means that the diffusion effects for the DBT HDS reaction can be neglected.
For the gas-phase toluene HYD reaction, the bulk diffusion coefficient was calculated by the Fuller correlation [29] to be 0.02 cm2 s–1. The Knudsen diffusion coefficient inside the catalyst pores was estimated according to simple kinetic theory [30] to be 0.008 cm2 s–1. Thus, Knudsen diffusion controls the transport inside the mesopores, as was to be expected for a gas-phase reaction inside relatively small pores. Taking the effective diffusion coefficient to be an order of magnitude lower, i.e. 8 × 10–4 cm2 s–1, a catalyst pellet diameter of 1.3 mm, and a high value of the first-order hydrogenation rate constant of 4.4 h–1 measured for the Ni–W/SBA-15 catalyst [17], we calculated a Thiele modulus of 0.03. Thus, in this case, too, the effectiveness factor was equal to unity, and no diffusion limitations were present.
4.2 SO4–ZrO2/SBA-15
The sulfation of a disordered ZrOx phase inside the SBA-15 nanotubes after CSD, followed by calcination at 873 K, yielded an acidic catalytic material in which the tetragonal zirconia had a crystal size of 3.5 nm [19]. The sulfur content and acidity (NH3 adsorption) of this composite were about twofold higher than the reference bulk sulfated zirconia with a surface area of 107 m2 g–1 and a crystal size of 10 nm. The sulfated zirconia CP stabilized inside SBA-15 nanotubes exhibited a higher catalytic activity than the bulk sulfated zirconia (Table 3). With the OMS composite, conversions of isopropanol and methanol/tert-butanol mixture to diisopropylether and methyl tertiary-butyl ether (MTBE), respectively, gave 1.5–2.2 times (depending on ZrO2 loading) higher product yields than those obtained with the bulk reference catalyst. This effect may be attributed to the higher capacity of the small 3.5-nm nanocrystals of tetragonal zirconia CP stabilized in OMS matrix for surface sulfate anions compared with that of bulk ZrO2 with a substantially higher crystal size. The same selectivities in iso-propanol dehydration measured with the reference bulk SO4–ZrO2 and SO4–ZrO2/SBA-15 host/guest composites were evident for the same types of acid sites in the bulk and supported materials.
Performance of sulfated ZrO2/SBA-15 in acid-catalyzed reactions measured after a reaction time of 2 h [19]
| Catalyst | ZrO2 crystal size (Nm) | ZrO2 loading (wt.%) | Isopropanol dehydration, 473 K | MTBE yield (%) in t-BuOH/MeOH condensation, 398 K | |
| Conversion (%) | Diisopropyl ether selectivity (%) | ||||
| SO4–ZrO2 (commercial) | 10 | 96.8 | 6.2 | 83.0 | 22.6 |
| SO4–ZrO2 /SBA-15 | 3.5 | 48.1 | 9.2 | 84.5 | 32.3 |
| SO4–ZrO2 /SBA-15 | 3.5 | 58.0 | 13.2 | 85.0 | 46.2 |
Since the kinetic parameters of the above-mentioned reactions were not known, the Weisz modulus Φ [31] was used as a criterion for the importance of pore diffusion. This approach was used to estimate the diffusion limitations in the condensation of t-butanol and MeOH and the dehydration of iso-propanol performed by Landau et al. [19] with SO4–ZrO2/SBA-15 catalysts of particle size <50 μm. Taking a rate of 1.3 mol h–1 l–1 for condensation (0.87 mol h–1 l–1 for dehydration), a t-butanol concentration of 5.2 mol l–1 (13.4 mol l–1 of iso-propanol) and an effective diffusion coefficient of 10–6 cm2 s–1 – the regular order of magnitude in liquid phase – we obtained Φ << 1 for both reactions. This value indicates negligible diffusion limitations. Similar results were obtained for the batch alkylation of toluene with benzyl alcohol at 383 K with Al-containing SBA-15 powder [32].
4.3 TiO2(ZrO2)/SBA-15
Platinum supported on titania (anatase) is an efficient catalyst in the combustion of VOCs, where fragmentation of organic molecules at the TiO2 surface is the rate-limiting step [33]. At a relatively low temperature of 523 K, a series of TiO2(ZrO2)/SBA-15 catalysts containing 0.3 wt.% Pt demonstrated a linear dependence of the rate constant for ethyl acetate combustion on the specific surface area of the CP, TiO2 or ZrO2 [16] (Fig. 6). The TiO2-based catalysts displayed substantially higher activity. Stabilizing the TiO2 or ZrO2 nanocrystals inside the SBA-15 nanotubes at loadings of > 50 wt.% yielded catalytic materials with two to three times higher activity than catalysts based on bulk Ti- and Zr-oxides, with crystal diameters of 12 nm. A similar effect was obtained with TiO2/SBA-15 catalysts in the gas-phase oxidation of methoxytoluene at 623 K: the methoxytoluene conversion increased linearly with increasing TiO2 surface area in the catalytic material, and a substantial increase in methoxybenzoic acid selectivity was obtained at the smaller TiO2 crystal size of ≤ 8.5 nm.
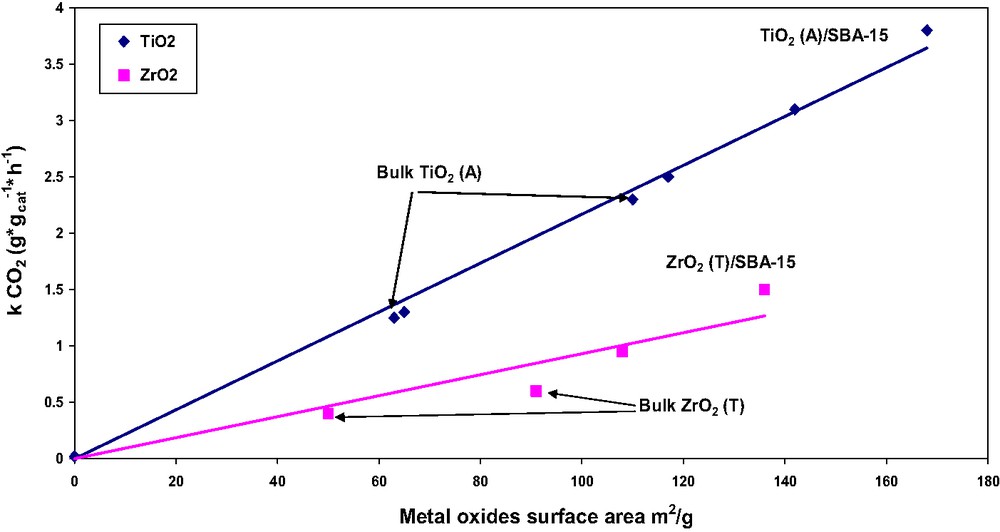
Effect of surface area of bulk and SBA-15-embedded crystalline Ti(Zr)O2 phases on the activity of supported Pt catalysts in ethyl acetate combustion at 523 K [16].
The absence of diffusion limitations in a relatively fast gas-phase reaction of ethyl acetate combustion at 500–550 K with Pt–TiO2(ZrO2)/SBA-15 catalysts was demonstrated by the linear increase of the rate constant with increasing specific surface area of embedded CP (Fig. 6) [16]. This finding reflected the equal specific activity of CP inside the OMS mesopores measured at different loadings. In addition, the specific activity of the CP in CP/OMS composites was equal to that measured with bulk Pt/TiO2(ZrO2) catalysts, where diffusion limitations were excluded by selecting the appropriate pellet size [16].
4.4 Al2O3/MCM-41
Introduction of alumina clusters into wide-pore MCM-41 by successive MG changed the surface chemical functionality of the mesostructured silica to that of pure alumina [26]. The embedded alumina phase displayed unusually high acidity and catalytic activity in the alkylation of phenol with methanol. At an alumina loading of close to 40 wt.%, the reaction rate was higher by a factor of 2.2 with this Al2O3/MCM-41 composite than that with bulk alumina of high surface area of 460 m2 g–1 (Fig. 7). As was demonstrated by 27Al FAM (II)-MQMAS NMR, this effect was a result of stabilizing very small alumina clusters inside mesostructured silica nanotubes, yielding a high (~10%) concentration of penta-coordinated alumina sites together with the large abundance of tetrahedral coordinated sites that increase the acidity of the material [27].

Effect of alumina loading in MCM-41 on the rate of phenol alkylation with methanol at 623 K [26].
4.5 Tungstophosphoric heteropolyacid and salt catalysts
Grafting the silica surface with alumina species produced efficient anchoring sites for H3PW12O40 heteropolyacid, with no apparent leaching in polar media [34]. Formation of a bulk alumina phase decreased the acidity of the anchored heteropolyacid, and therefore as the alumina loading increased, the performance of such a composite catalyst produced by the MG method reached a maximum at the point corresponding to a saturated monolayer of grafted alumina species. This is demonstrated in Fig. 8 for H3PW12O40–Al2O3/SiO2 composites based on a regular mesoporous silica-gel with a surface area of 362 m2 g–1 and on SBA-15 with a surface area of 922 m2 g–1 in the t-BuOH + MeOH → MTBE condensation reaction. Due to the superior alumina capacity of SBA-15 over silica-gel, the former matrix anchored a 1.5-fold higher amount of heteropolyacid and demonstrated proportionally higher activity in MTBE production.

Effect of alumina content in alumina-grafted SiO2 gel (AGSG) and SBA-15 OMS (AGSBA) on the H3PW12O40 loading by adsorption (shown next to the data points) and MTBE yield at 383 K.
The use of the Cs-salt at the optimal Cs/W ratio of 0.2 resolved the solubility problem of the H3PW12O40 heteropolyacid [35]. However, colloidization of the material in polar media and the relatively large crystal size of 10–15 nm limits the application of Cs2.5H0.5PW12O40 salt in acid-catalyzed reactions. Combination of grafting the surface of SBA-15 with CsPrO and deposition of Cs2O at the external surface of SBA-15 microcrystals yielded a composite with 65–70 wt.% loading of Cs2.5 H0.5PW12O40 at different locations in the matrix after adsorption of H3PW12O40 heteropolyacid [20]. In addition to 7.5-nm nanocrystals of the Cs2.5H0.5PW12O40 salt stabilized inside the nanotubular channels (Fig. 5, lower left), up to 35 wt.% of that CP was located at the external surface of SBA-15 particles as 50–150 nm agglomerates of primary nanocrystals (Fig. 9). This high-loaded Cs2.5H0.5PW12O40/SBA-15 material demonstrated high resistance to colloidization in polar solvents and 1.2–1.7 times superior activity over the bulk Cs2.5H0.5PW12O40 salt, on a catalyst weight basis, in acid-catalyzed reactions (Fig. 10). This seems to be the first successful attempt to increase the activity of Cs2.5H0.5PW12O40 catalyst developed as long ago as 1980 [35] by its deposition on a suitable support.

Disintegrated 30–150 nm secondary aggregates of 13-nm Cs2.5H0.5PW12O40 salt nanocrystals at the external surface of SBA-15 particles [20].
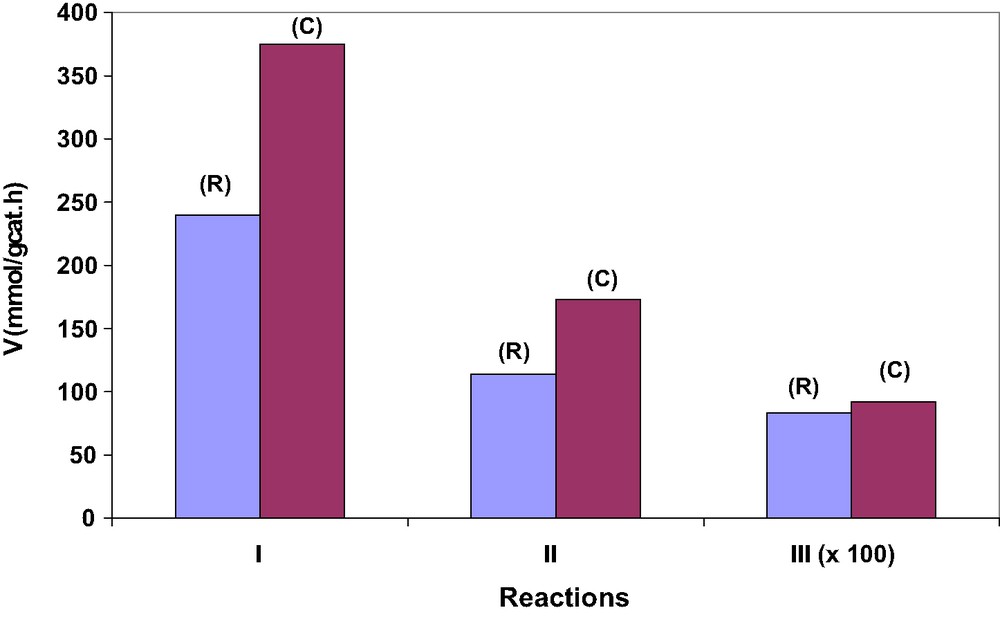
Reaction rates measured with the bulk reference Cs2.5H0.5PW12O40 salt (R) and the same salt deposited on SBA-15 OMS with mixed CP location (C) in the condensation of t-BuOH with MeOH to yield MTBE at 383 K (I), propyonylation of anisole with propyonic anhydride at 333 K (II), and alkylation of cathechol with t-BuOH at 373 K (III); adapted from Ref. [20].
It is thus the unique properties of OMS as catalyst supports that not only increase the specific activity of the CP per weight but also, in combination with high CP loading, compensate for the effect of CP dilution with the inert silica. The insertion of a CP inside the OMS matrix does not limit the accessibility of active sites. The only problem here could be complete plugging the OMS mesopores, which would exclude part of CP from catalytic process. This portion may be estimated qualitatively on the basis of NSA values, as shown above.