

1 Introduction
Tertiary phosphine ligands are ubiquitous in the realms of coordination chemistry and organometallic chemistry [1]. This is largely due to the wide range of steric and electronic properties that are accessible through variation of the substituents. The range of tertiary phosphines that are available provides a degree of control over the properties of transition metal complexes, and facilitating the ability to tune the activity and selectivity of a catalyst. A recent protocol for developing stereoelectronic maps for phosphorus ligands concluded that bulky electron-poor phosphines were largely unknown, so would be useful targets [2]. Such ligands have the potential to stabilise coordinatively unsaturated metals in low oxidation states [3].
We recently reported the synthesis of a range of N-carbazolyl phosphines PPh3–n(NC12H8)n (n = 1, 1; n = 2, 2; n = 3, 3) and their coordination chemistry with rhodium(I), palladium(II) and palladium(0) [4]. Some of these phosphines have also been reported by Jackstell et al. [5], who used them as ligands in Rh-catalysed hydroformylation reactions. The carbonyl stretching frequencies in the IR spectra of the [Rh(acac)(CO){PPh3–n(NC12H8)n}] complexes suggested that the N-carbazolyl phosphines have similar electronic properties to their N-pyrrolyl phosphine analogues PPh3–n(NC4H4)n [6], though the presence of the N-carbazolyl groups ensures the ligands are much larger.
In this paper we report the crystal structures of 2 and 3. The 1JPSe coupling constants in the phosphine selenides provide a means of assessing phosphine electronic properties, so the synthesis and 31P{1H} NMR spectra of the selenide derivatives SePPh3–n(NC12H8)n (n = 1, 4; n = 2, 5; n = 3, 6) are also detailed, along with the crystallographic characterisation of the co-crystal 0.65 SeP(NC12H8)3·0.35 P(NC12H8)3 7. Finally, we describe the reaction of the unsymmetrical diphosphine Ph2PCH2P(NC4H4)2 with sulphur to form Ph2P(S)CH2P(NC4H4)2 8.
2 Crystal structures of 2·MeC(O)Me and 3
Slow evaporation of an acetone solution of PPh(NC12H8)2, 2, gave crystals suitable for X-ray crystallography. Analysis revealed the compound to be the acetone solvate 2·MeC(O)Me, the structure of which is shown in Fig. 1. Selected bond distances and angles are presented in Table 1. The phosphine adopts a propeller-like conformation [7] in the solid state and the nitrogen atoms are both planar, with the respective sums of the angles around the nitrogen atoms (Σ∠N) having values of 358° and 359°. The P–N bonds in 2 are longer than those in P(NC4H4)3 [1.677(8)–1.710(8) Å] [8], which is likely to be a consequence of the increased steric requirements of the N-carbazolyl groups. Analysis of the supramolecular structure reveals the presence of C–H···π interactions between the carbazolyl and phenyl rings on neighbouring molecules.
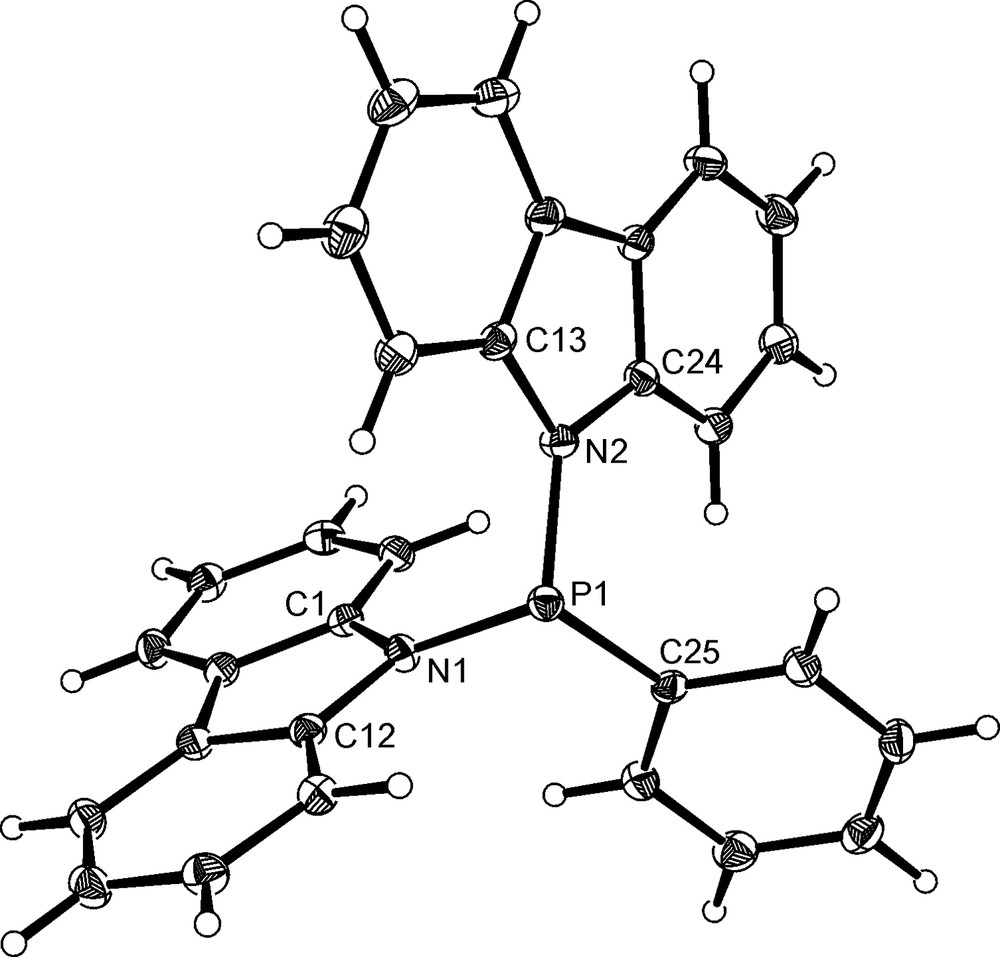
The molecular structure of PPh(NC12H8)2·MeC(O)Me, 2·MeC(O)Me, with the acetone molecule omitted for clarity. Thermal ellipsoids are shown at the 30% probability limit.
Selected bond lengths (Å) and angles (°) for 2·MeC(O)Me and 3
| 2 | 3 | ||
| P(1)–N(1) | 1.7216(14) | P(1)–N(1) | 1.7476(16) |
| P(1)–N(2) | 1.7320(15) | P(1)–N(2) | 1.7159(16) |
| P(1)–C(25) | 1.8153(18) | P(1)–N(3) | 1.7159(16) |
| C(1)–N(1)–C(12) | 106.67(13) | C(1)–N(1)–P(1) | 116.67(12) |
| C(1)–N(1)–P(1) | 133.38(12) | C(1)–N(1)–C(12) | 105.44(14) |
| C(12)–N(1)–P(1) | 119.29(12) | C(12)–N(1)–P(1) | 118.26(12) |
| C(24)–N(2)–C(13) | 107.13(14) | C(13)–N(2)–P(1) | 115.53(12) |
| C(24)–N(2)–P(1) | 134.43(12) | C(24)–N(2)–C(13) | 105.94(15) |
| C(13)–N(2)–P(1) | 116.33(12) | C(24)–N(2)–P(1) | 135.08(13) |
| N(1)–P(1)–N(2) | 102.89(7) | C(25)–N(3)–P(1) | 132.87(13) |
| N(1)–P(1)–C(25) | 103.33(8) | C(25)–N(3)–C(36) | 106.62(15) |
| N(2)–P(1)–C(25) | 103.90(8) | C(36)–N(3)–P(1) | 118.11(13) |
| N(2)–P(1)–N(3) | 104.89(8) | ||
| N(2)–P(1)–N(1) | 100.94(8) | ||
| N(3)–P(1)–N(1) | 101.07(8) |
Crystals of P(NC12H8)3, 3, suitable for crystallographic analysis were obtained by the slow diffusion of hexane into a dichloromethane solution. The structure of 3 is shown in Fig. 2 and selected bond angles and distances are given in Table 1. In contrast with the structure of P(anthracenyl)3 [9], 3 deviates from a propeller-like arrangement due to unequal rotations of the carbazolyl rings about the P–N bonds. The plane of the carbazolyl ring containing N(1) is almost perpendicular to the planes of the carbazolyl rings containing N(2) and N(3).
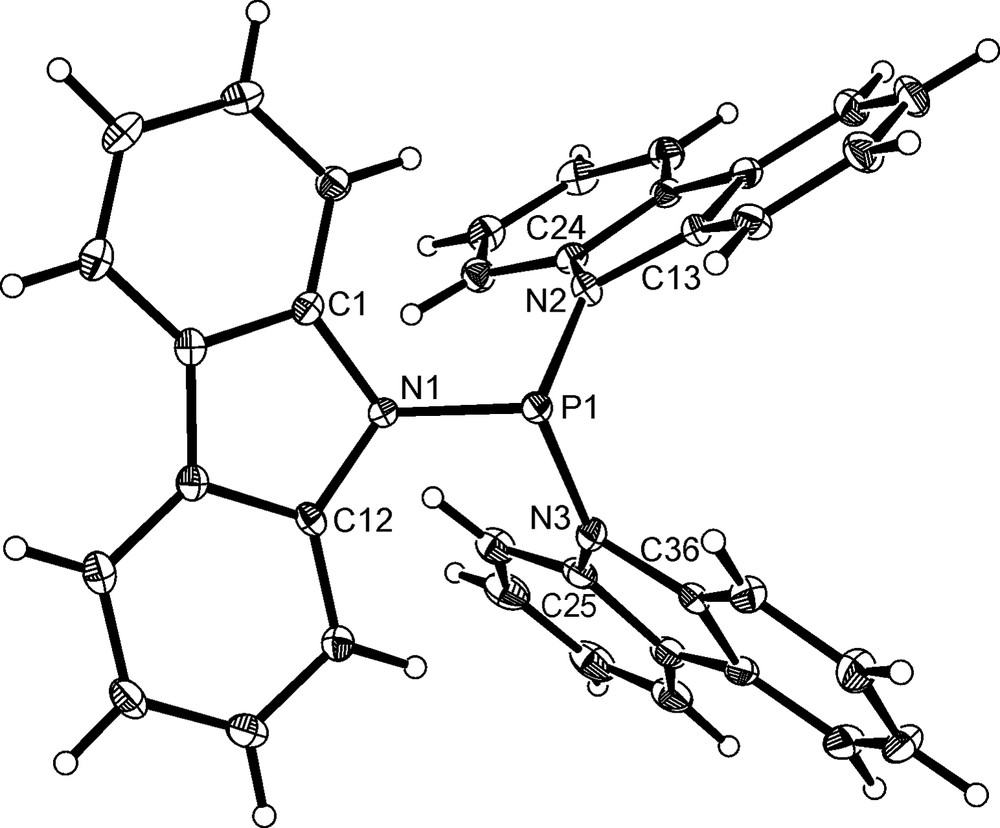
The molecular structure of P(NC12H8)3 3. Thermal ellipsoids are shown at the 30% probability limit.
The most striking feature in the structure of 3 is the deviation in N(1) from the planarity expected for an sp2-hybridised nitrogen atom. The sum of the angles (Σ∠N) around N(1) is only 340°, whereas for N(2) and N(3) the sums are 357° and 358°, respectively. N(1) is oriented such that its lone pair is anti to that of the phosphorus atom. These structural features are in contrast to those observed for tri(N-pyrrolyl)phosphine, P(NC4H4)3, and tri(N-indolyl)phosphine, P(NC8H6)3, both of which only contain nitrogen atoms that are virtually planar. The presence of a pyramidal nitrogen in 3 is reminiscent of the structures of many tris(dialkylamino)phosphines and their metal complexes [6,10]. For example, in the structure of P(NMe2)3 the phosphorus atom is bonded to two nearly planar nitrogen atoms (Σ∠N 353–359°) and one pyramidal nitrogen atom (Σ∠N 337 and 339° in the two independent molecules) [11]. In P(NMe2)3, the P–N bond involving the pyramidal nitrogen is longer (0.03–0.05 Å) than those to the planar nitrogen atoms. A similar increase in the P–N bond distance is observed for 3, with P(1)–N(1) [1.7476(16) Å] approximately 0.03 Å longer than the two other P–N bonds, and also longer than the P–N distances in P(NC4H4)3 [1.677(8)–1.710(8) Å] [8] and P(NC8H6)3 [1.698–1.728 Å] [12].
The supramolecular structure of 3 reveals the presence of both C–H···π and π···π interactions. These interactions are shown in Fig. 3 which contains views approximately parallel and orthogonal to the plane of the carbazolyl ring containing N(1). Hydrogen–aromatic ring distances lie in the range 2.64–2.76 Å, whereas the closest distance between parallel carbazolyl rings is 3.54 Å. The pyramidal distortions in 3 facilitate these intermolecular interactions by placing the N-carbazolyl groups in the appropriate positions.
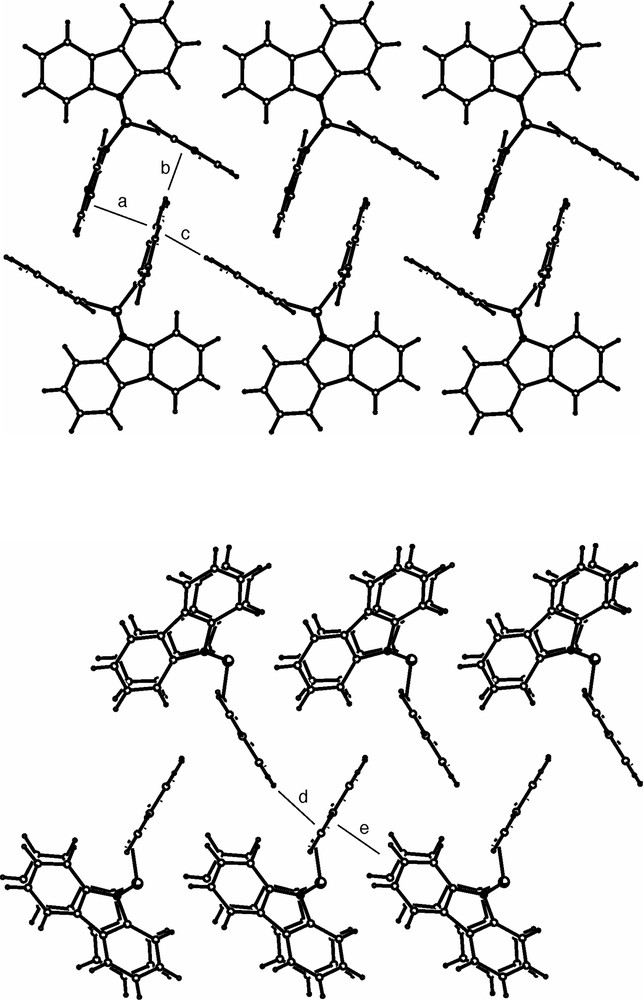
Intermolecular π···π (a) and C–H···π (b-e) interactions within the crystal structure of 3. Shortest distance from C or H to mean carbazolyl plane (a) 3.48, (b) 2.67, (c) 2.64, (d) 2.76, (e) 2.65 Å.
3 Phosphine selenides
In order to further investigate the electronic properties of 1–3, the seleno derivatives SePPh3–n(NC12H8)n (n = 1, 4; n = 2, 5; n = 3, 6) were prepared by reaction of the phosphines with an excess of selenium powder in toluene. The conditions required for the reaction to reach completion become increasingly severe with more N-carbazolyl substituents, hence PPh2(NC12H8) reacts at room temperature while P(NC12H8)3 requires 48 h at reflux. This trend is consistent with the increase in the electron-withdrawing nature of PPh3–n(NC12H8)n with increasing n, and the accompanying decrease in the availability of the phosphorus lone pair. Compounds 4–6 were isolated in good yield as crystalline solids.
The 31P{1H} NMR spectra of compounds 4–6 consist of singlets with 77Se satellites, and the spectral data are presented in Table 2 along with that for related compounds. There is a well established correlation between 1JPSe and the electronic properties of the parent phosphorus ligands, which takes the form of an inverse relationship with the σ-basicity [13–15]. Thus, as expected, the values of the 1JPSe coupling constant decrease in the order 6 > 5 > 4 which is consistent with the anticipated decrease in σ-basicity of 1–3 as the number of carbazolyl substituents at the phosphorus atom increases. Further comparison shows that 6 and SeP(N-indolyl-3-CH3)3 have similar 1JPSe values, which are appreciably smaller than that for SeP(NC4H4)3, suggesting that 3 is a better σ-donor than P(NC4H4)3. This contrasts with the 31P{1H} NMR and IR data observed for the compounds [Rh(acac)(CO)(L)] [L = 3, P(NC4H4)3], indicating that 3 and P(NC4H4)3 have similar electronic properties, though differences in the π-acceptor character may counterbalance those in the σ-basicity.
NMR parameters for selected phosphine selenide compounds
| δP | 1JPSe/Hz | ΔδPa | References | |
| SePMe3 | 8.0 | 684 | +70.0 | [22,23] |
| SePMe2Ph | 15.1 | 710 | +61.1 | [22,23] |
| SePMePh2 | 22.3 | 725 | +50.3 | [22,23] |
| SePPh3 | 34.1 | 736 | +41.5 | [22,23] |
| SeP(NMe2)3 | 81.2 | 805 | –40.3 | [22,23] |
| SePPh2(NC12H8) 4 | 54.4 | 811 | +21.7 | this work |
| SePPh(NC12H8)2 5 | 50.3 | 873 | –2.6 | this work |
| SeP(NC12H8)3 6 | 30.1 | 942 | –47.5 | this work |
| SeP(N-indolyl-3-CH3)3 | 28.2 | 943 | –36.6 | [14] |
| SeP(OMe)3 | 77.5 | 963 | –62.5 | [22,23] |
| SeP(NC4H4)3 | 43.0 | 970 | –36.6 | [6,14] |
| SeP(OPh)3 | 58.6 | 1027 | –67.8 | [23,24] |
| SeP{(NC4H3)3CH} | 28.3 | 1032 | –8.4 | [13] |
a ΔδP = δP(SePR3) – δP(PR3).
For the phosphines 1–3, the trend in chemical shift is straightforward, and an increase in chemical shift accompanies the increase in the number of electron-withdrawing N-carbazolyl substituents. Thus, δP is 32.7, 52.9 and 77.6 for 1–3, respectively. This can be rationalised on electronic grounds as simply being a consequence of increased deshielding of the phosphorus atom with the increasing number of N-carbazolyl groups, although steric effects leading to an increase the energy of the excited state, and hence the paramagnetic contribution, may also be a factor. However, the trend for the phosphine selenides is opposite: as shown in Table 2, the chemical shift decreases with the increasing number of N-carbazolyl substituents. For phosphine selenides such as SePMe3 and SePPh3, the difference in chemical shift between the selenide and the free phosphine (ΔδP) is large and positive, whereas for 6, ΔδP is large and negative. Similar observations have been made for the selenides of other electron-withdrawing phosphines such as P(NC4H4)3 and P(N-indolyl-3-CH3)3 [13,14], and since P(NC4H4)3 is similar in size to PPh3 [6,16], the origin must be electronic rather than steric. A possible explanation for this lies in the relative importance of the R3P=Se and R3P+–Se– resonance forms, since a decrease in the importance of the ionic resonance form for 6 would lead to reduced deshielding of the phosphorus nucleus, hence the observed upfield shift. However, the paramagnetic contribution may also be important as the R3P=Se resonance form would better stabilise the ground state than the R3P+–Se– form.
4 Crystal structure of 0.65 SeP(NC12H8)3·0.35 P(NC12H8)3 7
Co-crystals of the phosphine selenide SeP(NC12H8)3 and the phosphine P(NC12H8)3 were grown from slow diffusion of hexane into a dichloromethane solution containing both 3 and 6. On the basis of the X-ray structural analysis, the crystalline product was formulated as 0.65 SeP(NC12H8)3·0.35 P(NC12H8)3, 7, with the two compounds disordered. The phosphine selenide component of 7 is shown in Fig. 4 and selected bond distances and angles are given in Table 3. In contrast to 3, the molecule adopts a propeller-like conformation with no strong pyramidal distortions at any of the nitrogen atoms; Σ∠N is in the range 354–360°. This contrasts with the structures of SeP(NC4H8)3 and SeP(NMe2)3, both of which contain one nitrogen atom with a strong pyramidal distortion (Σ∠N 342–344°), in a similar way to the free phosphines and their metal complexes. The absence of any pyramidal distortion in 7 is consistent with its observation in 3 being a consequence of the intermolecular interactions, rather than being intrinsic to the compound. In line with this argument, there are no π···π interactions present in the supramolecular structure of 7. Instead, the extended structure is dominated by C–H···π interactions, as shown in Fig. 5.
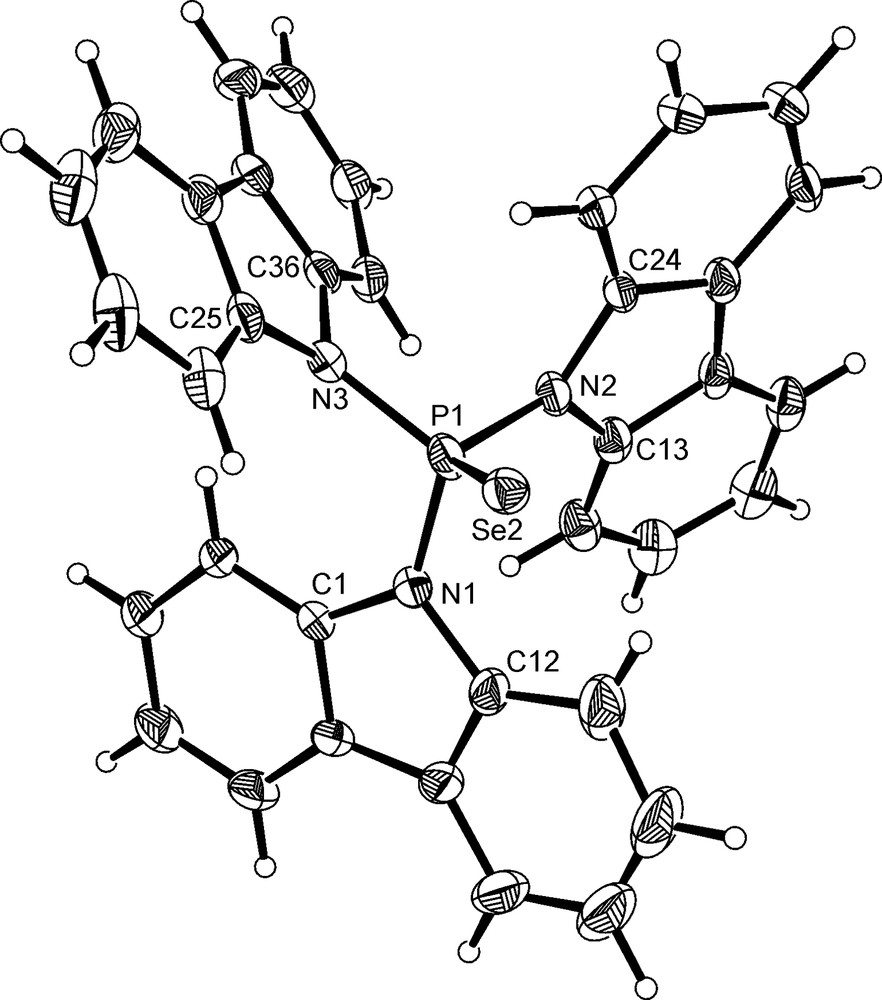
Molecular structure of 0.65 SeP(NC12H8)3·0.35 P(NC12H8)3, 7, with only the phosphine selenide component shown for clarity.
Thermal ellipsoids are shown at the 30% probability limit.
Selected bond lengths (Å) and angles (°) for 7
| P(1)–N(1) | 1.683(3) |
| P(1)–N(2) | 1.705(3) |
| P(1)–N(3) | 1.701(3) |
| P(1)–Se(2) | 2.0369(10) |
| C(1)–N(1)–C(12) | 106.7(3) |
| C(1)–N(1)–P(1) | 132.8(2) |
| C(12)–N(1)–P(1) | 120.5(2) |
| C(24)–N(2)–C(13) | 107.4(2) |
| C(24)–N(2)–P(1) | 118.9(2) |
| C(13)–N(2)–P(1) | 131.4(2) |
| C(36)–N(3)–C(25) | 106.1(3) |
| C(36)–N(3)–P(1) | 125.7(2) |
| C(25)–N(3)–P(1) | 121.8(2) |
| N(1)–P(1)–N(3) | 106.47(13) |
| N(1)–P(1)–N(2) | 103.92(14) |
| N(3)–P(1)–N(2) | 101.57(13) |
| N(1)–P(1)–Se(2) | 110.48(10) |
| N(3)–P(1)–Se(2) | 119.10(10) |
| N(2)–P(1)–Se(2) | 113.89(10) |
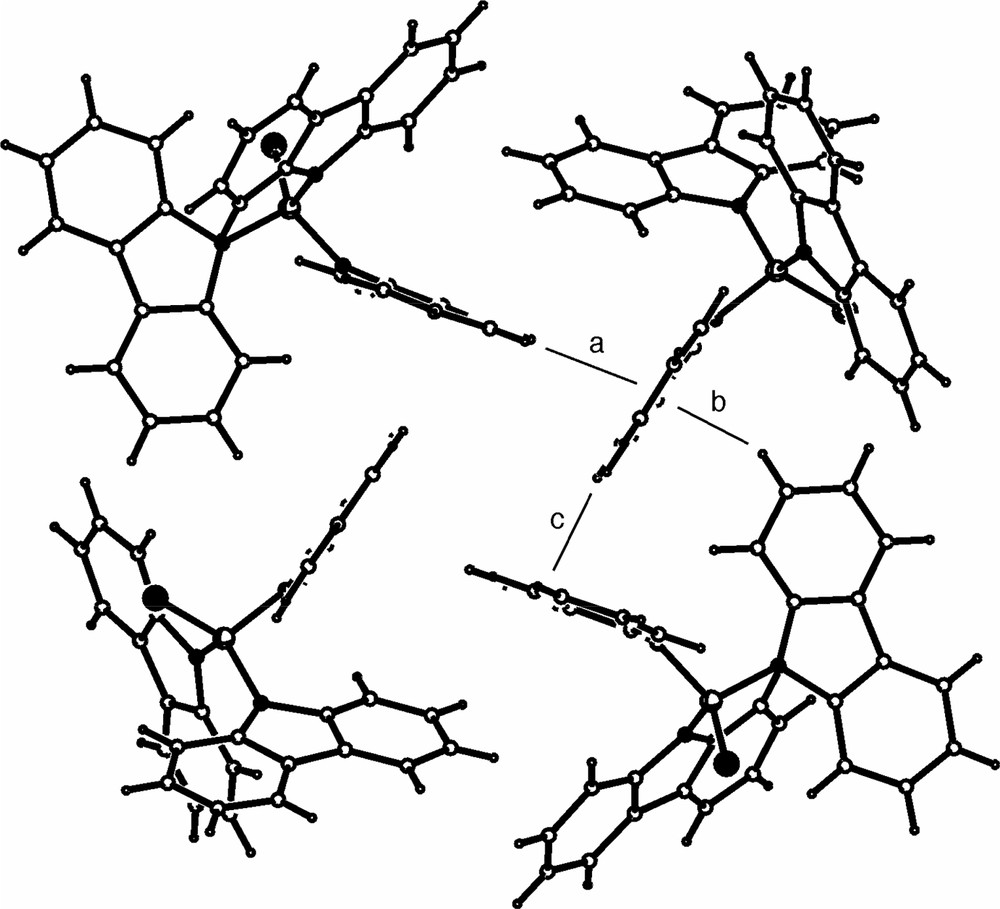
Intermolecular C–H···π interactions within the crystal structure of 7. Shortest distance from H to mean carbazolyl plane (a) 2.88, (b) 2.58, (c) 2.71 Å.
The P–Se bond length in 6 of 2.0369(10) Å is one of the shortest observed between these atoms. A search of the Cambridge Structural Database [17] revealed the presence of 146 compounds containing P–Se bonds in which the selenium is not bonded to another atom, corresponding to 217 independent P–Se bond lengths. Of these, the average P–Se bond length is 2.100 Å. There is a correlation between the electronic nature of the substituents and the P–Se distance, with electron donating groups leading to larger bond lengths than electron-withdrawing groups. As with the chemical shift data, this can be understood on the basis of the two resonance forms R3P=Se and R3P+–Se–. The ionic resonance form is stabilised by electron donating substituents, but is less important with electron-withdrawing groups such as N-carbazolyl. The short P–Se bond observed in 7 is therefore a consequence of the greater contribution of the R3P=Se resonance form.
5 Reaction of Ph2PCH2P(NC4H4)2 with sulphur
The diphosphine Ph2PCH2P(NC4H4)2 contains two electronically different phosphino groups, with the di(N-pyrrolyl)phosphino group more electron-withdrawing than the diphenylphosphino group. Studies with late transition metals have shown that the reactivity of these groups differs, and the di(N-pyrrolyl)phosphino group is more readily displaced from a metal centre than the diphenylphosphino group [18]. In order to determine whether the electronic difference influences oxidation of the phosphorus atoms, the reaction between Ph2PCH2P(NC4H4)2 and sulphur was investigated. The compounds were stirred together in dichloromethane for 4 h, and following removal of excess sulphur, the compound Ph2P(S)CH2P(NC4H4)2 8 was isolated by recrystallisation from dichloromethane–hexane.
The 31P{1H} NMR spectrum of 8 consists of doublets at δ 56.9 and δ 35.6, with a mutual coupling constant of 85 Hz. These resonances can be assigned on the basis of chemical shift and peak width to the phosphorus(III) and phosphorus(V) centres, respectively. The broader resonance at δ 56.9 is relatively close to that for the di(N-pyrrolyl)phosphino group in Ph2PCH2P(NC4H4)2 (δ 66.1) [18], and the large peak width is a consequence of the quadrupolar nitrogen nucleus [6]. The doublet at δ 35.6 has shifted by Δδ +60.1 from that of the diphenylphosphino group in Ph2PCH2P(NC4H4)2, consistent with oxidation of this phosphorus atom.
Crystals of 8 suitable for X-ray analysis were obtained by the slow diffusion of hexane into a dichloromethane solution. The asymmetric unit contains two independent molecules, though there are only minor differences between them. The structure of 8 is shown in Fig. 6, and selected bond lengths and angles are given in Table 4. The structure concurs with the NMR data in that only the diphenylphosphino group has been oxidised. Both N-pyrrolyl groups contain planar nitrogen atoms, with the sums of the angles around both N(1) and N(2) being 360°. Intramolecular π···π interactions are present between one of the pyrrolyl and one of the phenyl rings. Further intermolecular interactions between the two phosphino groups give rise to fourfold embraces in the supramolecular structure [19].
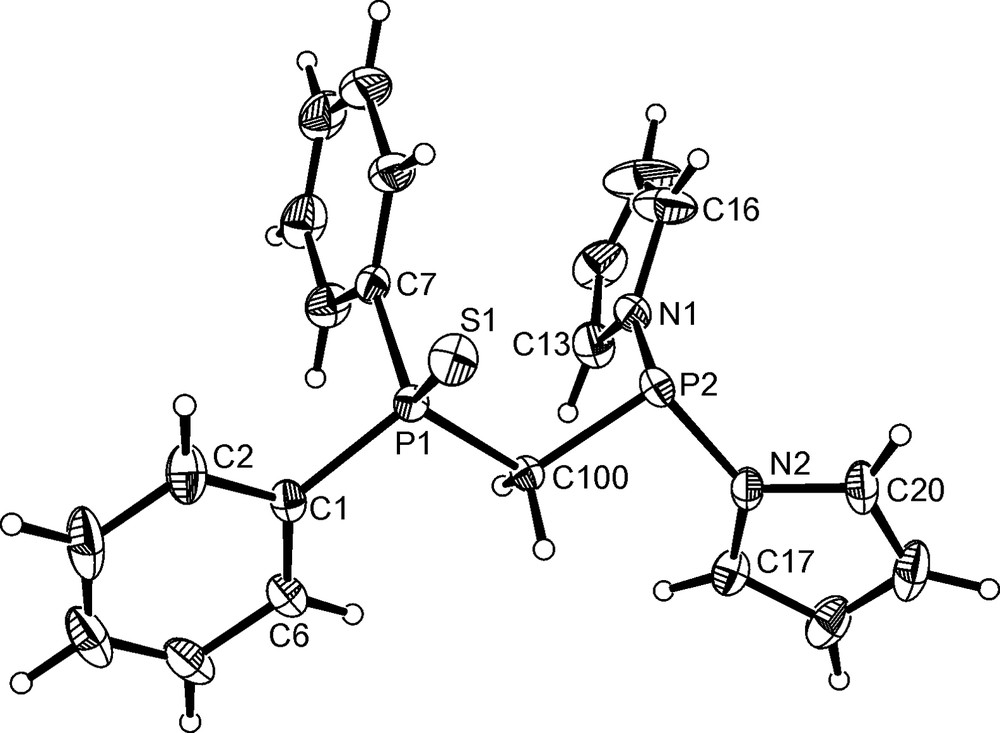
Molecular structure of Ph2P(S)CH2P(NC4H4)3 8. Only one of the two independent molecules in the asymmetric unit is shown. Thermal ellipsoids are shown at the 30% probability limit.
Selected bond lengths (Å) and angles (°) for 8
| S(1)–P(1) | 1.9528(6) | S(2)–P(3) | 1.9523(6) |
| P(2)–N(1) | 1.7126(18) | P(4)–N(3) | 1.7173(18) |
| P(2)–N(2) | 1.7287(17) | P(4)–N(4) | 1.7312(17) |
| C(16)–N(1)–C(13) | 106.9(2) | C(36)–N(3)–C(33) | 107.06(18) |
| C(16)–N(1)–P(2) | 121.64(18) | C(36)–N(3)–P(4) | 130.86(14) |
| C(13)–N(1)–P(2) | 131.08(15) | C(33)–N(3)–P(4) | 122.67(16) |
| C(17)–N(2)–C(20) | 107.60(18) | C(37)–N(4)–C(40) | 107.42(18) |
| C(17)–N(2)–P(2) | 132.12(14) | C(37)–N(4)–P(4) | 131.83(13) |
| C(20)–N(2)–P(2) | 120.19(16) | C(40)–N(4)–P(4) | 120.74(16) |
| C(7)–P(1)–C(1) | 105.30(8) | C(21)–P(3)–C(27) | 104.92(8) |
| C(7)–P(1)–C(100) | 105.72(8) | C(21)–P(3)–C(200) | 106.54(8) |
| C(1)–P(1)–C(100) | 106.65(8) | C(27)–P(3)–C(200) | 107.22(9) |
| C(7)–P(1)–S(1) | 114.15(7) | C(21)–P(3)–S(2) | 112.08(6) |
| C(1)–P(1)–S(1) | 111.91(6) | C(27)–P(3)–S(2) | 113.35(7) |
| C(100)–P(1)–S(1) | 112.51(6) | C(200)–P(3)–S(2) | 112.21(6) |
| N(1)–P(2)–N(2) | 102.00(8) | N(3)–P(4)–N(4) | 101.93(8) |
| N(1)–P(2)–C(100) | 101.14(8) | N(3)–P(4)–C(200) | 100.55(8) |
| N(2)–P(2)–C(100) | 98.48(7) | N(4)–P(4)–C(200) | 98.42(7) |
6 Conclusion
Analysis of the NMR spectra of the phosphine selenides 4–6 has revealed correlations of both the 1JPSe coupling constants and the δP chemical shifts with the electronic character of the parent phosphine. The pyramidal distortion observed around one of the nitrogen atoms in the crystal structure of 3 is not observed in the phosphine selenide 6. This suggests that, in contrast to tris(dialkylamino)phosphines, it is not intrinsic to the phosphine, but rather a consequence of the intermolecular interactions. The reaction of Ph2PCH2P(NC4H4)2 with sulphur to form 8 confirms that the diphenylphosphino group is easier to oxidise than the di(N-pyrrolyl)phosphino group.
7 Experimental
7.1 General experimental
Reactions were routinely carried out using Schlenk-line techniques under pure dry dinitrogen or argon, using dry dioxgen-free solvents unless noted otherwise. Microanalyses (C, H and N) were carried out by Mr. Alan Carver (University of Bath Microanalytical Service). NMR spectra were recorded on JEOL EX-270, Varian Mercury 400 and Bruker Avance 300 spectrometers referenced to TMS or 85% H3PO4. Compounds 1–3 [4] and Ph2PCH2P(NC4H4)2 [18] were prepared as previously reported.
7.2 Synthesis of SePPh2(NC12H8) 4
A mixture of 1 (0.146 g, 0.41 mmol) and selenium powder (0.120 g, 1.50 mmol) in toluene was stirred overnight. The solution was filtered and the solvent eliminated under reduced pressure. The resulting white powder was recrystallised from dichloromethane–hexane. Yield: 0.144 g (81%). Calc. for C24H18NPSe: C, 67.0; H, 4.22; N, 3.25. Found: C, 67.3; H, 4.27; N, 3.30%. 31P{1H} NMR (121.5 MHz, CDCl3): δ 54.4 (s, 1JPse 811 Hz). 1H NMR (300.2 MHz, CDCl3): δ 8.07–8.00 (m, 6H, Ho, H4,5), 7.64–7.59 (m, 2H, Hp), 7.55–7.49 (m, 4H, Hm), 7.26–7.22 (m, 2H, H3,6), 7.11–7.05 (m, 2H, H2,7), 6.68 (d, 2H, 3JHH 9 Hz, H1,8). 13C{1H} NMR (75.5 MHz, CDCl3): δ 141.1 (d, 2JCP 4 Hz, C10,13), 132.7 (d, 2JCP 12 Hz, Co), 132.5 (d, 2JCP 3 Hz, Ci), 130.1 (s, Cp), 128.8 (d, 3JCP 14 Hz, Cm), 126.8 (d, 3JCP 5 Hz, C11,12), 125.6 (s, C2,7), 121.7 (s, C3,6), 119.7 (s, C4,5), 115.2 (d, 3JCP 3 Hz, C1,8).
7.3 Synthesis of SePPh(NC12H8)2 5
As for 4 using 2 (0.92 g, 0.21 mmol) and selenium powder (0.049 g, 0.63 mmol) in toluene heated at reflux for 24 h. Yield: 0.102 g (93%). 31P{1H} NMR (121.5 MHz, CDCl3): δ 50.3 (s, 1JPse 873 Hz). 1H NMR (300.2 MHz, CDCl3): δ 8.18–8.10 (m, 2H, Ho), 8.03 (bd, 4H, J 9.0 Hz, H4,5) 7.75–7.69 (m, 1H, Hp), 7.59–7.52 (m, 2H, Hm), 7.29–7.16 (m, 8H, H3,6, H2,7), 7.11–6.99 (m, 4H, H1,8). 13C{1H} NMR (75.5 MHz, CDCl3): δ 140.9 (d, 2JCP 6 Hz, C10,13), 134.2 (d, 2JCP 12 Hz, Co), 131.8 (d, 2JCP 4 Hz, Ci), 130.1 (s, Cp), 129.2 (d, 3JCP 14 Hz, Cm), 127.1 (d, 3JCP 6 Hz, C11,12), 122.8 (s, C2,7), 120.0 (s, C4,5), 115.7 (s, C1,8).
7.4 Synthesis of SeP(NC12H8)3 6
As for 4 using 3 (0.125 g, 0.24 mmol) and selenium powder (0.880 g, 1.1 mmol) in toluene heated at reflux for 48 h. Yield: 0.110 g (75%). 31P{1H} NMR (121.5 MHz, CDCl3): δ 30.1 (s, 1JPse 942 Hz). 1H NMR (300.2 MHz, CDCl3): δ 8.09–8.04 (m, 6H, H4,5), 7.31–6.91 (m, 18H, H1,8, H2,7, H3,6). 13C{1H} NMR (75.5 MHz, CDCl3): δ 138.2 (d, J 5 Hz), 125.3 (d, J 7 Hz), 119.8 (s), 118.2 (s), 117.8 (s), 114.3 (s).
7.5 Synthesis of Ph2P(S)CH2P(NC4H4)2 8
Sulphur (0.089 g, 2.8 mmol) was added to a dichloromethane solution of Ph2PCH2P(NC4H4)2 (0.089 g, 0.25 mmol), and the mixture was stirred for 4 h. The solution was filtered to remove excess sulphur, and the volume of solvent halved under reduced pressure. On standing at –20 °C, yellow crystals of sulphur were obtained. These were separated by filtration, and the filtrate was recrystallised from dichloromethane–hexane to give a colourless powder, which was dried under reduced pressure. Yield 0.080 g (82%). Calc. for C21H20N2P2S·CH2Cl2: C, 55.1; H, 4.63; N, 5.84. Found: C, 55.1; H, 4.54; N, 6.00%. 31P{1H} NMR (161.8 MHz, CDCl3): δ 56.9 (d, 2JPP 85 Hz, PIII), 35.6 (d, 2JPP 85 Hz, PV). 1H NMR (399.8 MHz, CDCl3): δ 7.74-7.68 (m, 4H, Hm), 7.47–7.36 (m, 6H, Ho, Hp), 6.87 (ps. quin, 4H, Hα), 6.17 (ps. t, 4H, Hβ), 3.66 (dd, 2H, 2JHP 13.2, 1.2 Hz, CH2). 13C{1H} NMR (75.5 MHz, CDCl3): δ 131.7 (s), 131.5 (s), 130.7 (d, J 10 Hz), 128.5 (d, J 12 Hz), 123.4 (d, J 17 Hz, Cα), 112.3 (d, J 3 Hz, Cβ), 37.4 (dd, 1JCP 50, 26 Hz, CH2).
7.6 Crystallography
Single crystals of compounds 2·CH3C(O)CH3, 3, 7 and 8 were analysed using a Nonius Kappa CCD diffractometer and molybdenum radiation throughout. Details of the data collections, solutions and refinements are given in Table 5. The structures were solved using SHELXS-97 [20] and refined using SHELXL-97 [21]. Absorption corrections (semi-empirical from equivalent reflections) were applied to data for 3, 7 and 8. [max./min. transmission factors 1.06 and 0.92, 0.88 and 0.81, and 0.90 and 0.83, respectively].
Crystallographic information for compounds 2·MeC(O)Me, 3, 7 and 8
| Compound | 2·MeC(O)Me | 3 | 7 | 8 |
| Formula | C33H27N2OP | C36H24N3P | C36H24N3PSe0.65 | C21H20N2P2S |
| M | 498.54 | 529.55 | 580.88 | 394.39 |
| T/K | 150(2) | 170(2) | 150(2) | 150(2) |
| λ/Å | 0.71073 | 0.71073 | 0.71073 | 0.71073 |
| Crystal system | monoclinic | monoclinic | monoclinic | monoclinic |
| Space group | P21 | P21/n | P21/c | P21/n |
| a/Å | 8.7170(1) | 9.8160(2) | 16.5450(3) | 13.0640(1) |
| b/Å | 15.2970(3) | 7.8210(1) | 9.3500(2) | 14.2480(2) |
| c/Å | 10.4140(2) | 33.8940(6) | 18.2990(4) | 22.5130(3) |
| β/° | 114.318(1) | 93.507(1) | 102.669(1) | 104.201(1) |
| U/Å3 | 1265.43(4) | 2597.20(8) | 2761.86(10) | 4062.42(8) |
| Z | 2 | 4 | 4 | 8 |
| ρcalc/g cm–3 | 1.308 | 1.354 | 1.397 | 1.290 |
| μ/mm–1 | 0.139 | 0.138 | 0.988 | 0.324 |
| Crystal size/mm | 0.20 × 0.20 × 0.20 | 0.50 × 0.40 × 0.20 | 0.40 × 0.10 × 0.10 | 0.60 × 0.50 × 0.40 |
| Reflections collected | 24,788 | 37,362 | 42,557 | 48,958 |
| Independent reflections |
|
|
|
|
| R1, wR2 [I > 2σ(I)] | 0.0372, 0.0810 | 0.0483, 0.1013 | 0.0476, 0.1044 | 0.0479, 0.1335 |
| R indices (all data) | 0.0509, 0.0864 | 0.1027, 0.1194 | 0.0635, 0.1093 | 0.0591, 0.1448 |
| Flack parameter | –0.10(7) |
Compound 7 is an average of 65% SeP(NC12H8)3 and 35% P(NC12H8)3. Compound 8 crystallises with two independent molecules in the asymmetric unit.
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (CCDC 271732–271735) and can be obtained by contacting the CCDC.
Acknowledgements
The EPSRC is thanked for financial support.