

1 Introduction

Diamino-substituted phosphorus compounds have been used as good precursors of transition metal phosphenium complexes [1–9] as well as of metal-free phosphenium compounds [1–4,10]. Transition metal complexes bearing P(NMeCH2)2(R′) (R′ = alkoxy, amino) have been converted into the corresponding phosphenium complexes for Cr, Mo, W, Mn, Fe, and Ru complexes by the reaction with a Lewis acid, such as BF3·OEt2 or Me3SiSO3CF3 (TMSOTf) (Eq. (1)) [3,4]. In these reactions, an R′ group on the phosphorus is abstracted as an anion by a Lewis acid. In contrast, in the reaction of a molybdenum complex having a hydride ligand and P(NMeCH2)2(OMe), [Cp*Mo(H)(CO)2{P(NMeCH2)2(OMe)}], with TMSOTf, a preferential H− abstraction followed by OTf− coordination on the Mo center takes place to give [Cp*Mo(CO)2(OTf){P(NMeCH2)2(OMe)}] rather than OMe− abstraction at the P center (Eq. (2)) [11].
Many complexes bearing a diamino-substituted phosphorus ligand have been synthesized for several kinds of transition metals and have been subjected to the reaction with a Lewis acid. However, a platinum complex with P(NMeCH2)2(R′) has not been reported to date.
With platinum complexes, it should be noted that: (i) platinum phosphenium complexes have been reported in the reaction of a Pt(0) complex with a metal-free phosphenium or with an N-heterocyclic carbene adduct of a phosphenium [12] and (ii) LnPt–R (R = H [13], Me [14], Ph [15]) shows R/OTf exchange in the reaction with R′′OTf (R′′ = H, Me, Me3Si) to give LnPt–OTf (Eq. (3)).
Herein, we report the reactions of PtR2(cod) (R2 = Me, p-tol, Cl, I) with P(NMeCH2)2R′ (R′ = OMe, NEt2), crystal structures of Pt complexes with P(NMeCH2)2R′ thus prepared, and the reactivity of the Pt complexes with a Lewis acid because R abstraction from the Pt and R′ abstraction from the P are conceivable.
2 Results and discussion
A dialkyl- or diaryl-platinum complex, PtR2(cod) (cod = η2, n2-1,5-cyclooctadiene, R = Me, p-tol) [16], reacted with a diamino-substituted P ligand, P(NMeCH2)2(R′) (R′ = OMe [17], NEt2 [18]), at room temperature to produce a neutral complex, cis-[Pt(R)2{P(NMeCH2)2(R′)}2] (R′ = OMe, R = Me: 1, p-tol: 2, R′ = NEt2, R = Me: 3, p-tol: 4) in appropriate to excellent yields [Eq. (4)]. The 31P{1H} NMR spectra of 1–4 showed the singlet flanked by 195Pt satellites at δ 125.07 (1JPPt = 2860 Hz) for 1, 118.56 (1JPPt = 2809 Hz) for 2, 120.13 (1JPPt = 2860 Hz) for 3, and 107.40 (1JPPt = 2688 Hz) for 4.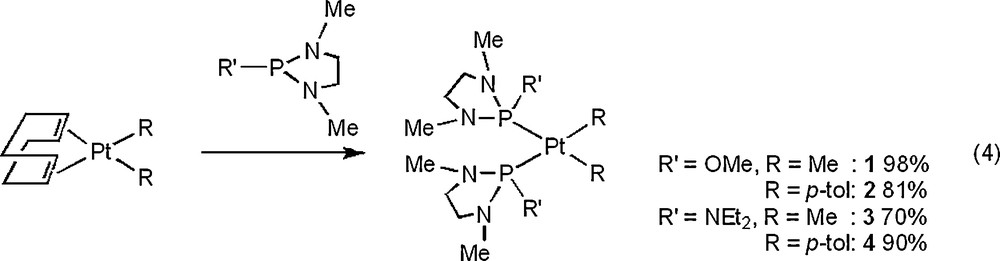
The structures of 1–3 were determined by single-crystal X-ray diffraction analyses. The molecular structures of 1–3 are shown in Figs. 1–3. Crystal data and the selected bond lengths and angles are listed in Tables 1 and 2. As two independent molecules of 1 and 3 crystallized in the unit cell, only one molecule (Pt1) is shown for 1 and 3 with the atom numbering scheme in Figs. 1 and 3. Complexes 1–3 have a typical square-planar configuration: the platinum has two methyl or p-tol ligands and two P ligands. These P ligands are situated mutually in a cis position. The Pt–P bond distances of 1–3 resemble those of previously reported cis-PtMe2L2 (L = monodentate tertiary phosphorus ligand) (2.252–2.344 Å) [14(a),19,20]. The Pt–C bond distances (2.097(7)–2.144(8) Å for 1, 2.067(5), 2.078(6) Å for 2, and 2.119(5), 2.122(4) Å for 3) are similar to those of analogous complexes (2.075–2.132 Å for cis-[Pt(Me)2(PR3)2] (R3 = Me(C2F5)2 [14(a)], Et3, PhMe2, Ph2Me, (pyrl)3, and Cy3 [19]) and 2.057(12), 2.057(9) Å for cis-[Pt(p-tol)2(PEt3)2] [20]).
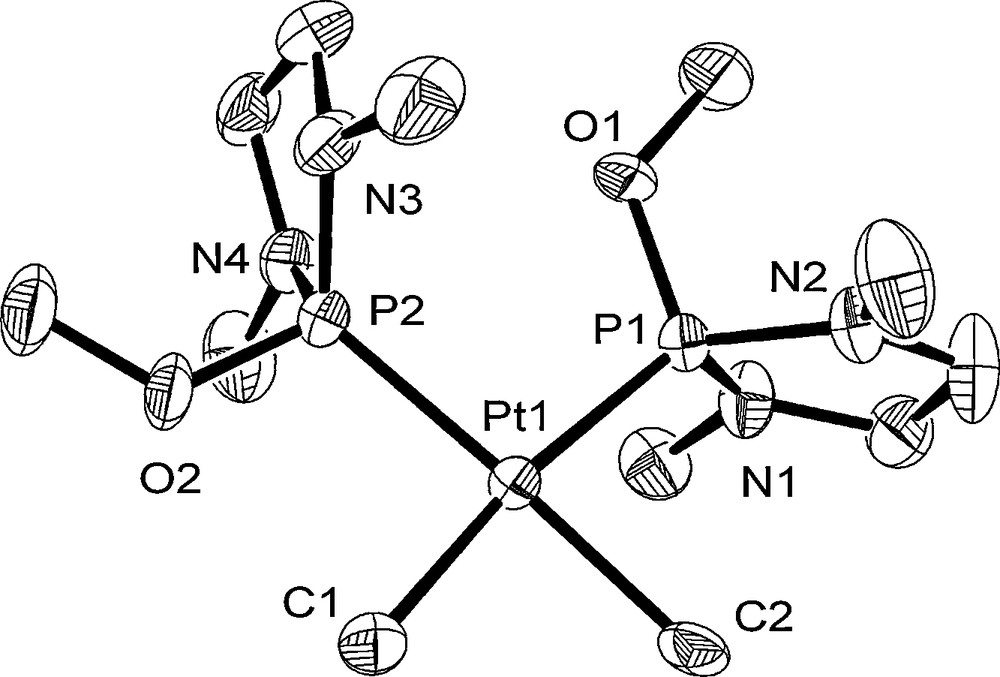
Crystal structure of Pt1 molecule of 1 at the 50% ellipsoidal level. The hydrogen atoms are omitted for simplicity.

Crystal structure of 2 at the 50% ellipsoidal level. The hydrogen atoms are omitted for simplicity.

Crystal structure of Pt1 molecule of 3 at the 50% ellipsoidal level. The hydrogen atoms are omitted for simplicity.
Crystal data and experimental parameters used for the intensity data collection of 1–3. Procedure and final results of the structure determination.
| Empirical formula | C12H32N4O2P2Pt 1 | C24H40N4O2P2Pt 2 | C18H46N6P2Pt 3 |
| Formula weight | 521.45 | 673.63 | 603.64 |
| T (K) | 203(2) | 110(1) | 110(1) |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21/c | Pbca | C2/c |
| a (Å) | 9.3500(5) | 13.4206(5) | 35.942(3) |
| b (Å) | 29.4100(17) | 14.4097(5) | 9.3194(7) |
| c (Å) | 14.3700(9) | 27.8523(11) | 23.0164(17) |
| β (°) | 95.000(3) | 90.521(4) | |
| Volume (Å3) | 3936.5(4) | 5386.3(3) | 7709.2(10) |
| Z | 8 | 8 | 12 |
| ρcalcd (mg m–3) | 1.760 | 1.661 | 1.560 |
| μ (cm–1) | 7.300 | 5.356 | 5.600 |
| F(000) | 2048 | 2688 | 3648 |
| Crystal size (mm3) | 0.22 × 0.05 × 0.03 | 0.20 × 0.18 × 0.06 | 0.10 × 0.08 × 0.08 |
| Reflections collected | 30204 | 38829 | 29063 |
| R(int) | 5218 (0.032) | 6063 (0.0407) | 8701 (0.0412) |
| R (I > 2σ(I)) | 0.056 | 0.0477 | 0.0427 |
| wR2 | 0.091 | 0.1049 | 0.0745 |
| Goodness of fit | 1.085 | 1.524 | 1.163 |
Selected bond lengths (Å) and bond angles (°) for 1-3.
| 1 | 2 | 3 | |
| Pt1–C1 | 2.109(8) | 2.067(5) | 2.119(5) |
| Pt1–C2(C8 for 2) | 2.144(8) | 2.078(6) | 2.122(4) |
| Pt1–P1 | 2.2512(19) | 2.2737(13) | 2.2781(12) |
| Pt1–P2 | 2.2568(19) | 2.2627(13) | 2.2753(12) |
| P1–O1(N3 for 3) | 1.615(5) | 1.625(4) | 1.672(4) |
| P2–O2(N6 for 3) | 1.619(6) | 1.625(4) | 1.668(4) |
| C1–Pt1–C2(C8 for 2) | 86.7(3) | 84.8(2) | 81.3(2) |
| P1–Pt1–P2 | 96.21(7) | 98.66(5) | 100.56(4) |
The reaction of the dihalogeno platinum complex, PtX2(cod), with P(NMeCH2)2(OMe) formed a cationic triphosphite platinum complex, [PtX{P(NMeCH2)2(OMe)}3]X (X = Cl: 5, I: 6) [21], as a white solid in high and excellent yields [Eq. (5)]. Complexes 5 and 6 were formed in high yields when PtX2(cod) and P(NMeCH2)2(OMe) were treated in the 1:3 molar ratio. The same complexes were formed even if they were treated in the 1:2 ratio and PtX2{P(NMeCH2)2(OMe)}2 (X = Cl, I) were not obtained. Roulet et al. and Mézailles et al. reported analogous reactions of PtX2(PMe3)2 (X = Cl, Br) with PMe3 to produce [PtX(PMe3)3]X [22(a)] and of PtCl2(cod) with (Mes)P = CH(NMe2) (Mes = 2,4,6-Me3C6H2) to produce [PtCl{(Mes)P = CH(NMe2)}3]Cl [22(b)]. The 31P{1H} NMR spectra of 5 and 6 show the two signals with 195Pt satellites at δ 71.24 (t, 2JPP = 13 Hz, 1JPPt = 5375 Hz, trans to Cl) and 99.17 (d, 2JPP = 13 Hz, 1JPPt = 3633 Hz, cis to Cl) for 5 in the 1:2 peak area ratio, and 70.43 (t, 2JPP = 15 Hz, 1JPPt = 5158 Hz, trans to I) and 93.63 (d, 2JPP = 15 Hz, 1JPPt = 3512 Hz, cis to I) for 6 in the 1:2 peak area ratio.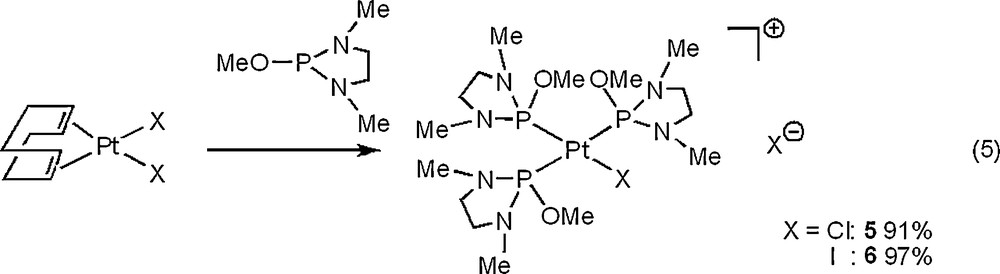
The analogous cationic complexes 7 and 8 were obtained in high yields in the reactions of a methyl(halogeno) platinum complex, PtMeX(cod) (X = Cl, I), with P(NMeCH2)2(OMe) in the 1:3 molar ratio [Eq. (6)]. In this case, Pt(Me)(X){P(N(MeCH2)2(OMe))}2 (X = Cl, I) were not obtained as in [Eq. (5)] even in the reaction of PtMeX(cod) with P(NMeCH2)2(OMe) in the 1:2 molar ratio. In contrast, the reaction of PtMeX(cod) (X = Cl, I) with a diamino-substituted phosphine ligand having NEt2 group on the P atom, P(NMeCH2)2(NEt2), gave the natural bis(phosphine) platinum complexes 9 and 10 [Eq. (7)]. In the 1H NMR spectra of 7 and 8, the methyl group on the Pt center showed a doublet (3JPH = 8.8 Hz) of triplets (3JPH = 7.3 Hz) with 195Pt satellites (2JPtH = 56.0 Hz) at δ 0.31 for 7 and 0.22 for 8, indicating the presence of three phosphite ligands coordinated to the Pt center. The 31P{1H} NMR data of 9 indicate the presence of trans and cis isomers in ca. 2:1 ratio. With cis-9, the 195Pt satellite value (1JPPt = 6037 Hz) of the doublet at δ 82.84 (assignable to P trans to Cl) was larger than the corresponding value (1JPPt = 2651 Hz) of the doublet at δ 116.60 (assignable to P trans to Me). This tendency is similar to that of analogous cis-PtMeClL2 (L = monodentate tertiary phosphorus ligand) [23]. With 10, the trans isomer was observed exclusively. The difference between the cis/trans ratio in 9 and 10 may derive from the steric bulkiness of a halogeno ligand on the Pt center [20].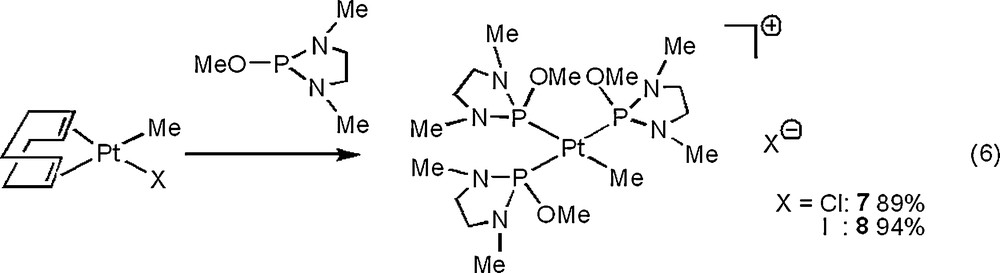
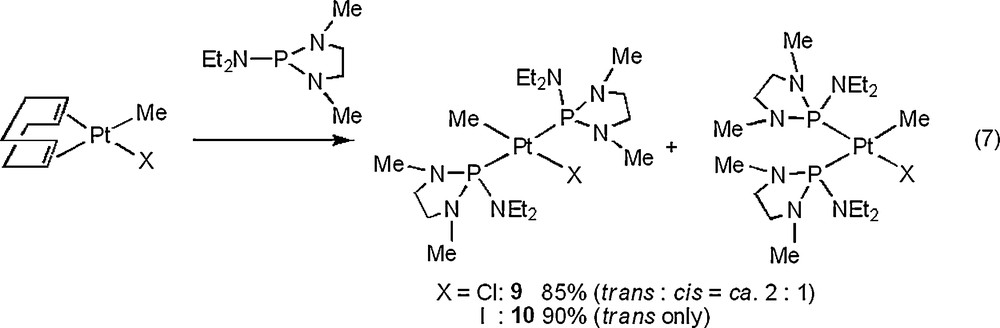
The molecular structures of 6, 8, and 10 are depicted in Figs. 4–6 with the atomic numbering scheme. Table 3 summarizes the crystallographic data. The selected bond lengths and angles are listed in Table 4. The Pt centers of 6, 8, and 10 have a distorted square-planar geometry. With 6, the distance of Pt–P bond trans to I (2.2343(11) Å) is shorter than those of trans to phosphite ligand (2.3211(12), 2.2987(13) Å) due to weak trans influence of an iodo ligand. On the other hand, three Pt–P bond distances of 8 (2.2882(16) Å for trans to Me, 2.2864(17), 2.2883(16) Å for trans to the phosphite ligand) are almost the same. The difference of Pt–P bond distances between 6 and 8 probably stems from the stronger trans influence of methyl ligand than of iodide ligand. Complex 10 has Me and I ligands in a trans position. The Pt–P and Pt–C bond distances of 10 (Pt–P = 2.300(3), 2.294(3) Å and Pt–C = 2.072(11) Å) are comparable to those of cis-[Pt(Me)2{P(NMeCH2)2(NEt2)}2] 3 (Pt–P = 2.2781(12), 2.2753(12) Å and Pt–C = 2.119(5), 2.122(4) Å).

Crystal structure of a cation part of 6·CH2Cl2 with 50% thermal ellipsoidal plots. An anion part, hydrogen atoms, and CH2Cl2 were omitted for simplicity.
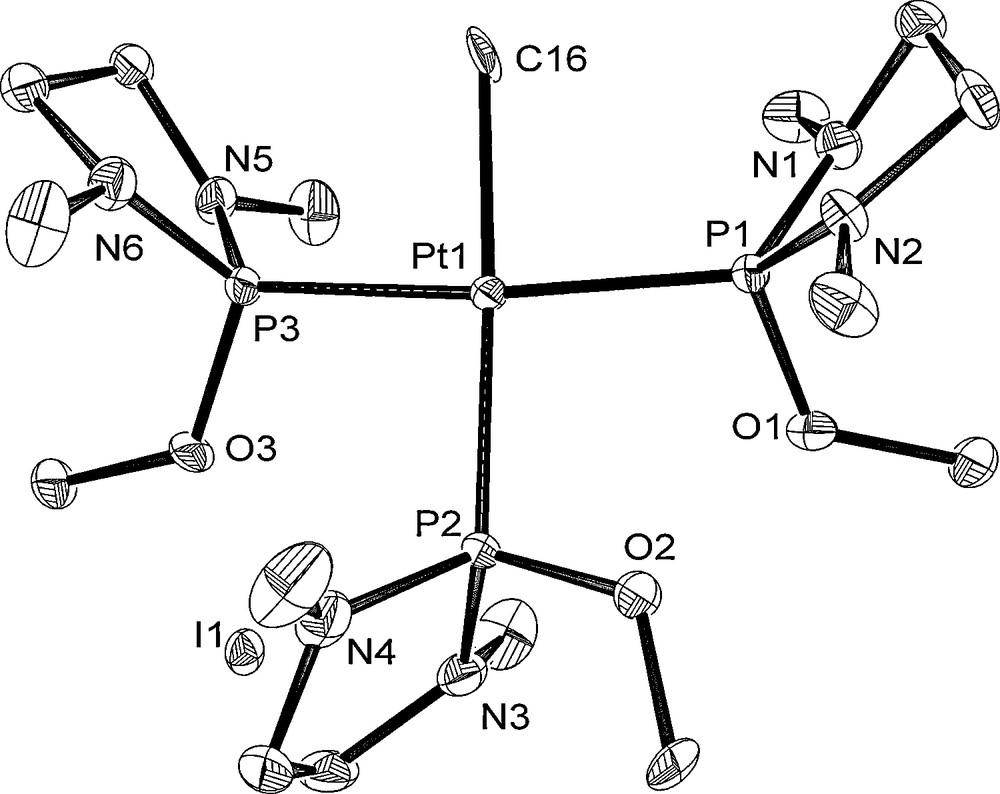
Crystal structure of 8 at the 50% ellipsoidal level. The hydrogen atoms are omitted for simplicity.
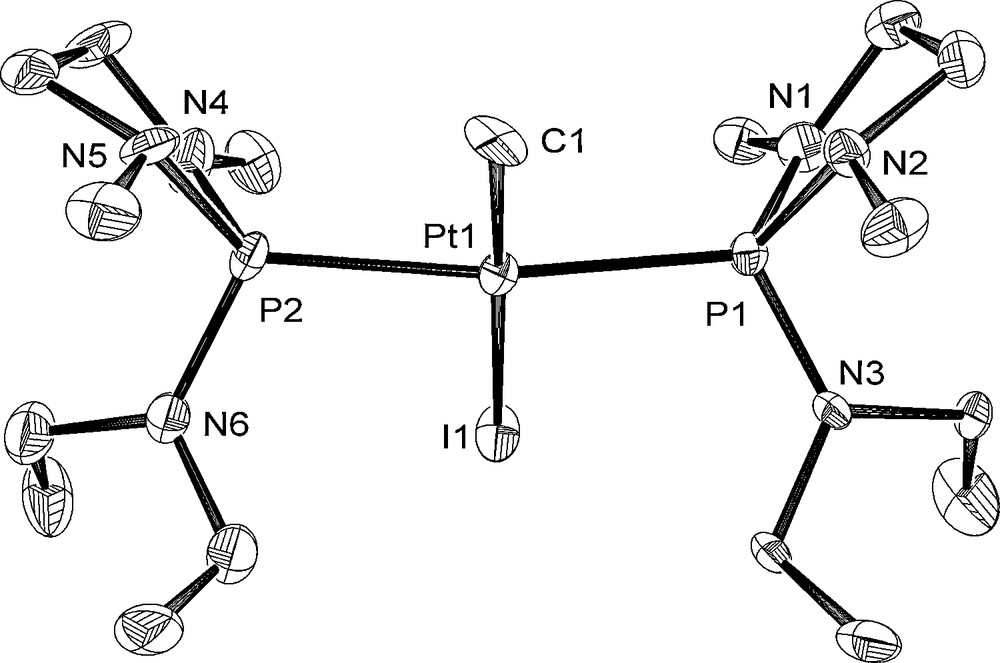
Crystal structure of 10 at the 50% ellipsoidal level. The hydrogen atoms are omitted for simplicity.
Crystal data and experimental parameters used for the intensity data collection of 6, 8, and 10. Procedure and final results of the structure determination.
| Empirical formula | C16H41N6O3P3Cl2I2Pt 6 | C16H42N6O3P3IPt 8 | C17H43N6P2IPt 10 |
| Formula weight | 978.25 | 781.46 | 715.50 |
| T (K) | 203(2) | 110(2) | 110(1) |
| Crystal system | Orthorhombic | Monoclinic | Orthorhombic |
| Space group | P212121 | P21/n | Pcab |
| a (Å) | 7.3640(2) | 11.6851(7) | 14.7786(16) |
| b (Å) | 19.2021(8) | 9.9735(6) | 14.7576(17) |
| c (Å) | 22.5145(9) | 24.0650(15) | 23.059(2) |
| β (°) | 96.246(3) | ||
| Volume (Å3) | 3183.6(2) | 2787.9(3) | 5029.0(10) |
| Z | 4 | 4 | 8 |
| ρcalcd (mg m–3) | 2.041 | 1.862 | 1.890 |
| μ (cm–1) | 6.693 | 6.340 | 6.950 |
| F(000) | 1864 | 1520 | 2784 |
| Crystal size (mm3) | 0.38 × 0.08 × 0.05 | 0.30 × 0.20 × 0.10 | 0.30 × 0.20 × 0.02 |
| Reflections collected | 24980 | 20607 | 37063 |
| R(int) | 7141 (0.0263) | 6185 (0.0281) | 5740 (0.0809) |
| R (I > 2σ(I)) | 0.0253 | 0.0379 | 0.0953 |
| wR2 | 0.0598 | 0.1141 | 0.1439 |
| Goodness of fit | 1.008 | 1.425 | 1.545 |
Selected bond lengths (Å) and bond angles (°) for 6, 8, and 10.
| 6 | 8 | 10 | |
| Pt1–P1 | 2.3211(12) | 2.2864(17) | 2.300(3) |
| Pt1–P2 | 2.2343(11) | 2.2882(16) | 2.294(3) |
| Pt1–P3 | 2.2987(13) | 2.2883(16) | |
| Pt1–I1 | 2.6586(3) | 2.7352(9) | |
| Pt1–C16 (C1 for 10) | 2.136(7) | 2.072(11) | |
| P1–O1(N3 for 10) | 1.589(4) | 1.608(5) | 1.662(11) |
| P2–O2(N6 for 10) | 1.589(3) | 1.596(5) | 1.666(11) |
| P3–O3 | 1.596(4) | 1.593(5) | |
| P1–Pt1–P2 | 94.61(5) | 94.48(6) | 170.37(12) |
| P2–Pt1–P3 | 95.42(5) | 94.57(6) |
As several Pt complexes with diamino-substituted phosphorus compounds were obtained, reactions of 1–10 with a Lewis acid were examined. To a CH2Cl2 solution of 1–10 in an NMR tube was added 2- or 3-fold molar excess amount of HOTf, Me3SiOTf, MeOTf, or BF3·OEt2 at room temperature, and then the 31P{1H} NMR spectra were recorded at 25 °C. In all cases except 7 and 8, several signals were observed, indicating that some complicated reactions took place. However, there is no signal in the region of a phosphenium ligand [4]. In the reactions of 7 and 8 with BF3·OEt2, a relatively clean reaction took place. Fig. 7 depicts the 31P{1H} NMR spectra of 8 and a solution containing 8 and BF3·OEt2. After 2 h, a doublet at 105.90 ppm (2JPP = 37 Hz, 1JPPt = 4070 Hz, trans to phosphite ligand) and a triplet at 122.60 ppm (2JPP = 37 Hz, 1JPPt = 2818 Hz, trans to I) of the starting complex 8 disappeared and new two signals at 93.63 ppm (d, 2JPP = 15 Hz, 1JPPt = 3508 Hz) and 70.43 ppm (t, 2JPP = 15 Hz, 1JPPt = 5170 Hz) were observed. The 195Pt satellite value of the triplet (1JPPt = 5170 Hz) of the product was larger than the corresponding value of the starting complex 8 (1JPPt = 2818 Hz). This large Pt–P coupling constant suggests the substitution of a methyl group for a ligand having weaker trans influence.

The 31P{H} NMR spectra of 8 (a) and 8 with BF3OEt2 (after 2 h) (b) at 25 °C in CH2Cl2. Peaks with asterisks are impurity.
We tried to isolate complexes formed in the reaction of complexes 7 and 8 with BF3·OEt2. A methyl complex, [PtMe{P(NMeCH2)2(OMe)}3]X (X = Cl: 7, I: 8), was treated with an equimolar amount of BF3·OEt2 at room temperature for 2 h and then with NaBPh4 in CH2Cl2 to afford the halogeno complex [PtX{P(NMeCH2)2(OMe)}3]BPh4 (X = Cl: 11, I: 12) as a result of the Me/X substitution reaction on the Pt center [Eq. (8)].
X-ray structure analyses of 11 and 12 were undertaken. The ORTEP drawings of 11 and 12 are displayed in Figs. 8 and 9, respectively. Crystal data and selected bond distances and angles are summarized in Tables 5 and 6. Both platinum complexes take a normal square-planar geometry with three P(NMeCH2)2(OMe) and one X ligands (X = Cl for 11, I for 12). Although the structure of a cationic part of 12 is similar to that of 6, the anion part is composed of a tetraphenylborate anion. The P–OMe bond distances of 12 (1.598(9)–1.614(10) Å) are similar to those of 6 (1.589(3)–1.596(4) Å) and of the corresponding complexes reported previously (1.59–1.6387 Å) [3,4,6,11,24]. The X-ray analyses revealed that preferential Me substitution on the Pt center takes place and the OMe group on the phosphite remains intact.

Crystal structure of 11·CH2Cl2 with 50% thermal ellipsoidal plots. The hydrogen atoms and CH2Cl2 were omitted for simplicity.

Crystal structure of 12·CH2Cl2 at the 50% ellipsoidal level. The hydrogen atoms are omitted for simplicity.
Crystal data and experimental parameters used for the intensity data collection of 11 and 12. Procedure and final results of the structure determination.
| Empirical formula | C40H61N6O3BCl3P3Pt 11 | C40H61N6O3BCl2IP3Pt 12 |
| Formula weight | 1079.11 | 1170.56 |
| T (K) | 100(1) | 100(1) |
| Crystal system | Triclinic | Triclinic |
| Space group | P-1 | P-1 |
| a (Å) | 13.0976(18) | 13.1133(17) |
| b (Å) | 13.6179(18) | 13.7843(18) |
| c (Å) | 14.6613(19) | 14.4299(19) |
| α (°) | 102.513(2) | 101.684(4) |
| β (°) | 102.661(4) | 103.378(4) |
| γ (°) | 104.213(4) | 103.742(4) |
| Volume (Å3) | 2370.0(5) | 2371.6(5) |
| Z | 2 | 2 |
| ρcalcd (mg m–3) | 1.512 | 1.639 |
| μ (cm–1) | 3.273 | 3.865 |
| F(000) | 1092 | 1164 |
| Crystal size (mm3) | 0.10 × 0.10 × 0.02 | 0.31 × 0.20 × 0.10 |
| Reflections collected | 18440 | 18573 |
| R(int) | 10288 (0.0501) | 10305 (0.0674) |
| R (I > 2σ(I)) | 0.0673 | 0.0787 |
| wR2 | 0.1269 | 0.2192 |
| Goodness of fit | 1.186 | 1.063 |
Selected bond lengths (Å) and bond angles (°) for 11 and 12.
| 11 | 12 | |
| Pt1–P1 | 2.3000(19) | 2.287(3) |
| Pt1–P2 | 2.2756(17) | 2.262(3) |
| Pt1–P3 | 2.274(2) | 2.310(3) |
| Pt1–Cl1 (I1 for 12) | 2.232(5) | 2.5870(13) |
| P1–O1 | 1.603(5) | 1.614(10) |
| P2–O2 | 1.611(5) | 1.598(9) |
| P3–O3 | 1.585(6) | 1.605(8) |
| P1–Pt1–P2 | 95.35(6) | 95.67(11) |
| P2–Pt1–P3 | 95.76(7) | 95.65(10) |
Two plausible reaction pathways from 7, 8 to 11, 12 are shown in Scheme 1. Along Path A, BF3 reacts with X− to give XBF3− (A), and then a metathesis reaction between the Pt–Me and X–BF3− bonds takes place (B → C). The counter anion MeBF3− is exchanged by BPh4− to give the final product (C → D). Alternatively in Path B, the Me– group on the Pt center instead of the OMe− group on the P atom is abstracted by BF3 to generate a dicationic complex and MeBF3− (E → F). Then a halogeno anion coordinates to the Pt center (F → C), and a subsequent pathway is similar to Path A. Path A is highly likely because of the advantage of avoiding the formation of the unstable dicationic intermediate F. However, Path B cannot be ruled out.

Plausible pathway of formation of [PtX{P(NMeCH2)2(OMe)}3]BPh4 (X = Cl: 11, I: 12).
Through this work it was found that the platinum complexes with diamino-substituted phosphorus compounds were not converted into the corresponding phosphenium complexes in the reaction with a Lewis acid, whereas some other transition metal complexes were. The role of the platinum is not clear now, and is under investigation.
3 Conclusion
We found that a reaction of platinum complex with P(NMeCH2)2(R’) (R’ = OMe, NEt2) produces the corresponding cationic complex or neutral cis- or trans-complex depending on the substrate on the Pt center and the bulkiness of diamino-substituted phosphorus ligand. Reactions of cationic methyl platinum complexes 7 and 8 with BF3·OEt2 clearly showed that the predominant Me/X substitution on the Pt center takes place and the OMe group on the P atom remains intact.
4 Experimental section
4.1 General considerations
All reactions were carried out under an atmosphere of dry nitrogen by using standard Schlenk tube techniques. Solvents except CH2Cl2 were distilled from sodium metal and were stored under nitrogen atmosphere. CH2Cl2 was distilled from CaH2 under dry nitrogen prior to use. [PtMe2(cod)] [16(a)], [Pt(p-tol)2(cod)] [16(b)], [PtMeX(cod)] (X = Cl, I [16(b)]), [PtI2(cod)] [21], and [P(NMeCH2)2(R’)] (R’ = OMe [17], NEt2 [18]) were synthesized according to the reported procedures. NMR spectra (1H, 13C, 31P) were measured on JEOL JNM-AL400 spectrometer at 25 °C. 1H and 13C NMR data were referred to residual peaks of solvent as an internal standard. Peak position of the 31P NMR spectrum was referenced to an external 85% H3PO4.
4.2 Syntheses of complexes 1–12
4.2.1 cis-[Pt(Me)2{P(NMeCH2)2(OMe)}2]: 1
A benzene solution of Pt(Me)2(cod) (300 mg, 0.90 mmol) was slowly added to [P(NMeCH2)2(OMe)] (296 mg, 2.00 mmol) at 25 °C. The mixture was stirred for 1 h at ambient temperature. Volatile materials were removed under reduced pressure, and the resulting residue was washed with a small amount of hexane, collected by filtration, and dried in vacuo to give a white solid of 1 (463 mg, 0.88 mmol, 98%). Single-crystals of 1 were obtained by solvent diffusion over a few days from an acetone layer containing 1 and an overlayer of hexane. 1H NMR (400 MHz, CDCl3, δ, ppm): 0.42 (apparent t, JPH = 7.3 Hz, JPtH = 67.4 Hz, 6H, PtMe), 2.49 (d, JPH = 10.7 Hz, 12H, NMe), 3.18 (dt, JPH = 5.9 Hz, JHH = 8.3 Hz, 4H, NCH2), 3.30 (d, JPH = 10.7 Hz, 6H, OMe), 3.37 (br, 4H, NCH2). 13C{1H} NMR (100.4 MHz, CDCl3, δ, ppm): 1.27 (dd, JPC = 12.5, 138.7 Hz, JPtC = 563.8 Hz, PtMe), 33.49 (apparent t, JPC = 7.5 Hz, JPtC = 10.8 Hz, NMe), 50.83 (apparent t, JPC = 5.0 Hz, JPtC = 15.0 Hz, OMe), 52.42 (apparent t, JPC = 1.7 Hz, JPtC = 11.6 Hz, NCH2). 31P{1H} NMR (161.7 MHz, CDCl3, δ, ppm): 125.07 (s, JPtP = 2860 Hz). Elemental Analysis. Calcd. For C12H32N4O2P2Pt: C, 27.64; H, 6.19; N, 10.74; Found: C, 27.31; H, 6.08; N, 10.45%.
4.2.2 cis-[Pt(p-tol)2{P(NMeCH2)2(OMe)}2]: 2
In a procedure analogous to that outlined above, Pt(p-tol)2(cod) (346 mg, 0.71 mmol) and P(NMeCH2)2(OMe) (259 mg, 2.42 mmol) gave a gray powder of 2 (389 mg, 0.58 mmol, 81%). Single-crystals of 2 were obtained from an acetone-hexane solution containing 2. 1H NMR (400 MHz, CDCl3, δ, ppm): 2.12 (s, 6H, PhMe), 2.53 (d, JPH = 10.3 Hz, 12H, NMe), 2.82 (dt, JPH = 5.4 Hz, JHH = 7.8 Hz, 4H, NCH2), 3.22 (d, JPH = 10.3 Hz, 6H, OMe, and overlapped 4H of NCH2), 6.71 (d, JHH = 6.8 Hz, 4H, m-Ph), 7.04 (t, JHH = 7.3 Hz, JPtH = 56.6 Hz, 4H, o-Ph). 13C {1H} NMR (100.4 MHz, CDCl3, δ, ppm): 21.20 (s, PhMe), 33.09 (apparent t, JPC = 7 Hz, NMe), 50.07 (apparent t, JPC = 5 Hz, OMe), 51.83 (s, NCH2), 126.72 (t, JPC = 4 Hz, JPtC = 63 Hz, o-Ph), 129.67 (s, p-Ph), 137.63 (t, JPC = 2 Hz, JPtC = 38 Hz, m-Ph), 157.77 (dd, JPC = 18, 164 Hz, JPtC = 830 Hz, PtC). 31P {1H} NMR (161.7 MHz, CDCl3, δ, ppm): 118.56 (s, JPPt = 2809 Hz). Elemental Analysis. Calcd. For C24H40N4O2P2Pt: C, 42.79; H, 5.99; N, 8.32; Found: C, 42.74; H, 6.04; N, 8.00%.
4.2.3 cis-[Pt(Me)2{P(NMeCH2)2(NEt2)}2]: 3
In a procedure analogous to that outlined above, Pt(Me)2(cod) (284 mg, 0.85 mmol) and [P(NMeCH2)2(NEt2)] (322 mg, 1.70 mmol) gave a gray powder of 3 (359 mg, 0.60 mmol, 70%). Single-crystals of 2 were obtained by a saturated hexane solution at −20 °C. 1H NMR (400 MHz, CDCl3, δ, ppm): 0.36 (m, JPtH = 66.4 Hz, 6H, PtMe), 0.95 (t, JHH = 7.3 Hz, 12H, NCH2CH3), 2.47 (d, JPH = 11.2 Hz, 12H, NMe), 2.95 (d, JPH = 5.9 Hz, JPtH = 17.6 Hz, 4H, NCH2), 3.07 (dq, JHH = 7.3 Hz, JPH = 9.8 Hz, 8H, NCH2CH3), 3.18 (br, 4H, NCH2). 13C {1H} NMR (100.4 MHz, CDCl3, δ, ppm): 4.13 (dd, JPC = 11.5, 129 Hz, JPtC = 567 Hz, PtMe), 14.32 (s, NCH2CH3), 34.76 (apparent t, JPC = 5 Hz, NMe), 38.87 (m, NCH2CH3), 52.14 (s, JPtH = 4 Hz, NCH2). 31P {1H} NMR (161.7 MHz, CDCl3, δ, ppm): 120.13 (s, JPtP = 2860 Hz). Elemental Analysis. Calcd. For C18H46N6P2Pt: C, 35.82; H, 7.68; N, 13.92; Found: C, 36.09; H, 7.72; N, 13.79%.
4.2.4 cis-[Pt(p-tol)2{P(NMeCH2)2(NEt2)}2]: 4
In a procedure analogous to that outlined above, Pt(p-tol)2(cod) (238 mg, 0.49 mmol) and [P(NMeCH2)2(NEt2)] (315 mg, 1.66 mmol) gave a white solid of 4 (327 mg, 0.44 mmol, 90%). 1H NMR (400 MHz, CDCl3, δ, ppm): 1.02 (t, JHH = 7.3 Hz, 12H, NCH2CH3), 2.07 (s, 6H, PhMe), 2.24 (d, JPH = 7.3 Hz, 12H, NMe), 3.03 (s, 8H, NCH2), 3.22 (dq, JHH = 7.3 Hz, JPH = 3.4 Hz, 8H, NCH2CH3), 6.61 (d, JHH = 6.8 Hz, 4H, m-Ph), 7.03 (d, JHH = 5.9 Hz, JPtH = 55.2 Hz, 4H, o-Ph). 13C {1H} NMR (100.4 MHz, CDCl3, δ, ppm): 14.16 (s, NCH2CH3), 21.13 (s, PhMe), 33.81 (apparent t, JPC = 5 Hz, NMe), 38.93 (apparent t, JPC = 5 Hz, NCH2CH3), 51.21 (s, NCH2), 126.77 (t, JPC = 3 Hz, JPtC = 66 Hz, o-Ph), 128.43 (t, JPC = 9 Hz, p-Ph), 137.57 (t, JPC = 17 Hz, m-Ph), 156.44 (dd, JPC = 15, 143 Hz, JPtC = 804 Hz, PtC). 31P {1H} NMR (161.7 MHz, CDCl3, δ, ppm): 107.40 (s, JPtP = 2688 Hz). Elemental Analysis. Calcd. For C30H54N6P2Pt: C, 47.67; H, 7.20; N, 11.12; Found: C, 47.68; H, 7.21; N, 10.94%.
4.2.5 [PtCl{P(NMeCH2)2(OMe)}3]Cl: 5
In a procedure analogous to that outlined above, PtCl2(cod) (224 mg, 0.60 mmol) and [P(NMeCH2)2(OMe)] (266 mg, 1.79 mmol) gave a white powder of 5 (387 mg, 0.54 mmol, 91%). 1H NMR (400 MHz, CDCl3, δ, ppm): 2.75 (m, 6H, NMe), 2.80 (t, JPH = 5.9 Hz, 12H, NMe), 3.24 (m, 2H, NCH2), 3.39 (t, JPH = 3.4 Hz, 3H, OMe), 3.41-3.48 (m, 6H, OMe and 6H, NCH2), 3.51 (m, 4H, NCH2). 13C{1H} NMR (100.4 MHz, CDCl3, δ, ppm): 33.00 (m, overlapped NMe of three P ligands), 50.62 (d, JPC = 4 Hz, OMe on assignable P trans to Cl), 51.26 (t, JPC = 4 Hz, OMe on assignable P cis to Cl), 52.21 (m, overlapped NCH2 of three P ligands). 31P {1H} NMR (161.7 MHz, CDCl3, δ, ppm): 71.24 (t, JPP = 13 Hz, JPtP = 5375 Hz, trans to Cl), 99.17 (d, JPP = 13 Hz, JPtP = 3633 Hz, cis to Cl). Elemental Analysis. Calcd. For C15H39N6O3P3Cl2Pt: C, 25.36; H, 5.53; N, 11.83; Found: C, 23.34; H, 5.90; N, 10.68%.
4.2.6 [PtI{P(NMeCH2)2(OMe)}3]I: 6
In a procedure analogous to that outlined above, PtI2(cod) (557 mg, 1.00 mmol) and [P(NMeCH2)2(OMe)] (445 mg, 3.00 mmol) gave a white solid of 6 (868 mg, 0.97 mmol, 97%). Single-crystals of 6 were obtained by solvent diffusion over a few days from a CH2Cl2 layer containing 6 and an overlayer of hexane. 1H NMR (400 MHz, CDCl3, δ, ppm): 2.62 (apparent t, JPH = 6.0 Hz, 12H, NMe), 2.66 (d, JPH = 11.2 Hz, 6H, NMe), 3.17 (m, 2H, NCH2), 3.29 (m, 4H, NCH2), 3.34 (d, JPH = 12.4 Hz, OMe, 3H), 3.35 (apparent t, JPH = 6.0 Hz, 6H, OMe), 3.39 (m, 4H, NCH2), 3.45 (m, 2H, NCH2). 13C{1H} NMR (100.4 MHz, CDCl3, δ, ppm): 32.68 (m, JPtC = 159 Hz, NMe of assignable P cis to I), 32.77 (m, NMe of assignable P trans to I), 50.23 (m, OMe on assignable P cis to I), 51.15 (m, OMe of assignable P trans to I), 51.63 (m, overlapped NCH2 of three P ligands). 31P{1H} NMR (161.7 MHz, CDCl3, δ, ppm): 70.43 (t, JPP = 15 Hz, JPPt = 5158 Hz, trans to I), 93.63 (d, JPP = 15 Hz, JPPt = 3512 Hz, cis to I). Elemental Analysis. Calcd. For C15H39N6O3P3I2Pt: C, 20.17; H, 4.40; N, 9.41; Found: C, 20.44; H, 4.38; N, 9.06%.
4.2.7 [Pt(Me){P(NMeCH2)2(OMe)}2]3Cl: 7
In a procedure analogous to that outlined above, PtMeCl(cod) (175 mg, 0.50 mmol) and [P(NMeCH2)2(OMe)] (220 mg, 1.49 mmol) gave a gray powder of 7 (304 mg, 0.44 mmol, 89%). 1H NMR (400 MHz, CDCl3, δ, ppm): 0.31 (dt, JPH = 7.3, 8.8 Hz, JPtH = 56.0 Hz, 3H, PtMe), 2.69 (d, JPH = 11.2 Hz, 6H, NMe), 2.73 (t, JPH = 5.9 Hz, 12H, NMe), 3.17 (m, 2H, NCH2), 3.30–3.33 (m, 3H, OMe and 6H, NCH2), 3.42 (t, JPH = 5.9 Hz, 6H, OMe), 3.57 (m, 4H, NCH2). 13C{1H} NMR (100.4 MHz, CDCl3, δ, ppm): −3.00 (dt, JPC = 106 Hz, JPC = 9 Hz, JPtC = 443 Hz, PtMe), 32.75 (t, JPC = 7 Hz, NMe), 33.12 (d, JPC = 14 Hz, JPtC = 9 Hz, NMe), 50.83 (d, JPC = 13 Hz, OMe), 51.40 (apparent t, JPC = 9 Hz, OMe), 51.63 (s, JPtC = 10 Hz, NCH2), 52.20 (s, JPtC = 18 Hz, NCH2). 31P {1H} NMR (161.7 MHz, CDCl3, δ, ppm): 106.06 (d, JPP = 37 Hz, JPtP = 4070 Hz, cis to Me on the Pt), 122.78 (t, JPP = 37 Hz, JPtP = 2818 Hz, trans to Me on the Pt). Elemental Analysis. Calcd. For C16H42N6O3P3ClPt: C, 27.85; H, 6.14; N, 12.18; Found: C, 26.80; H, 6.30; N, 11.51%.
4.2.8 [Pt(Me){P(NMeCH2)2(OMe)}3]I: 8
In a procedure analogous to that outlined above, PtMeI(cod) (146 mg, 0.32 mmol) and [P(NMeCH2)2(OMe)] (145 mg, 0.98 mmol) gave a gray powder of 8 (237 mg, 0.30 mmol, 94%). Single-crystals of 8 were obtained by solvent diffusion over a few days from a CH2Cl2 layer containing 8 and an overlayer of hexane. 1H NMR (400 MHz, CDCl3, δ, ppm): 0.22 (dt, JPH = 7.3, 8.8 Hz, JPtH = 56.0 Hz, 3H, PtMe), 2.63 (d, JPH = 24.4 Hz, 6H, NMe), 2.71 (t, JPH = 4.8 Hz, 12H, NMe), 3.15 (m, 2H, NCH2), 3.23 (d, JPH = 11.6 Hz, 6H, OMe and overlapped 1H of NCH2), 3.42 (d, JPH = 5.2 Hz, 3H, OMe and overlapped 1H and 4H of NCH2), 3.56 (br, 4H, NCH2). 13C {1H} NMR (100.4 MHz, CDCl3, δ, ppm): 3.27 (dt, JPC = 8, 106 Hz, JPtC = 442 Hz, PtMe), 32.48 (t, JPC = 7 Hz, JPtC = 10 Hz, NMe), 32.85 (d, JPC = 8 Hz, JPtC = 15 Hz, NMe), 50.60 (d, JPC = 12 Hz, JPtC = 15 Hz, OMe), 51.26 (d, JPC = 5 Hz, JPtC = 22 Hz, OMe), 51.33 (s, JPtC = 8 Hz, NCH2), 51.85 (s, JPtC = 19 Hz, NCH2). 31P {1H} NMR (161.7 MHz, CDCl3, δ, ppm): 105.97 (d, JPP = 37 Hz, JPPt = 4003 Hz, cis to Me on the Pt), 122.73 (t, JPP = 37 Hz, JPPt = 2818 Hz, trans to Me on the Pt). Elemental Analysis. Calcd. For C16H42N6O3P3IPt: C, 24.59; H, 5.42; N, 10.75; Found: C, 24.76; H, 5.38; N, 10.55%.
4.2.9 cis-[Pt(Me)(Cl){P(NMeCH2)2(NEt2)}2] and trans-[Pt(Me)(Cl){P(NMeCH2)2(NEt2)}2]: 9
In a procedure analogous to that outlined above, PtMeCl(cod) (365 mg, 1.03 mmol) and [P(NMeCH2)2(NEt2)] (391 mg, 2.07 mmol) gave a white powder of 9 (550 mg, 0.88 mmol, 85%, trans: cis = ca. 2:1). 1H NMR (400 MHz, CDCl3, δ, ppm): 0.47 (apparent t, JPH = 6.8 Hz, JPtH = 86.4 Hz, 3H, PtMe), 0.71 (dd, JPH = 2.0, 8.8 Hz, JPtP = 49.3, 3H, PtMe), 0.99 (t, JPH = 7.3 Hz, 12H, NCH2CH3), 1.05 (t, JPH = 7.3 Hz, 12H, NCH2CH3), 2.47 (d, JPH = 11.7 Hz, 4H, NCH2), 2.65 (d, JPH = 10.3 Hz, 4H, NCH2), 2.72 (t, JPH = 5.4 Hz, 16H, NCH2CH3), 2.98–3.29 (m, 8H, NCH2 and 24H, NMe). 13C{1H} NMR (100.4 MHz, CDCl3, δ, ppm): –17.92 (t, JPtC = 678 Hz, JPC = 8 Hz, PtMe trans to chloride), 9.80 (dd, JPC = 6, 123 Hz, JPtC = 455 Hz, PtMe, trans to phosphine), 14.50 (d, JPC = 2 Hz, NCH2CH3), 14.81 (d, JPC = 2 Hz, NCH2CH3), 15.11 (s, NCH2CH3), 34.63 (t, JPC = 4 Hz, NMe), 35.02 (d, JPC = 8 Hz, NMe), 35.53 (d, JPC = 9 Hz, NMe), 39.69 (d, JPC = 7 Hz, NCH2CH3), 39.79 (d, JPC = 5 Hz, NCH2CH3), 40.70 (d, JPC = 7 Hz, NCH2CH3), 50.95 (s, NCH2), 51.67 (d, JPC = 2 Hz, NCH2), 52.07 (d, JPC = 3 Hz, NCH2). 31P{1H} NMR (161.7 MHz, C6D6, δ, ppm): 82.84 (d, JPP = 22 Hz, JPtP = 6037 Hz, trans to Cl), 102.20 (s, JPtP = 3938 Hz, trans to P), 116.60 (d, JPP = 22 Hz, JPtP = 2651 Hz, trans to Me). Elemental Analysis. Calcd. For C17H43N6P2ClPt: C, 32.72; H, 6.95; N, 13.47; Found: C, 32.10; H, 6.83; N, 13.06%.
4.2.10 trans-[Pt(Me)(I){P(NMeCH2)2(NEt2)}2]: 10
In a procedure analogous to that outlined above, PtMeI(cod) (482 mg, 0.97 mmol) and [P(NMeCH2)2(NEt2)] (367 mg, 1.94 mmol) gave a gray powder of 10 (627 mg, 0.88 mmol, 90%). Single-crystals of 10 were obtained by solvent diffusion over a few days from a CH2Cl2 layer containing 10 and an overlayer of hexane. 1H NMR (400 MHz, C6D6, δ, ppm): 0.73 (t, JPH = 6.4 Hz, JPtH = 83.5 Hz, 3H, PtMe), 1.05 (t, JHH = 6.8 Hz, 12H, NCH2CH3), 2.74 (t, JPH = 5.4 Hz, 12H, NMe), 3.15–3.23 (m, 8H, NCH2 and 8H, NCH2CH3). 13C{1H} NMR (100.4 MHz, C6D6, δ, ppm): −9.09(t, JPC = 8 Hz, JPtC = 678 Hz, PtMe), 14.72 (s, NCH2CH3), 35.80 (t, JPC = 4 Hz, NMe), 39.72 (t, JPC = 5 Hz, NCH2CH3), 51.10 (s, NCH2). 31P {1H} NMR (161.7 MHz, C6D6, δ, ppm): 103.24 (s, JPtP = 3850 Hz). Elemental Analysis. Calcd. For C17H43N6P2Pt: C, 28.16; H, 5.98; N, 11.45; Found: C, 28.54; H, 6.06; N, 11.75%.
4.2.11 [PtCl{P(NMeCH2)2(OMe)}3]BPh4: 11
BF3·OEt2 (38 μl, 0.3 mmol) was slowly added to a CH2Cl2 solution of 7 (207.0 mg, 0.3 mmol) at 25 °C. The mixture was stirred for 1 h at room temperature, and NaBPh4 (102.7 mg, 0.3 mmol) was added. After stirred for 1 h at room temperature, this mixture was filtrated. Volatile materials in the filtrate were removed under reduced pressure to give a colorless oil. Single-crystals of 11 (134.2 mg, 0.14 mmol, 45%) were obtained by solvent diffusion over a few days from a CH2Cl2 layer containing a colorless oil and an overlayer of hexane. 1H NMR (400 MHz, CDCl3, δ, ppm): 2.63 (t, JPH = 10.7 Hz, 6H, NMe), 2.68 (t, JPH = 5.9 Hz, 9H, NMe), 2.75 (t, JPH = 5.9 Hz, 3H, NMe), 3.02–3.07 (m, 2H, NCH2), 3.21(m, 4H, NCH2), 3.24 (d, JPH = 11.7 Hz, 3H, OMe), 3.29–3.30 (m, 2H, NCH2), 3.32 (t, JPH = 5.9 Hz, 6H, OMe), 3.43 (m, 4H, NCH2), 6.93 (t, JHH = 7.3 Hz, 4H, p-Ph), 7.08 (t, JHH = 7.3 Hz, 8H, m-Ph), 7.44 (br, 8H, o-Ph). 13C{1H} NMR (100.4 MHz, CDCl3, δ, ppm): 32.74 (apparent t, JPC = 7 Hz, NMe), 33.12 (d, JPC = 15 Hz, NMe), 51.27 (m, OMe), 51.50 (m, OMe), 52.16 (t, JPC = 10 Hz, NCH2, and overlapped other NCH2), 121.64 (s, p-Ph), 125.50 (q, JBC = 3 Hz, o-Ph), 136.37 (s, m-Ph), 164.33 (q, JBC = 49 Hz, ipso-Ph). 31P{1H} NMR (161.7 MHz, CDCl3, δ, ppm): 71.35 (t, JPP = 22 Hz, JPPt = 5342 Hz, trans to Cl), 99.09 (d, JPP = 22 Hz, JPPt = 3583 Hz, cis to Cl). Elemental Analysis. Calcd. For C39H59BClN6O3P3Pt: C, 44.52; H, 5.70; N, 7.79; Found: C, 44.97; H, 5.87; N, 7.73%.
4.2.12 [PtI{P(NMeCH2)2(OMe)}3]BPh4: 12
BF3·OEt2 (38 μl, 0.3 mmol) was slowly added to a CH2Cl2 solution of 8 (234.4 mg, 0.3 mmol) at 25 °C. The mixture was stirred for 2.5 h at room temperature, and NaBPh4 (102.7 mg, 0.3 mmol) was added. After stirred for 1 h at room temperature, this mixture was filtrated. Volatile materials in the filtrate were removed under reduced pressure to give a yellow oil. Single-crystals of 12 (136.8 mg, 0.13 mmol, 42%) were obtained by solvent diffusion over a few days from a CH2Cl2 layer containing a yellow oil and an overlayer of hexane. 1H NMR (400 MHz, CDCl3, δ, ppm): 2.63 (d, JPH = 11.7 Hz, 6H, NMe), 2.68 (t, JPH = 5.9 Hz, 12H, NMe), 2.93–3.03 (m, 4H, NCH2), 3.24 (d, JPH = 13.7 Hz, 3H, OMe), 3.28 (t, JPH = 6.4 Hz, 6H, OMe), 3.34–3.46 (m, 8H, NCH2), 6.92 (t, JHH = 7.3 Hz, 4H, p-Ph), 7.07 (t, JHH = 7.3 Hz, 8H, m-Ph), 7.43 (br, 8H, o-Ph). 13C{1H} NMR (100.4 MHz, CDCl3, δ, ppm): 32.85 (d, JPC = 12 Hz, NMe), 33.20 (t, JPC = 7 Hz, NMe), 50.64 (d, JPC = 3 Hz, OMe), 50.98 (m, OMe), 52.26 (s, overlapped NCH2 of three P ligands), 121.96 (s, p-Ph), 125.78 (q, JBC = 3 Hz, o-Ph), 136.60 (s, m-Ph), 164.54 (q, JBC = 49 Hz, ipso-Ph). 31P{1H} NMR (161.7 MHz, CDCl3, δ, ppm): 70.54 (t, JPP = 15 Hz, JPPt = 5173 Hz, trans to I), 93.68 (d, JPP = 15 Hz, JPPt = 3524 Hz, cis to I). Elemental Analysis. Calcd. For C39H59BIN6O3P3Pt: C, 43.15; H, 5.48; N, 7.74; Found: C, 43.53; H, 5.64; N, 7.41%.
4.3 X-ray crystallography measurements
X-ray intensity data were collected on a Rigaku/MSC Mercury CCD diffractometer with graphite monochromated Mo-Kα radiation. Calculations were performed with the CrystalClear software package of Molecular Structure Corporation. The structures were solved by direct methods and expanded using Fourier techniques. The structures were refined by full matrix least-squares technique using the program SHELXL-97 [25]. The non-hydrogen atoms were refined anisotropically. All hydrogen atoms were placed in calculated positions. Complexes 1–3, 6, 8, and 10–12 have been deposited with the Cambridge Crystallographic Data Centre under CCDC 761300, 761301, 761302, 761303, 761304, 761305, 761306, and 761307, respectively. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.html (or from the Cambridge Crystallographic Data Centre, 12, Union Road, Cambridge CB2 1EZ, UK; tel.; +44 1223 336408; fax: +44 1223 336033; email: deposit@ccdc.cam.ac.uk).
Acknowledgements
This work was supported by a Grant-in-Aid for Science Research on Priority Areas (No. 20036043, Synergistic Effect of Elements), by a Challenging Exploratory Research (No. 21655022), and by a Grant-in-Aid for Young Scientists (B) (No. 20750049) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, by Innovation Promotion Program in 2009 from New Energy and Industrial Technology Development Organization (NEDO) of Japan, and by a Research for Promoting Technological Seeds from the Japan Science and Technology Agency (JST). M.I. also acknowledges support from the Iketani Science and Technology Foundation (0211033-A).