

[60]Fullerene is a molecule with a remarkable electron acceptor character which has been skilfully used for the preparation of a wide variety of different electroactive systems [1]. According to theoretical calculations, it exhibits a triply degenerated LUMO (t1u) comparatively low in energy. Therefore, C60 behaves like an electronegative molecule which reversibly accepts up to six electrons in solution [2]. The cyclic voltammetry and the electron affinity measured for [60]fullerene clearly confirm that it is a moderate electron acceptor comparable to other organic molecules such as benzo- and naphtho-quinones. [3] This electron-accepting ability of C60 to form stable multianions [4] is in sharp contrast to the ability to generate the very unstable cationic species [5].
It is well known that most of the [60]fullerene derivatives present poorer electron acceptor properties than the parent C60 as a consequence of the saturation of a double bond of the C60 framework that raises the LUMO energy [6]. Therefore, different strategies have been followed for increasing the electron acceptor character in fullerene derivatives, which have been mainly focused on (i) the presence of electronegative atoms directly linked to the C60 core, (ii) electron acceptor groups, (iii) periconjugative effect, (iv) pyrrolidinium salts, (v) the presence of heteroatoms as constituents of the fullerene framework (heterofullerenes) and (vi) fluorofullerenes [7]. Some of these aspects have previously been reviewed [1] and, therefore, in this paper we will highlight the most remarkable examples reported during the last years.
A structurally simple type of electron acceptor organofullerenes is [60]fullerene-p-benzoquinone systems (1a–c, 2) which have been prepared from substituted o-quinodimethanes by reaction with C60 (Fig. 1) [8,9]. These cycloadducts (1a–c, 2) show three or four reduction waves at potential values similar to those of C60, which should be attributed to the fullerene moiety [8,9b, c]. The reduction waves, as expected, are shifted to more negative values in comparison to the parent C60, due to the saturation of a double bond [10]. In addition, another reduction wave appears at potential values very close to the first reduction potential of the C60 moiety. The CV values were rationalised on the basis of theoretical calculations [9b] and, according to the molecular orbital distribution, the attachment of the first electron in the reduction process takes place either in the p-benzoquinone fragment or in the C60 cage. Thus, whereas the introduction of two bromine atoms linked to the C60 moiety in 1c causes the stabilization of the LUMO of this moiety, the presence of a benzene ring fused to the quinone moiety in 1b, in contrast, produces a destabilization of the LUMO of the organic addend [11] which now lies above those of the C60 core. By comparison, the first reduction potential found for 2 [9c] can be considered as the sum of two different effects: the presence of the bromine atoms which shifts the reduction potential towards more positive values (stabilization of the LUMO) and the fused benzene ring which raises the LUMO energy [11].
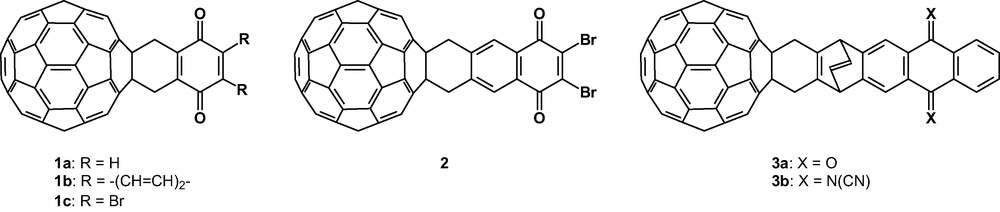
Examples of [60]fullerene-p-benzoquinone (1a–c, 2, 3a) and [60]fullerene–DCNQI (3b) acceptors.
Therefore, depending upon the nature of the substituents on the p-benzoquinone ring, it is possible to address the attachment of the first electron in the reduction process either to the C60 core or to the organic addend [9b].
Diekers et al. [12] reported the synthesis and electrochemical study of other Diels–Alder C60–acceptor dyads bearing either an anthraquinone (3a) or a dicyano-p-quinonediimine DCNQI (3b) fragment which contain a rigid linkage between the redox active centres. The electrochemical study of these compounds shows that the reduction processes are highly localised either on the fullerene core or on the addend, depending on the relative electron affinities. Thus, in the case of 3a, the first reduction is fullerene based, while for 3b the attachment of the first electron is addend-based. ESR spectroscopic data allowed unambiguous assignment of the location of each reduction step being consistent with the electrochemical observations, with the exception of that for the penta-anion of 3b, which shows that spin localization occurs exclusively on the anthraquinone moiety. This observation suggests that a diamagnetic tetra-anion is present on the fullerene group and an unpaired spin is localised on the addend, contrary to the electrochemically predicted electron distribution with a fullerene trianion and a quinone dianion.
Other [60]fullerene-based electron acceptors have also been prepared by following Prato's procedure [13,14].
The interesting redox properties of compounds 4–6 can also be understood in terms of the LUMO's energies as described above for the related systems. Thus, the attachment of the first electron in the reduction process takes place in the organic addend in compounds 4, 5 and 6b–c, while for the anthraquinone derivative 6a the first reduction process is fullerene-based.
Dyads 2 (Fig. 1) and 6b (Fig. 2) have been studied in the search for a possible electron transfer from the photoexcited fullerene moiety to the covalently linked electron-acceptor [15]. Formation of the fullerene triplet excited state rather than an intramolecular electron-transfer was observed, which can be accounted for by the inability of C60 to form stable cationic species [5,16].
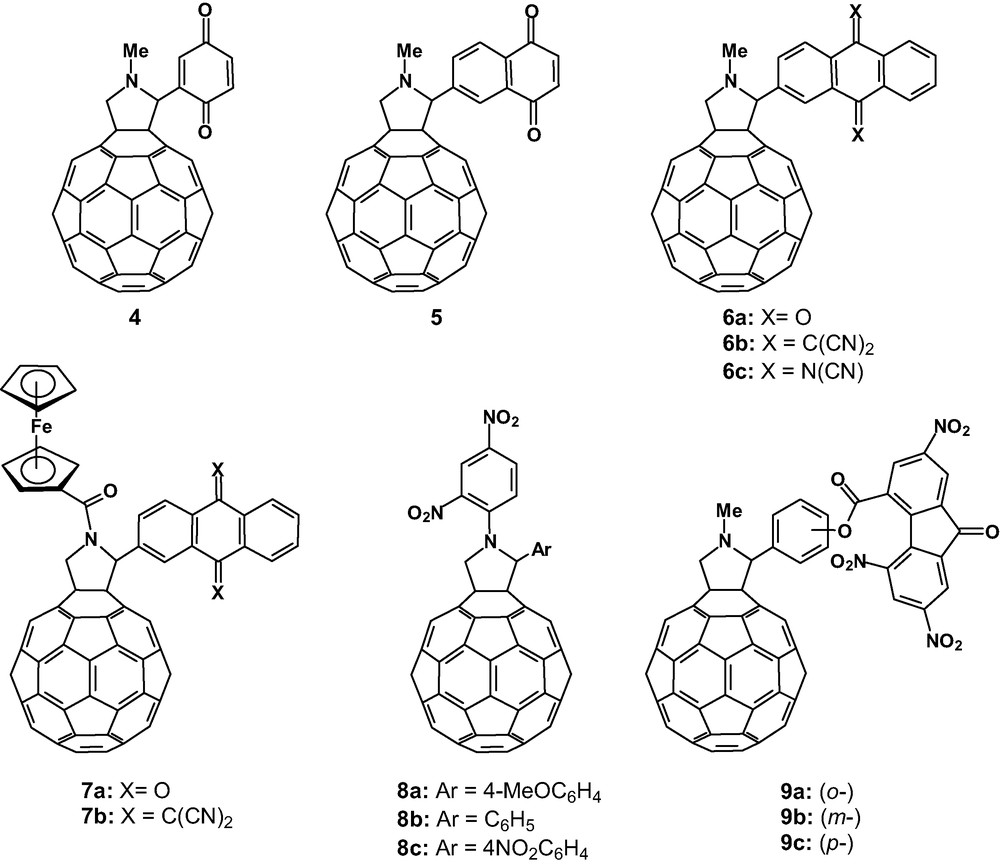
Different fulleropyrrolidines endowed with strong electron-accepting moieties.
Anthraquinone (AQ) and 11,11,12,12-tetracyano-9,10-anthraquinodimethane (TCAQ) were also used as acceptor moieties for the design of C60-based triads constituted by fullerene and AQ (7a) or TCAQ (7b) as acceptor units, which are weaker and stronger acceptors, respectively, relative to the parent C60, and ferrocene as donor unit [17]. The electrochemical study of these triads shows the presence of three one-electron reduction steps, all of them taking place at the [60]fullerene moiety, at potential values quite similar to those found for the parent C60. A possible reasoning for this observation stems from a combination of, first, the carbonyl group linked to the pyrrolidine nitrogen [18] and, second, the electronic effect of the electron-acceptor unit bound to the pyrrolidine ring.
The photophysical study of these triads (7a–b) revealed that only a reaction between Fc as donor and C60 as acceptor takes place, despite the fact that the reduction potential of TCAQ in triad 7b renders to be easier than the fullerene first reduction [17].
In compounds 8a–c, the presence of the nitro substituents in the N-phenyl ring has a striking influence on the reduction potential values [19]. Derivative 8c, containing a second electron-withdrawing 4-nitrophenyl group, resulted to be a slightly better acceptor than the parent C60. The electrochemical behaviour observed for 8a–c has been rationalised on the basis of PM3 calculations finding a correlation of the first reduction potential with the LUMO level of these compounds.
The synthesis of novel trinitrofluorenone–C60 (TNF–C60) acceptors in which the two subunits are placed at different distances (9a–c) has been recently reported [20]. The electrochemical study of these dyads shows a first reduction potential which is TNF-based and appears 220–260 mV anodically shifted in comparison with reference compound N-methylfulleropyrrolidine. The photorefractive properties of these compounds as sensitisers in poly(N-vinylcarbazole)-based composite materials have also been studied, finding some advantages with respect to the use of the parent C60.
A simple procedure to improve electron affinity of [60]fullerene derivatives in comparison to the parent C60 consists on the incorporation of electronegative atoms or electron-deficient carbon atoms directly connected to the C60 core. In this sense, apart from isoxazolofullerenes (see Fig. 3), triazolino and pyrazolino[60]fullerenes (see Fig. 4) have also been reported. Other electron-withdrawing substituents directly connected to the C60 cage [21] or through a methano bridge [21c] also present stronger acceptor properties than the parent C60.

Examples of isoxazolofullerenes as electron acceptors.

Examples of pyrazolino[60]fullerenes (15, 16) and triazolino[60]fullerenes bearing TTF (17) and exTTF (18a,b) units.
The introduction of an acceptor substituent as the pentafluorophenyl group in compounds 10a–b also led to an anodic shift of the reduction potentials of these compounds of 40–70 mV relative to the naked C60 [22]. To study the substituents' effects on the redox properties of isoxazolofullerenes, Irngartinger et al. [23] prepared a series of these derivatives. Thus, for compound 11a, with R = H, the electrochemical study showed a shift of 50 mV to more positive values compared to C60 for the first reduction potential, due to the strong electron-withdrawing effect of the heterocycle. A remarkable anodic shift of the reduction potential is also observed for diester derivative 11b (80 mV relative to pristine C60). For other substituents studied, the authors concluded that, not only the donor or acceptor characteristics of the substituent, but also the distances or geometries have an influence on the redox properties of the adducts, as shown by CV experiments. Thus, for example, compound 11c shows a redox potential which is 40 mV cathodically shifted relative to 11a. This behaviour can be explained in terms of through-space interactions between the heterocycle and the fullerene surface, which are favoured by the orientation of the addend.
In the same way, donor–acceptor dyads 12a–b show an anodic shift of 50–70 mV of the reduction potential values in comparison to the parent C60 [24].
The enhanced electron accepting properties of isoxazolo[4′,5′:1,2][60]fullerenes related to C60 prompted us to synthesise derivatives 13–14 bearing strong electron acceptor addends [14] (Fig. 3).
In contrast to the above mentioned pyrrolidino[3′,4′:1,2][60]fullerenes endowed with acceptor moieties (Fig. 2), these isoxazolofullerenes exhibit reduction potentials which are anodically shifted (70–110 mV) in comparison with the parent C60, which indicates that they are stronger electron acceptors than [60]fullerene. This finding can be accounted for by the presence of the oxygen atom directly linked to the C60 cage as well as by the electronic effect of the organic addend covalently attached to the isoxazole ring.
Similar effects on the reduction potentials can be observed in pyrazolino[60]fullerenes (15–16), which are easily prepared in one pot by 1,3-dipolar cycloaddition reaction from suitably functionalised hydrazones (Fig. 4) [25].
These pyrazolino derivatives present a remarkable advantage over the isoxazolino[60]fullerenes mentioned above to obtain electroactive compounds due to the possibility of preparing donor–donor–C60 or donor–acceptor–C60 triads by modification of the nature of the substituents linked to the sp3 nitrogen atom. All compounds 15a–c and 16a–c possess better electron affinity than the corresponding pyrrolidino[60]fullerene (by up to 170 mV), and all except 16b present an anodically shifted first reduction potential (up to 80 mV) in comparison with the parent C60. The inductive effect of the substituents must be one of the most important factors in determining the observed redox behaviour [25b].
Fullerotriazolines (17–18) [26,27] can also be utilised to form fullerene derivatives with enhanced electron accepting properties. These compounds are thermally labile intermediates in the reaction of C60 with azides [28]. As intermediates, they are of interest as precursors by thermal nitrogen extrusion of azafulleroids (opened 5,6-adducts) and fulleroaziridines (closed 6,6-adducts) [29].
The voltammogram of compound 17 shows, apart from the two reversible one-electron oxidation waves yielding the radical cation and dication of the TTF moiety [E1ox = 0.57 V and E2ox = 0.70 V (vs. SCE)], four quasireversible one-electron steps on the reduction side. These reduction waves correspond to the fullerene reductions [–0.57, –0.97, –1.52 and –2.00 V (vs. SCE)] and are remarkably shifted to more positive values (~100 mV) relative to other 1,2-dihydrofullerenes such as pyrrolidino[3′,4′:1,2][60]fullerenes as well as to the parent C60 (–0.60 mV vs. SCE). This shift can be explained by the electronegative character of the two nitrogen atoms covalently linked to the C60 core. Interestingly, the photophysical study of this compound shows than fullerotriazoline-TTF dyad gives rise to a reduced HOMO–LUMO energy gap (–5.12 eV) relative to previously reported fulleropyrrolidine–TTF dyads (–5.35 eV) [18,30]. This has a large effect on the electron transfer rates, which are similar to those observed in systems where the spatial separations between the donor and acceptor are markedly decreased [31]. Thus, increasing the electron accepting behaviour of the C60 moiety in C60-based dyads such as fullerotriazolines 17 and 18a–b can be used as a useful tool to improve (i) the dynamics of the forward electron transfer and (ii) the lifetime of the charge separated (CS) state. However, it should be stated that these advantages are only observed if the forward electron transfer is located in the normal Marcus region and the back electron transfer in the inverted Marcus region [32].
For dyads 18a–b the reduction potentials are also anodically shifted by 10–30 mV in comparison to pristine C60 [27]. Steady-state fluorescence experiments and transient absorption spectroscopy results in the formation of a radical pair, showing the characteristic fingerprint of C60●– and exTTF●+, with lifetimes in the range of hundred of nanoseconds, nearly two orders of magnitude larger than those found for 17.
A different approach for the preparation of electron acceptor organofullerenes is based on the periconjugative effect [33]. Thus, some quinone-type methanofullerenes were prepared (19–21) [34,35] (Fig. 5).
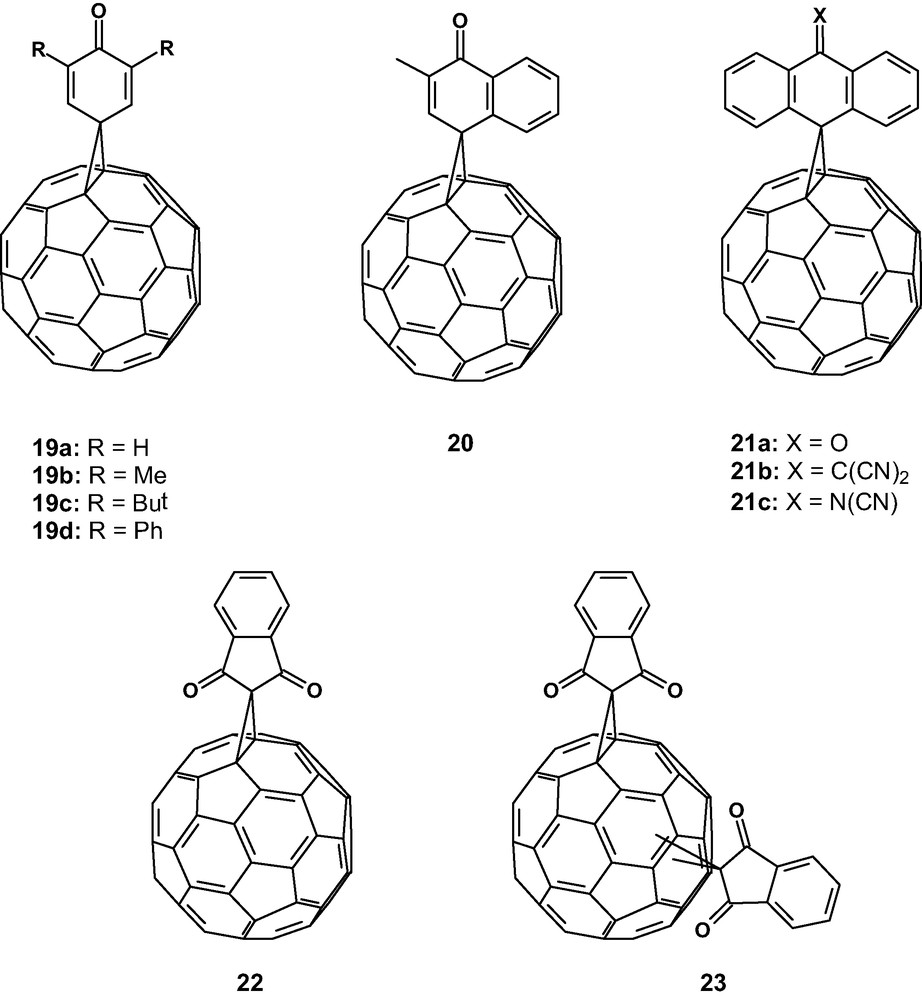
Spiro methanofullerene derivatives showing periconjugative effect.
In principle, the intramolecular electronic interaction (periconjugation) between the pz–π orbitals of the olefinic carbons of the quinone moiety and the adjacent carbon atoms of C60, separated by a spiro carbon atom, results in more extended conjugated molecules showing good acceptor properties. In agreement with these predictions, the CV data of compounds 19–20 showed a less negative first reduction potential than the parent C60, thus being stronger acceptor molecules than C60. In contrast, compounds 21 showed a more negative first reduction potential than naked C60, which has been accounted for by the steric hindrance between the “peri” hydrogens of the organic addend and the surface of the ball leading to a significant deviation from the orthogonality and hence to the loss of periconjugation.
Theoretical calculations on compounds 19a–c indicate that the attachment of the first electron causes the homolytic cleavage of one of the bonds connecting the addend to C60. This ring opening has been supported by EPR measurements and explain the irreversible electrochemical behaviour of these compounds [34b].
This bond cleavage leading to an irreversible electrochemical behaviour related these compounds with the retro-Bingel reaction, an electrochemical reduction reaction which efficiently removes the cyclopropane ring adduct from the fullerene surface resulting in the formation of the parent C60 [36]. Thus, we decided to study some of these derivatives (19a, d, 20, 21a, 22, 23) under controlled potential electrolysis (CPE) for potential adduct removal. It is remarkable that all these compounds except 21a show stronger electron-accepting properties that the parent C60 [37].
Reductive electrolysis of these methanofullerenes led to different results. Thus, CPE of 20, 21a, 22, and 23 in dichloromethane results in removal of the addend to form C60. An alternative reaction pathway was observed for 19a, d and 20 which leads to the formation of methanofullerene–CH2Cl products of unspecified structure. Reductive electrolysis in THF shows clean adduct removal for 20 and 21a, 22–23. Another reaction pathway was observed for 21a and 22 in THF, an intramolecular addend crossover reaction leading to bis-adducts (Fig. 6). The regioisomer distribution of these bis-adducts obtained by electrolysis was determined for compound 22 and resulted to be clearly different from that obtained by the regular synthetic route. The electrochemical formation of the cis-3 bis-adduct as one of the predominant regioisomers is particularly remarkable [38].
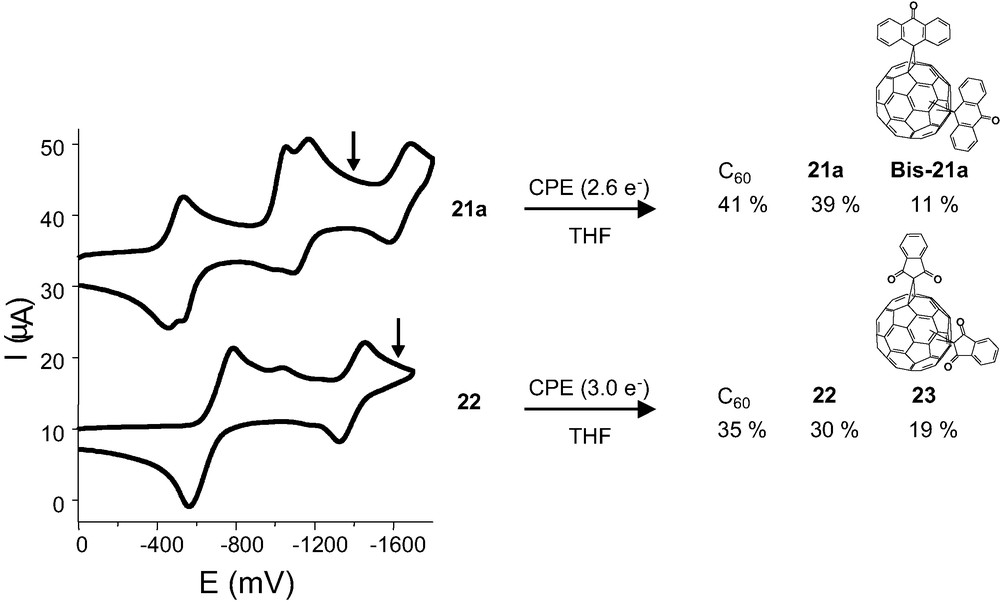
Cyclic voltammetry of compounds 21a and 22 in THF and CPE results.
In order to investigate more deeply these new electrochemically-induced reactions we prepared compounds 24–26 containing a nitrophenyl group which exhibits a strong and easily recognizable ESR signal when reduced [39] (Fig. 7).
Methanofullerenes endowed with an ESR active nitrophenyl group.
The electrochemistry of these products showed a similar behaviour for 24 and 25, exhibiting three fullerene reductions and one reversible nitrobenzene reduction. Compound 26 shows more chemical irreversibility due, probably, to the major extent in which the retro-Bingel reaction occurs (Fig. 8). Typically, each compound was subjected to CPE at a potential ca. 100–150 mV more cathodic than every reduction wave and then analysed by ESR spectroscopy. Reoxidation at 0 V was usually performed after bulk electrolysis at the third of fourth reduction potential followed by analysis of the products.
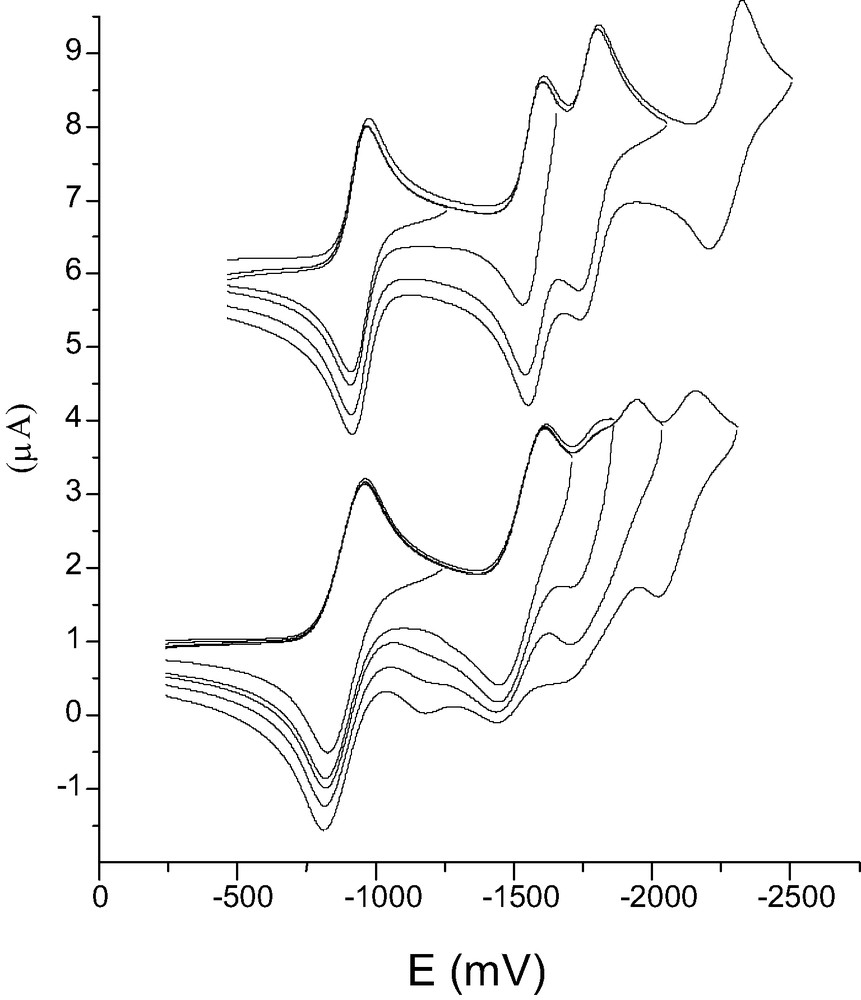
Cyclic voltammetry of compounds 25 and 26 in TFH vs. Fc.
CPE of methanofullerene 24 presented, after reoxidation and purification, analysis of the reaction mixture by HPLC and MALDI-TOF, the formation of a small amount of C60 (5%) and recovery of 40% of starting material. CPE for compounds 25 and 26 resulted in the recovery of C60 (23% for 25 and 52% for 26), formation of bisadducts (18% for 25 and 11% for 26) and the starting material (40% for 25) or an uncharacterised material (37% for 26).
ESR spectroscopy allowed unambiguous assignment of the sites of reduction and monitoring of the reactive intermediates resulting from subsequent reductions. ESR spectra for the reduction of 26 in THF is shown if Fig. 9. The ESR signals during the different stages of the electrolysis showed how spin localization changed from fullerene to nitrobenzene-based and allowed the measurement of electronic effects induced by electron withdrawing substituents. The simulated spectrum (Fig. 9, e) clearly shows that the radical generated contains a single spin 1/2 nucleus, most probably a proton, which exhibits a hyperfine coupling constant of 0.40 G. A possible structure for this radical is shown in Fig. 9. Exactly how this transformation takes place is not presently known in detail, but it fits well with the ESR observations and with the known formation of C60 via the retro-Bingel reaction as a final product after reoxidation.

Experimental and simulated ESR spectra of 26 in THF solution.
Fulleropyrrolidinium salt derivatives of C60 have also shown to have enhanced electron-accepting properties with respect to both the parent pyrrolidine derivatives and C60. Remarkably, CV measurements of compounds 27a–c (Fig. 10) performed at low temperatures and fast scan rates, using ultramicroelectrodes, have allowed the observation for the first time in fullerene derivatives, of six C60-centred reductions [40]. Since the chemistry of fulleropyrrolidines is very rich [18], formation of pyrrolidinium ions, especially in donor–bridge–acceptor dyads, is a valuable methodology to modulate the electronic properties of the C60 moiety.
Fulleropyrrolidinium salts with enhanced electron-accepting properties.
In order to promote water solubility and to preclude fullerene clustering in aqueous media, bis(pyrrolidinium) salts (28a–d) [41] were also investigated. Considering the reduction potential of C60 (–0.35 vs. SCE) and that of the monopyrrolidinium salt (–0.29 vs. SCE), reduction potentials of bis(pyrrolidinium) salts 28 are between –0.32 and –0.34 V vs. SCE. This enhanced electronegativity can be reasonably interpreted in terms of inductive effects which leads to a decrease of the electron density within the fullerene π-system.
For the development of novel donor–acceptor arrays that display improved performance, the use of a fulleropyrrolidinium acceptor clearly has a distinct advantage over fulleropyrrolidine (29–31) [42]. Interestingly, the fulleropyrrolidinium-based dyads reveal faster charge separation dynamics, relative to the fulleropyrrolidine analogues, a trend that parallels their better acceptor properties. Regarding charge recombination, formation of the zwitterionic fullerene π-radical anion appears to be a key factor for stabilizing the charge-separated state.
The CV study of a series of novel bisfulleropyrrolidines (32) and bisfulleropyrrolidinium ions (33) has also been reported [43]. The eight possible stereoisomers of each series were systematically investigated. The stabilizing effect of positive charges produced a significant enhancement of the electronegative properties in 33. The study evidenced that in both 32 and 33, the CV pattern and, in particular, the potential separation between the second and third reductions, changes significantly with the addition pattern. A sequential π-electron model that simulates the effect of subsequent reductions of C60 bis-adducts gives a good correlation (r > 0.96) with the cyclic voltammetry data when the molecules are divided in two sets dependent on the location of the addends in the same or in opposite hemispheres.
Bourgeois et al. ([44]) have reported positive shifts in the fullerene reduction potential (ΔE = 90 mV) of a C60–dibenzo–18-crown-6 conjugate (34) (Fig. 11) upon complexation of a potassium cation due to the electrostatic effect of K+ bound in close proximity to the carbon sphere. Similar effects have also been observed by D'Souza in compound 35 by complexation with different cations [45]. It is important to note, however, that in these systems (34, 35) the first reduction potential value of the fullerene unit is cathodically shifted in comparison with pristine C60 due to the saturation of two double bonds of the fullerene cage which raises the LUMO energy in these compounds. Similarly, the positive shift in potential observed for the reduction of 37 relative to the uncomplexed compound 36 is attributed to an electrostatic effect able to counterbalance the electronic interactions between the bis(2,9-diphenyl-1,10-phenanthroline)Cu(I) core and the C60 units that tend to negatively shift the reduction potentials [46].

Representative examples of fullerene complexes with better reduction potentials than pristine [60]fullerene.
An attractive alternative to isocyclic fullerenes, as components of multiredox and light-responsive systems, are heterofullerene building blocks (i.e. C59N) [47]. In azaheterofullerene derivatives, substitution of a carbon atom with nitrogen alters the redox potential and the excited state energies of the fullerene ([48]. Significantly, C59N derivatives render better electron acceptors than C60 and C60 derivatives that bear similar addends. The simplest azaheterofullerene can only be isolated as its corresponding dimer (38) (Fig. 12). The C–H bonded monomer, HC59N, has also been characterised [48]. The monomeric C59N+ cation, which is isoelectronic with C60, can be isolated as a carborane anion salt [C59N+] [Ag(CB11H6Cl6)–2] [49]. The synthesis involves a rare example of oxidation of an sp3–sp3 C–C bond to produce a carbenium ion.

Dimer (38) and monomers (39) azaheterofullerenes show good electron-acceptor properties.
Recently, Hauke et al. [50] have reported a novel series of fluorophore-heterofullerene conjugated (39) in which the flexibility of the acetyl-group linker opens the way for conformations with π–π stacking interactions between the chromophores. Photophysical steady-state and time-resolved measurements clearly show the photosensitization effect of the fluorophores that act as an antenna system and transmit their excited-state energy to the covalently attached C59N moiety. This approach offers a viable alternative for developing well-defined architectures, bearing a C59N core, which give rise to sequential energy and electron transfer processes [50].
Summary and perspectives
Although fullerene C60 is known to exhibit a good electron acceptor ability, the chemical modification of fullerenes has been skilfully used to enhance the electronegative character of substituted fullerenes. Different strategies have been developed mainly involving the presence of exohedral or constituent electronegative or electron withdrawing groups either as neutral or charged species and some of the most significant examples have been highlighted in this review. The remarkable negative inductive effect of these substituents on the fullerene network compensates the saturation of a double bond of the fullerene structure in the functionalization process resulting in stronger electron acceptors than the parent C60. Other different and more subtle strategy is based on the periconjugative effect. However, more work is needed in order to clarify the extent of this through space electronic interaction.
Improved C60-based electron acceptors have been covalently connected to different electron donor units and faster photoinduced charge separation processes have been observed. These remarkable photophysical features are of interest for the design of novel fullerene derivatives with improved properties for the preparation of photovoltaic devices.
In summary, highly efficient electron acceptor fullerenes are able to act as real electron-accepting sponges forming multianion species with theoretical and synthetic interest, which have only been scarcely studied. Furthermore, the synthesis of modified fullerenes showing stronger electron accepting character than pristine C60 is still a challenge for the design of new molecules of interest in fields such as artificial photosynthetic systems and photovoltaic devices where electron transfer from a donor unit to the fullerene core plays a key role.
Acknowledgements
Support of this work by MCYT of Spain (BQU-2002-00855) is gratefully acknowledged.