

1 Introduction
Nuclear magnetic resonance is recognized as one of the most useful and innovative techniques available to chemists and biologists for determining the structures of molecules. Indirect nuclear spin–spin coupling provides, via the J constant, key data for compound characterization, regardless of the nature (organic, organometallic) or the size (small molecules, pharmaceuticals, biomolecules) of the observed systems. For many years, this electron-mediated J-coupling has been commonly thought as only transmitted by covalently bonded “magnetic” atoms. However, a significant number of experimental and theoretical reports now bring to the fore the existence of J-couplings operating “through-space” (TS). They range from small (ca. 4 Hz) to very large values (higher than 60 Hz). They are often called “long-range” couplings in the literature, the corresponding nuclei being separated by at least four covalent bonds. However, this term is somewhat misleading since with the exception of fully conjugated systems, spin–spin transmission does not occur through the many bonds that link the two remote, interacting atoms. The “long-range” coupling is in fact a consequence of a direct nuclear–nuclear nonbonded, electron-mediated interaction. The present account aims at highlighting the “through-space” contribution of indirect nuclear spin–spin in various polyphosphane ligands, notably ferrocenyl phosphanes and phosphino cavitands. The NMR data will be correlated to solid state and computer modelled structures.
2 Background of “through-space” NMR spin coupling
2.1 Origin and scope of “through-space” coupling
Since Roberts et al. reported in the sixties the observation of “long-range” spin–spin coupling constants apparently transmitted via spin polarization of nonbonded-electron pairs (named later “through-space” couplings, TS) many papers were published reporting a wide variety of magnetic coupling situations related to this phenomenon [1]. It was clearly established that organic compounds containing two fluorine atoms separated by four or more bonds, but spatially close, exhibit large FF nuclear spin coupling constants J(F,F) [2]. This phenomenon, best designated as nuclear spin–spin coupling via nonbonded interactions, has been intensively studied for over thirty years by Mallory and co-workers, who produced a useful combination of experimental and theoretical works [3]. These “through-space” couplings first identified from FF J-constants in small organic molecules, were shown to be a general phenomenon encountered for nuclei as different as 14N, 31P [4], 77Se, 13C and even 1H when involved in hydrogen bonding. The studies reviewed by Contreras et al. provided a more theoretical and predictive background for the observed phenomenon. These are based on modelling and calculation approaches, which take into account the four terms contributing to J(X,X′), namely the Fermi contact (FC), spin dipolar (SD), paramagnetic spin–orbit (PSO) and diamagnetic spin–orbit (DSO) interactions [5,6].
2.2 Geometrical approach and simplified modelling
The studies conducted by Mallory showed that “through-space” couplings result from overlap interactions between lone-pair orbitals on the two crowded atoms. For instance, in the particular constrained geometry of the compounds sketched in Fig. 1 (top), the C–F/C–F, or C–F/C–N bonds, are coplanar and approximately parallel. As a consequence, the nonbonding distances d F⋯F, or d F⋯N, are short. In this orientation, the two lone-pair orbitals experience a σ-directed overlap. As displayed in Fig. 1 (bottom), the overlap between these lone-pair orbitals is expected to afford an in-phase and out-of-phase combination. As both orbitals are occupied (two-orbitals, four-electron interaction) no stabilization is observed. However, it provides an adequate pathway for transmitting spin–spin information between the coupled nuclei. The magnitude of J(F,F) depends on the extent in which the two lone-pair orbitals interact as a result of their overlap.

Bonding and anti-bonding orbitals generated by the overlap of two lone-pair orbitals on intramolecularly crowded nitrogen- and fluorine-containing constrained molecules.
This model, which initially was mainly qualitative [2,3a,3b], eventually allowed the so-called overlap interaction to be quantitatively estimated for a number of 1,8-difluoronaphthalene derivatives. This became possible once an exponential relationship between the ab initio calculated internuclear distances and the observed J(F,F) coupling was established. This work confirmed previous studies by Ernst and Ibrom, who came up with a similar valuable semi-quantitative relationship in a number of difluorocyclophanes [7].
2.3 General requirements for the occurrence of “through-space” nuclear spin–spin coupling
In the general approach, the nuclear spin–spin coupling constant J(X,X′) is expressed as the sum of three terms (Eq. (1)):
| (1) |
Empiric requirements for the detection of “through-space” coupling constants were detailed in studies dealing with FF [3a,3e,3g], NF [3d,3f] HF or CF [3b,3c], and occasionally with PX spin–spin couplings [3g,8a–d] (with X = P, Se, F). They can be expressed as follows: (i) chemically identical nuclei in interaction need to be anisochronous. In other words, the molecule must be in some way dissymmetric as regards the environment of the observed nuclei; (ii) the nuclei must be in close proximity in solution, as in Mallory's fluoroaromatic compounds (Fig. 1) and in related phosphinonaphthalenes, which both contain rigid backbones [4,9]. The same is also true for the P-containing polymetallic catenates reported by Rheingold and Fountain [10] as well as for some bis(ferrocenyl)diphosphanes [11]; (iii) finally, each of the interacting nuclei should usually bear a lone-pair for mutual overlap. However, recent findings contradict this last assertion (see below).
3 “Through-space” 31P31P spin–spin couplings in constrained diphosphane and polyphosphane ligands
3.1 Ferrocenyl tri- and tetraphosphanes and their coordination complexes
A number of polyphosphanes with interesting structural and catalytic properties appear every year in the literature. Among them the ferrocenyl tetraphosphane (1,1′,2,2′-tetrakis(diphenylphosphino)-4,4′-di-tert-butylferrocene (Fc(P)4tBu, Fig. 2 left)) was recently shown to promote various palladium-catalysed C–C cross-coupling reactions [12,13]. This was attributed to a conformationally blocked ferrocene backbone, which allows this ligand to bind the metal centre by three instead of only two phosphorus atoms, incidentally increasing the electron density on the metal. This behaviour contrasts with that of related ferrocenyl tetraphosphanes in which the Cp rings rotate freely (Fig. 2) [14].
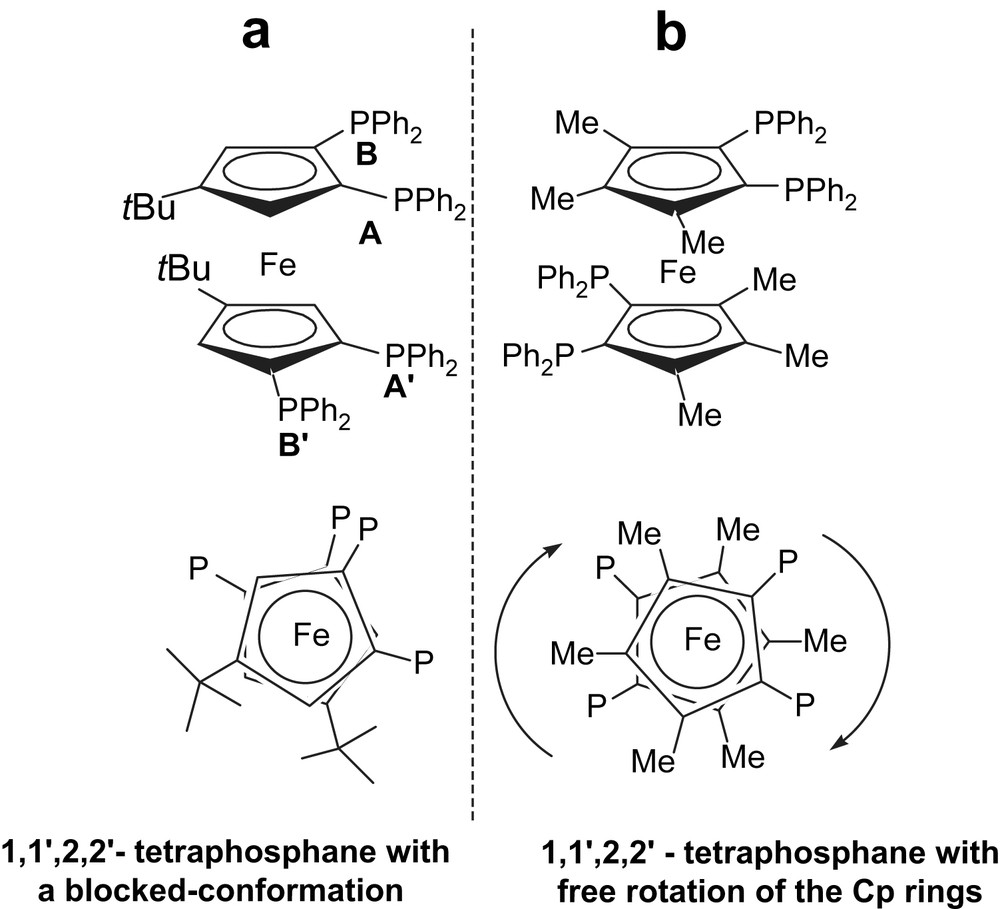
Ferrocenyl tetraphosphanes with different conformational properties: blocked (left) or flexible (right) ligands.
The conformational rigidity of Fc(P)4tBu was inferred from the corresponding 31P NMR spectrum which shows an AA‘BB’ system (Fig. 3). The occurrence of nonbonded nuclear spin couplings between phosphorus atoms was clear from the intense J(P,P) coupling constant (60 Hz) found between the heteroannular P atoms corresponding to A and A′ (see Fig. 2). From X-ray structure analysis a quasi eclipsed conformation is observed for these nuclei, with lone-pairs orientated towards each other (Fig. 3, internuclear distance determined in the solid state from X-ray measurements was d P⋯P′ = 3.728(2) Å). In keeping with Mallory's model, the phosphorus lone-pairs of Fc(P)4tBu undergo a strong overlap which is responsible for the observed direct “through-space” spin–spin interaction.
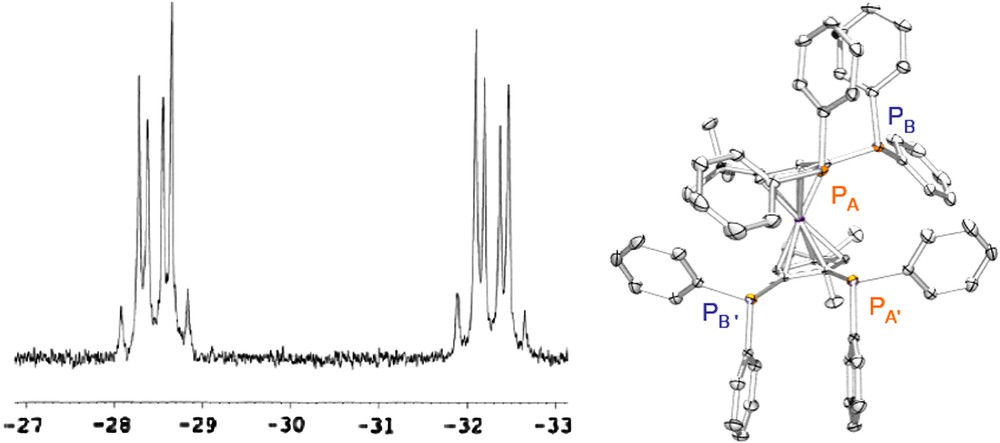
31P NMR spectrum showing the AA‘BB’ system and X-ray structure (POV-Ray) of Fc(P)4tBu. The phosphorus atoms A and A′ are eclipsed and their lone-pairs point towards each other. The large nonbonded J(P,P) coupling constant (60 Hz) originates from the overlapping P orbitals.
In the course of NMR-mechanistic studies on transition metal complexes derived from the tetraphosphane ligand Fc(P)4tBu [15], unusual features that can only be explained by the presence of TS spin couplings were detected in their 31P NMR spectra (See Fig. 4). In order to gain more in depth information on the nuclear spin–spin interactions in these coordinated tetraphosphanes, the palladium complexes [PdX2{Fc(P)4tBu}], [Pd2X4{Fc(P)4tBu}] (X = Cl or Br), as well as their nickel analogs [NiX2{Fc(P)4tBu}] and [Ni2X4{Fc(P)4tBu}] were synthesized and characterized by X-ray crystallography in the solid state and 31P NMR in solution [16]. The coordination complexes of Fc(P)4tBu keep the conformational rigidity of the free ligand in solution. As a result, the same type of spin–spin coupling seen in the ligand was detected in the complexes. Data presented in Table 1 clearly establish a correlation between the P⋯P′ separations between non-adjacent phosphorus atoms (determined from crystallographic data) and the corresponding coupling constants.

Example of a palladium dibromide tetraphosphane complex. X-ray structure (POV-Ray) and 31P NMR spectrum showing multiple J(P,P) couplings between the four anisochronous P nuclei giving rise to an ABMX spin system (G-NMR modelling of spectrum in mirror).
| P⋯P distances (Å)/J(P,P) constants (Hz) | [PdCl2{Fc(P)4tBu}] | [PdBr2{Fc(P)4tBu}] |
| d PA⋯PM/J(A,M) | 3.842(1)/24.0 | 3.6765(12)/25.6 |
| d PA⋯Px/J(A,X) | 4.698(2)/n.d. | 5.3971(13)/1.9 |
| d PB⋯PM/J(B,M) | 4.440(1)/6.4 | 4.4879(13)/5.8 |
| d PB⋯Px/J(B,X) | 6.292(2)/n.d. | 6.7628(13)/n.d. |
The collected data allowed to correlate geometric features found in the solid state with J(P,P) coupling constants (solution spectra), so that an expression of J(P,P) vs. d P⋯P′ in coordination complexes could be devised (see details in Section 5).
TS spin couplings were also observed for other members of this family of ligands and related to their structure in solution. The 31P NMR pattern of 1,1′,2-tris(diphenylphosphino)-4-tert-butylferrocene (Fig. 5, right) displays a triplet at −19.3 ppm and a doublet at −23.0 ppm in CDCl3, which were assigned to the 1′-PPh2 and to the 1,2-PPh2 groups, respectively (A2B spin system). The observed spin–spin coupling constant was found to be larger in C6D6, J(P,P) = 5.0 Hz, than in CDCl3 (J(P,P) = 2.8 Hz). The J(P,P) value of 5.0 Hz appears to be too high for a classical “through-bond” 4J(P,P) coupling constant (usually in ferrocenyl polyphosphane 4J(P,P) between anisochronous heteroannular phosphorus atoms is null); such a strong dependence on solvent is also unexpected for a “through-bond” coupling. Thus, only a “through-space” spin–spin coupling mediated by the ferrocenyl ring rotation and/or by changes in the relative orientation of the phosphino groups can explain such unusual values. To account for this TS coupling, the relative rotation of the cyclopentadienyl rings (which is not blocked in this case) has to be restricted to a certain degree around the conformation of higher symmetry (Cs) pictured in Fig. 5 [17], and possibly occurs rapidly to facilitate via space–spin interaction of phosphorus nuclei.

Molecular conformation of highest symmetry (Cs). A conformation of this type would explain a “through-space” 31P spin–spin coupling.
The interpretation of the NMR spectrum of another ferrocene derivative, namely triphosphane 1,1′,2-tris(diphenylphosphino)-3′-(tris-phenyl)methyl-4-tert-butyl ferrocene [18], also required a nonbonded coupling to be considered (see Fig. 6). The signals corresponding to the phosphorus atoms of the chelating pair, 1-P and 2-P, are centred at −18.8 and −24.9 ppm. They appear as doublet of doublets, due to a strong, but classical coupling constant of 3J(P,P) = 41 Hz, as well as to another coupling constant with the 1′-P phosphorus atom (in the following P′ designates a P atom belonging to the other Cp ring). The signal of the third phosphorus atom is a pseudo-triplet, thus reflecting nearly identical TSJ(P,P) coupling constants (TSJ(P,P) = 11 and 12 Hz, respectively). These intense spin–spin couplings between heteroannular phosphorus atoms most probably operate “through-space” and therefore result from a spatial proximity of the spin-active nuclei. Consequently, in the absence of X-ray, we assigned to this triphosphane the conformation depicted in Fig. 6. This assumption was confirmed by the NMR spectrum of the coordination complex which readily formed with [Pd(allyl)Cl]2. In this complex, all three phosphorus atoms are involved in binding, as deduced from the observed 31P chemical shifts (Fig. 7) [18]. Very interestingly, one of the phosphorus atoms (presumably 1′-P) is less strongly bonded, its signal appearing at −10 ppm, instead of an expected value near 25–30 ppm.

The triphosphane ligand 1,1′,2-tris(diphenylphosphino)-3′-(tris-phenyl)methyl-4-tert-butyl ferrocene and its ABC NMR spin system.

31P NMR spectrum of the [Pd(allyl)Cl]/triphosphane complex in CD2Cl2.
This example is the first in which NMR analysis allowed both to identify the existence of “through-space” J(P,P) coupling constants and, incidentally, to anticipate the tridentate coordination behaviour of a triphosphane [18]. In view of the TSJ(P,P) coupling constants inferred from the spectral analysis (10–12 Hz), we can reasonably assume that for each pair of phosphorus atoms the d P⋯P′ distance in solution is smaller than 4.3 Å.
3.2 Calixarene P(III)-podands
The TSJ(P,P) coupling constants observed in the aforementioned polyphosphanes and related to short spatial separations between phosphorus nuclei (d < 5.5 Å) are not restricted to ferrocene-derived P(III)-ligands. The successful preparation of a series of regioselectively phosphinated calix[4]arenes led Dieleman et al. to investigate the calixarenes 1–6 in which two proximal phenoxy rings are substituted by a –CH2PPh2 podand [19,20]. In these molecules the phosphorus atoms are separated by ten chemical bonds (Fig. 8). Calixarenes 2–6 are chiral, and were isolated either as optically pure compounds (2–4) or as a racemic mixture (5 + 6). Due to the difference between R1 and R2 groups the 31P NMR spectra of the latter display two distinct phosphorus signals. However, while the spectra of 2–4 are each characterized by clear singlets (with no J(P,P) couplings detected), the 31P NMR spectrum of the benzoyl-substituted enantiomers 5 and 6 displays a typical AB spin system with J(P,P) = 8 Hz, the corresponding doublets being centred at −20.1 ppm and −22.2 ppm, respectively [21].
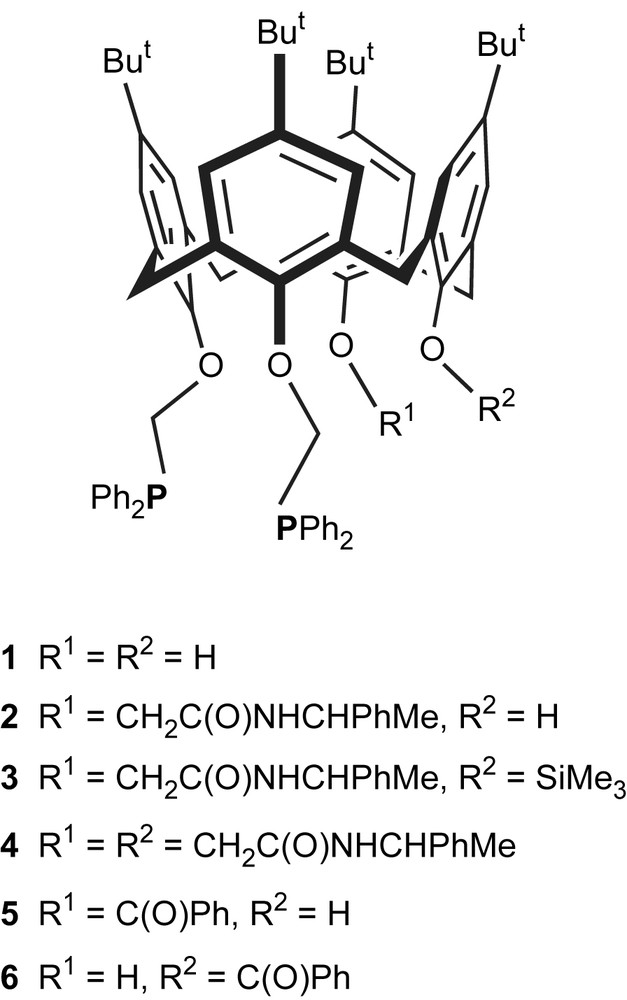
Proximally-diphosphinated calixarenes 1–6.
The spin coupling existing between the two phosphorus atoms, separated by ten bonds, in 5/6 was evidenced by measuring the spectrum in CDCl3 at different fields and also by carrying out a 2D homonuclear 31P COSY experiment. When the experiments were repeated in CD3OD, the magnitude of the coupling did not significantly change. The observed coupling constant was intuitively assigned to a “through-space” interaction rather than to a “through-bond” coupling since the phosphorus atoms of 5/6 are linked through a non-conjugated bond system. It is worth noting, that in a highly conjugated naphthyl-derived diphosphane, a 9J(P,P) of barely 4.0 Hz has been reported [22]. The solid state structure of the enantiomeric mixture (5 + 6) was determined by an X-ray diffraction study (Fig. 9).
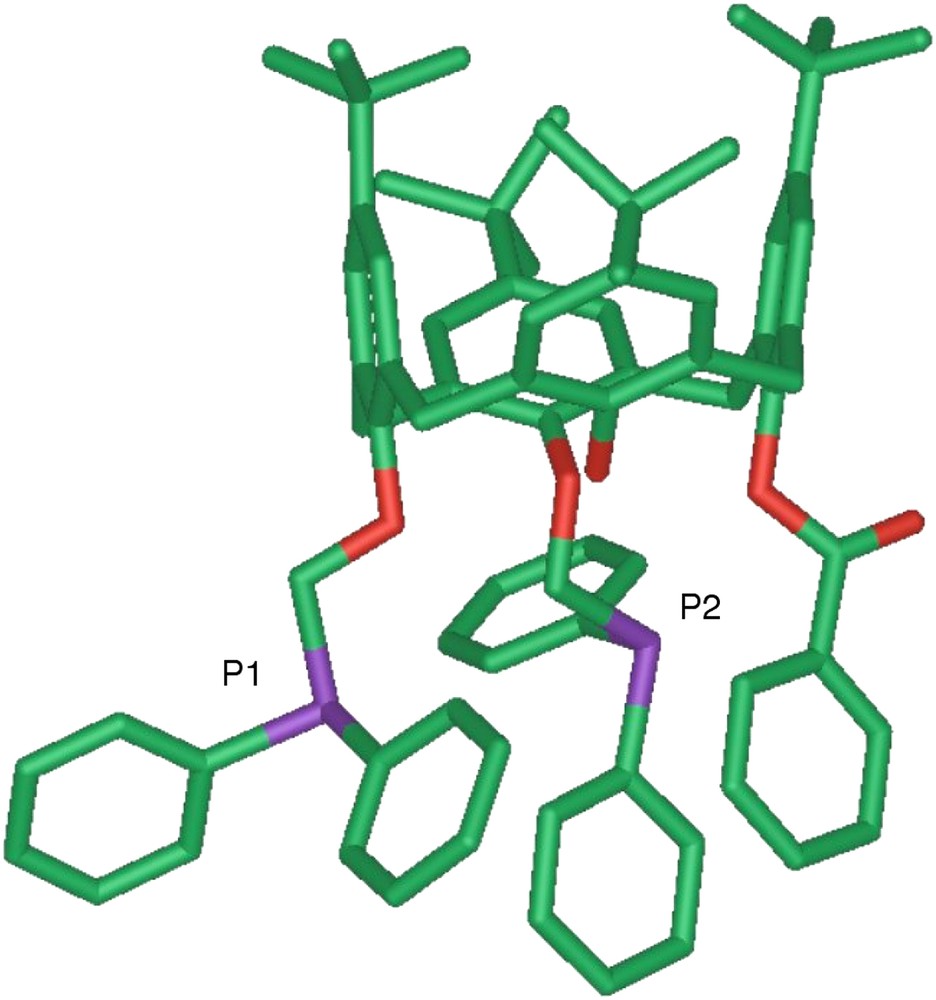
Molecular structure of diphosphino-calixarene 6.
In the solid state, the separation between the two phosphorus atoms was found to be of 5.333(1) Å, with an approximate alignment of the two phosphorus lone-pairs. It appears likely that in solution the P⋯P′ separation becomes even temporarily shorter. As revealed by a 2D ROESY experiments, the calixarene core of 6 displays a high flexibility, the unsubstituted phenolic ring flipping rapidly through the calixarene annulus. This fluxional behaviour combined with the mobility of the –OCH2PPh2 arms increases the probability of interaction of the phosphorus lone-pairs. The J(P,P) coupling constant value increases upon lowering the temperature (from 8.0 Hz at 283 K to 10.5 Hz at 183 K), an observation that is consistent with a statistically longer period of overlap of the lone-pairs as a result of slower molecular dynamics. The spatial proximity in solution of the phosphorus atoms of 6 was further substantiated by the easy formation of cis-chelate coordination complexes. Finally, the “through-space” J(P,P) coupling constant observed for the 5/6 couple was rather unexpected, and to date, there is no other example of chelating diphosphane with a ten bond P⋯P′ separation for which such an intense spin coupling is directly observable.
4 Rare “through-space” 31P13C spin couplings
Although virtually all spin-active nuclei are susceptible to display nonbonded nuclear spin couplings, nonbonded spin–spin nuclear couplings between phosphorus and carbon atoms are not very common. To the best of our knowledge, the first identification of such a coupling was made by Pascal et al.[23]. They observed J(P,C) couplings involving the phosphorus atom of a cyclophane phosphane and the six carbon atoms of an aromatic ring facing the P lone-pair. Despite the five-bond separation between each carbon atom and the P atom (see Fig. 10), J(P,C) coupling constants ranging from 3.5 to 7.5 Hz were observed, thus reflecting the spatial proximity of the coupling nuclei. As shown by an X-ray diffraction study, the corresponding P⋯C distances are of ca. 3.1 Å. More recent examples of such “through-space” couplings are outlined hereafter.

Pascal's constrained cyclophane phosphane.
4.1 31P13C spin couplings in ferrocenyl polyphosphanes
The proximity of the two cyclopentadienyl rings in the ferrocene unit, which is in part responsible for the PP “through-space” couplings discussed in Section 3, also gives rise to P⋯C spin couplings between atoms connected to distinct Cp rings. The ferrocenyl triphosphane shown in Fig. 11 provides a remarkable example of ligand in which such a coupling occurs [24].

Dissymmetric triphosphane and X-ray view.
The 13C NMR spectroscopic data revealed a neat PC spin–spin nuclear coupling (J(P,C) = 5.5 Hz) between the methyl carbon atoms of the tBu group (fast rotating methyl groups) and the PiPr2 phosphorus atom, as unambiguously demonstrated by selective 31P decoupling experiments (see Fig. 12). This means that the corresponding phosphorus atom is located close to the tBu group, and accordingly no J(P,P) coupling constant is observed from the A2X 31P NMR spectrum of this triphosphane (the lone-pair is involved in J(P,C) coupling). The five-bond separation between the P and C nuclei of concern cannot lead to a detectable 5J(P,C) if only “through-bond” spin coupling is operating. Further proof for a “through-space” coupling mechanism came from the absence of any other 4J(P,C) or 5J(P,C) coupling constant. A single crystal X-ray diffraction study confirmed the spatial proximity of the tBu and PiPr2 groups (see Fig. 11 left; shortest P⋯C separation: 3.64 Å). The staggered conformation imposed by the ferrocene backbone favours the observed P,C interaction. The P lone-pair is directed towards the carbon atoms of the tBu group, thus supporting the existence of an overlap between this orbital and a filled Csp3 orbital (or with the C–C bond of tBu group since no J(P,H) is observed).
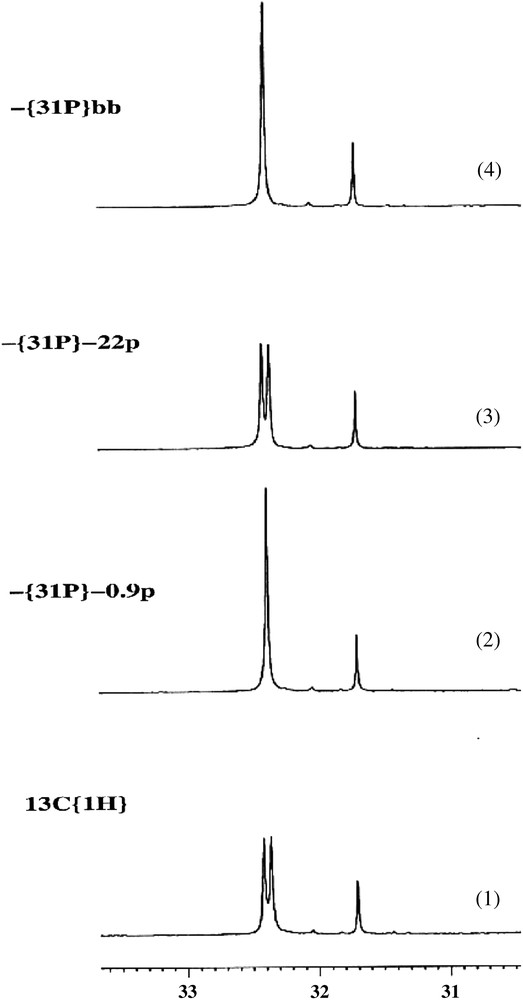
13C NMR spectra (CCH3 region) of a ferrocenyl triphosphane with selective phosphorus decoupling: (1) δ = 32.4 ppm (d, 3C, Me of tBu, J(C,P) = 5.5 Hz), no decoupling; (2) saturation of the P signal at −0.9 ppm (–P(iPr)2); (3) saturation of the signal at −22 ppm (–PPh2); (4) phosphorus broad band decoupling.
4.2 A trans-spanning cyclodextrin-diphosphane
Nonbonded PC spin–spin couplings are naturally not limited to ferrocenyl phosphanes. Such a phenomenon was also revealed in the rigid, C2-symmetrical cyclodextrin-derived TRANSDIP (Fig. 13). Here no PP spin--spin coupling can be simply detected in the 31P NMR spectrum, due to the magnetic equivalence of phosphorus nuclei [25]. However, the C-4, C-5 and C-6 atoms of the P-bridged A, B, D, E glucose units display spin--spin couplings with both phosphorus atoms in the 13C NMR spectrum, an observation that can only be rationalized by the existence of P,P orbital overlap. Moreover, a neat spin–spin coupling J(P,C) = 3.5 Hz between the C-6C atom (equivalent to C-6F) and both phosphorus atoms was also detected (see Fig. 14), thus indicating that both C-6 atoms lie in close proximity to the P atoms. The triplet observed reflects the equivalence of the two phosphorus atoms regarding the carbon atoms, and also evidences an unexpected extent of the orbital overlap.

Cyclodextrin-based diphosphane TRANSDIP.
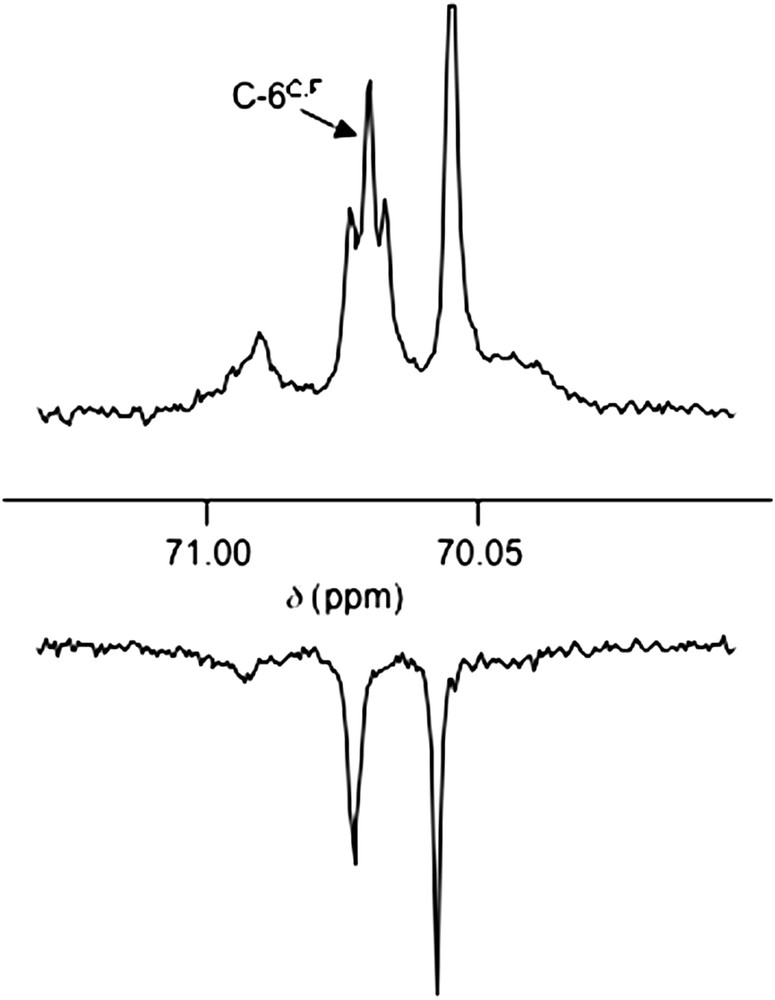
Resonances of the C6C,F atoms in TRANSDIP: 13C{1H} (top) and 13C{1H,31P} (bottom) NMR spectra recorded in CDCl3 at 125.8 MHz.
The “through-bond” contribution to PC spin-spin coupling is likely to be negligible given the eight-bond separation existing between the C6 atoms of glucose units C and F (Fig. 13) and the two phosphorus atoms. Molecular mechanics calculations (MM2), as well as X-ray crystal structures of TRANSDIP metal complexes [26], confirmed that the C-6C,F atoms are close to both phosphorus atoms (averaged P⋯C separation: 4.8 Å, see Fig. 15).
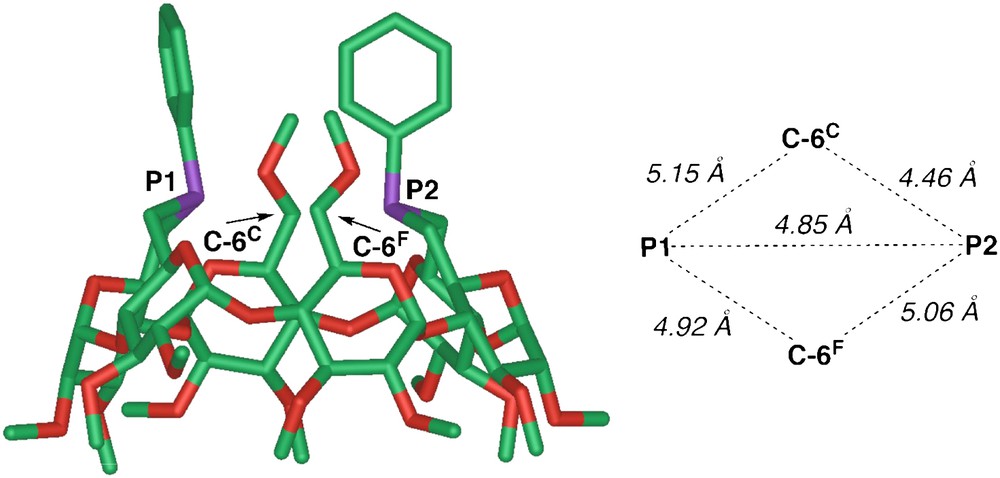
Minimized structure of TRANSDIP (left, MM2 using Spartan 1.0.3.) and the corresponding calculated P⋯P and P⋯C6C,F distances.
The J(P,C-6C,F) coupling constant of 3.5 Hz is somewhat smaller than the “through-space” J(P,C) value (5.5 Hz) observed for the aforementioned ferrocenyl triphosphane, suggesting a longer P⋯C separation in the case of TRANSDIP. The fact that only the C-6C,F carbon atoms of the cyclodextrin give rise to such a direct interaction is a further indication of the endo orientation of the P lone-pairs as well as of the ligand rigidity. Overall, the 13C{1H} NMR spectrum gives valuable information on the structural features of this rigid diphosphane.
5 Advances in modelling TS couplings: distance dependence and real-space function visualization
As mentioned in Section 2.2, the overlap interaction of fluorine lone-pairs has been quantitatively estimated for a number of constrained molecules. Computational methods were used for estimating the distances between the “through-space” coupled fluorine atoms. This allowed to establish exponential relationships between calculated internuclear distances and observed J(F,F) couplings [3g,7].
The first d P⋯P′/J(P,P) correlation between “through-space” interacting phosphorus nuclei was proposed recently in a publication dealing with complexes derived from Fc(P)4tBu (see Section 3.1). In this work, the model developed by Mallory [16] was extended to show that “through-space” nuclear spin couplings do not necessarily require the presence of two lone-pair orbitals. Indeed, a lone-pair with an appropriate orientation may interact with electrons of a P–M bond, a “through-space” interaction taking place within a three-centre [P,P′,M] system. This concept (see Fig. 16), which extends the model developed for purely organic compounds is also consistent with the existence of PC couplings, the interaction involving possibly a C–C or a C–H bond.

Extension of Mallory's lone-pair overlap interaction model to polyphosphane coordination complexes (left), and schematic orbital diagram involving a transition metal orbital contribution (right).
As in the case of the fluorine compounds [2], overlap between P-centred electron clouds generates significant Fermi contact (FC) interactions. It is worth noting that the predominant role of the Fermi contact in nonbonded coupling has been confirmed by density functions interpretation [5b].
The J(P,P) vs. d P⋯P′ correlation diagram shown in Fig. 17 was drawn using the data obtained for the above mentioned palladium complex (Table 1), as well as those corresponding to related nickel and palladium complexes [16,27]. The best fit was obtained for an exponential curve expressed by Eq. (2), with J(P,P) in Hz and d P⋯P′ in Å.
| (2) |

Plot of J(P,P) vs. d P⋯P′ “through-space” distances. The plot is fitted by the exponential relationship defined by Eq. (2) [27]. The dotted line corresponds to earlier results [16] and encircled values are examples from Table 1.
It must be specified that this correlation constitutes, strictly speaking, only an approximation in the absence of a more developed tool taking into account that these TS couplings depend both on the orientation of the lone-pair and on the molecular orbitals (shape, symmetry energy) involved in the interaction. However, the phosphorus nucleus being a statistical centre for the electron clouds concerned, the resulting approximation allows in this case an effective estimation of TS spin coupling from the internuclear distance.
Recent work by Malkina and Malkin constitutes an interesting approach for an accurate visualization of spin couplings transmission [28]. These authors introduced a model based on real-space functions in three-dimensional space, which is appropriate for both localized and delocalized bonding situations. The indirect spin–spin coupling is expressed as the energy splitting between states with parallel and antiparallel nuclear spins. These energies are then written as an integral over an energy density called the coupling energy density (CED). The integral of CED over all space is equal to the reduced coupling constant. CED is a real-space function and can be visualized easily in 3D space. The CED was calculated using a double finite perturbation theory for various molecules, notably C2H2(PH2)2 (Fig. 18). The visualization clearly established that the “through-space” interaction between the two phosphorus nuclei dominates over the “through-bond” pathway [28].

Visualization of the J(P,P) coupling density (CED) in the C2H2(PH2)2 molecule from DFT calculations (taken from Ref. [28]).
Calculations aimed at 3D visualization of the above mentioned ferrocenylphosphane ligands and their complexes are currently underway.
6 Conclusion and perspectives
Both from a theoretical and an experimental point of view, the analysis of high-resolution NMR parameters is a critical issue. A deeper understanding of the relationship between coupling constants and molecular structure will greatly broaden future applications of high-resolution spectroscopy for the elucidation of molecular structures [29]. The phosphinocalixarenes, phosphinocyclodextrins and ferrocenyl phosphanes presented herein may contribute to furthering our knowledge about nonbonded coupling. The important structural information provided by careful analysis of the corresponding NMR spectra should encourage researchers to systematically pay more attention to all those constrained molecules in which nonbonded spin–spin couplings are likely to occur.
Overall, it may be anticipated that future studies in this field will ultimately allow easier and non-ambiguous discrimination between “through-bond” and “through-space” contributions in every type of molecule.
Acknowledgements
Thanks are due to B. Hanquet for G-NMR modelling, and to P. Meunier and his group for their continuous support and trust in the studies developed around ferrocene functionalization and applications. The authors thank the CNRS, the Université de Bourgogne (and “Conseil Régional de Bourgogne”), and the Université de Strasbourg for financial support.