

1 Introduction
Kinetics plays a central role in catalysis and is not solely restricted to fundamental approaches providing new insight into reaction mechanisms. Indeed, recent investigations underlined the potential of kinetic modeling from a practical point of view [1]. For instance, the efficiency of lean NOx trap systems can be optimized by using a microkinetic model [2,3] with the objective to lower the noble metal content. Hence, there is presently a growing interest to develop robust mathematical models for predicting unsteady-state kinetic regimes in order to control more efficiently integrated after-treatment systems [1,4,5]. Different methodologies have been already envisioned which consist in integrating kinetic parameters previously determined on powder catalysts in a numerical model adapted for the prediction of outlet gas composition of Selective Catalytic Reduction (SCR) monolith reactors. This strategy was profitably used to highlight the inhibiting effect of ammonia and to define the optimal surface coverage at low temperature. From this approach, it seems possible to implement intelligent urea dosage strategies for lean-burn after-treatment systems. Zuckerman et al. [1] used the COMSOL Package environment to create a platform capable to simulate the dynamic behavior of coupled NOx storage/Reduction (NSR) and SCR after-treatment systems integrating separate kinetic models for ammonia formation and oxidation during the regenerative step of the NSR catalyst (Fig. 1). Based on these previous achievements, it seems obvious that such predictive mathematical tools can be adapted for more complex architectures of after-treatment systems combining Diesel oxidation catalysts (DOC), Diesel particulate filter (DPF), NOx storage reduction (NSR) and selective catalytic reduction (SCR) systems including chemical processes at the surface of the catalyst and the effects of transport phenomena which affect the catalyst efficiency.

(Color online.) A. θNO and θNH3 (the ratio between adsorbed NO and NH3 to catalyst capacity, respectively) in NOx storage unit and NH3-SCR unit, as a function of time (at experimental time and optimal time). B. NO Conversion and the ratio between reacted NH3 and adsorbed (stored) NH3 for two volume configurations.
Reproduced with permission from reference [1].
Returning to fundamental aspects, numerous investigations implemented kinetic approaches to a wide panel of applications under steady-state [6–14] or transient operating conditions [15–17]. Further developments of new advanced methodologies such as Steady-State Isotopic Transient Kinetic Analysis (SSITKA) utilizing labeled molecules [18–23] also contributed to the elucidation of complex surface chemical processes. Parallel to those developments, surface science investigations provided in the past three decades reliable information on the nature of elementary steps especially in the field of environmental catalysis in connection with the development of Three-Way Catalysts (TWC) containing noble metals (Pd, Pt and Rh). While those experiments have been currently performed over model catalysts and under Ultra-High-Vacuum (UHV), far from realistic pressure conditions, they provided useful kinetic data related to the structure sensitivity of the reduction of NO over noble metals [24–27]. These results have been further verified over polycrystalline catalysts as reported elsewhere [28].
The emergence of new experimental techniques also contributes to a better nano description of the catalysts. First, the strategy associated with the implementation of the Temporal Analysis of Products (TAP) reactors [28–33] can be considered as an intermediate approach between classical kinetic and surface science studies, characterizing real catalysts rather than single crystals and in pressure ranges higher than those currently encountered in this latter case. Interestingly, it was found that the kinetic parameters obtained from TAP measurements can correctly model steady-state kinetic experiments at higher pressure conditions [34,35] emphasizing the fact that the strategies developed by using this technique can be considered at the boundary between conventional kinetics and surface sciences. The development of microkinetic approaches is also intimately related to the implementation of spectroscopic techniques that can probe the surface under reaction conditions. This approach is more reliable since the construction of active sites will strongly depend on the nature of the gas composition under reaction conditions initiating surface reconstructions which can modify the final architecture of the active sites. Operando spectroscopic investigations are nowadays widespread. But one has to say that only few of them led to relevant information at the molecular level [36–39] because of the difficulty to identify accurately reactive intermediates involved in the catalytic cycle among all adsorbate species accumulated on the active sites and on the support.
In line with previous above-mentioned considerations, this contribution will tentatively offer a short overview of complementary kinetic and spectroscopic information over Natural Gas Vehicle (NGV) catalysts. The problematic is related to the simultaneous removal of NOx and methane. Particular attention will be paid to the cold start engine for which incomplete reduction of NOx to N2O is currently observed and may represent an important task for the near future with the adoption of new standard regulations including greenhouse gas such as N2O. In addition, several challenging aspects are addressed concerning the conversion of refractory atmospheric pollutants such as methane in the specific case of natural gas-fuelled engines. The efficiency of the catalysts in these conditions strongly depends on the nature of the reductive/oxidative atmosphere and usually results from complex chemical processes at the surface involving the participation of the metal/support interface especially for methane activation. In this context, it will be demonstrated that TAP measurements can provide original quantitative kinetic information.
2 The low temperature NO/H2 reaction over monometallic and bimetallic noble metal NGV catalysts
2.1 Brief summary of previous studies of the kinetics of the catalytic NO/H2 reaction
This reaction has been of investigated in the past three decades starting from fundamental approaches on model surfaces and coinciding with the commercialization of TWCs. Surface science investigations under UHV conditions provided interesting kinetic features in conditions far from those encountered in three-way converters operating at elevated temperature and atmospheric pressure. Monte Carlo simulations were also reported for modeling oscillations during the NO/H2 reaction over Pt(100) [40]. As a general trend, such an oscillation behavior occurs when a rapid bi-stable process is combined with a slower one related to adsorbate-induced surface restructuring, formation of surface or sub-surface oxides or diffusion limitation. Under UHV conditions, oscillations are often related to adsorbate-induced surface restructuring as previously pointed out for CO oxidation and necessitate high local CO or oxygen coverage. Similar behavior was highlighted by Tanaka and Sasahara [41] during the NO/H2 reaction over bimetallic single crystals of Rh/Pt(100), Rh/Pt(110), Pt/Rh(100) and Pt/Rh(110). Interestingly, they found that the NO/H2 reaction is sensitive to the crystallographic structure of single Pt or Rh surfaces according to the following sequence: Pt(100) > Pt(110) and Rh(110) > Rh(100). On the other hand, this reaction becomes structure insensitive over bimetallic surfaces since an inactive Pt(110) will become active after annealing a bimetallic Rh/Pt(110) surface in oxygen (Fig. 2). The same tendency is also observed on Pt/Rh(100) after surface reconstructions under oxygen exposure corresponding to the extraction of Rh atoms from the second layer forming highly active p(3 × 1) surface towards the NO/H2 reaction as illustrated in Fig. 3. Regarding model Pd catalysts, hysteresis phenomena were observed during the NO/H2 reaction on Pd(111) between RT and 577 °C at very low NO pressure (10−7 mbar) ascribed to the formation/decomposition of hydrides [42]. Interestingly, these authors also proved the formation of N2 and N2O desorbing from the surface to be independent of the surface coverage. For supported polycrystalline catalysts, the support may generate such oscillatory behaviors. By way of illustration, Machida and Ikeda observed oscillations during low temperature NO/H2/O2 reaction over Pt supported on TiO2-ZrO2 [43] exhibiting basic sites. Under net oxidizing conditions, nitrate species accumulate on the basic sites and block further NO adsorption inducing a loss of conversion. The authors established a correlation between the surface coverage of the nitrates and the oscillation period.

Catalytic activity of the reconstructed bimetallic surfaces (a) p(3 × 1) Rh/Pt(100)/O and c(2 × 2) Rh/Pt(110)/O; (b) p(3 × 1) Pt/Rh(100)/O and c(2 × 2) Pt/Rh(110)/O.
Reproduced with permission from reference [41].

Step-by-step heating of Pt0.25Rh0.75(100) surface in 1 × 10−7 mbar O2 for 5 min. Segregation of Rh and p(3 × 1) reconstruction occur at temperature higher than ca. 400 K.
Reproduced with permission from reference [41].
Today, the interest for this reaction is renewed due to the development of new varieties of catalysts, essentially silver and gold supported on alumina which were found much more selective than noble metals for the reduction of NO in the presence of hydrogen under net oxidizing conditions [44–47]. Presently, the role of hydrogen is not completely elucidated over such catalysts. Several explanations have been suggested, some of them are closely related to those earlier proposed for noble metals regarding the positive effect of hydrogen for destabilizing inactive nitrates species strongly adsorbed on silver sites and the generation of metal hydride well-recognized when palladium is used for such an application. Alternatively, Backman et al. [48] suggested on silver-based catalysts a reaction mechanism which would involve chemisorbed HNO intermediates formed on Ag particles via a reaction between chemisorbed NO molecules and H atoms. Such an interpretation seems closely related to the mechanisms previously proposed over noble metals involving a dissociation step of NO assisted by chemisorbed hydrogen atoms [49–55]. Hence, this explanation can be relevant to explain the significant gain in activity when H2 reduces NO at rather low temperature as compared to CO. This is particular true for Pt based catalysts where strongly chemisorbed CO species prevent NO adsorption [56]. On the other hand, over Rh, it is the strong adsorption of NO species that inhibits the dissociative H2 adsorption [10]. Similar behavior has also been evidenced on Pd. Modeled TPD curves using COMSOL in Fig. 4 show that NO displaces chemisorbed hydrogen at low temperature whereas desorption and reaction take place simultaneously at higher temperature leading to lower emissions of gaseous H2. This predicted behavior seems consistent with experimental evidences collected in Fig. 5 from transient experiments. As illustrated, when a H2-pre-adsorbed surface is exposed to NO pulses at 70 °C then the production of N2 and H2 evolves in the first stage of the experiments which emphasizes the fact that H2 is partly displaced by NO adsorption. It is also worthwhile to note that N2 production occurs more readily. This observation suggests that a fraction of chemisorbed H atoms would be involved in the NO bond scission. As a consequence, the recombination of chemisorbed N atoms to form N2 prevails while no ammonia formation takes place via successive hydrogenation steps of chemisorbed nitrogen atoms. Afterwards, the formation of N2O appears presumably coinciding with a reduced surface coverage of hydrogen. All those fundamental information are of practical interests especially to improve the efficiency of NSR catalysts at low temperature during the cold start engine. Indeed, it was demonstrated that nitrates can be converted at much lower temperature than that requested for their thermal decomposition which suggests that hydrogen dissociated over noble metal would spill-over onto the surface to react with N-containing species at the metal/support interface [57]. These observations lead to practical developments, especially for the regeneration step of NSR catalysts with the injection of a reformate (CO/H2 mixture) upstream the trap [58]. As previously argued, these findings can open new practical developments associated with significant breakthrough related to the production and the use of ammonia in coupled NSR + SCR systems [59].
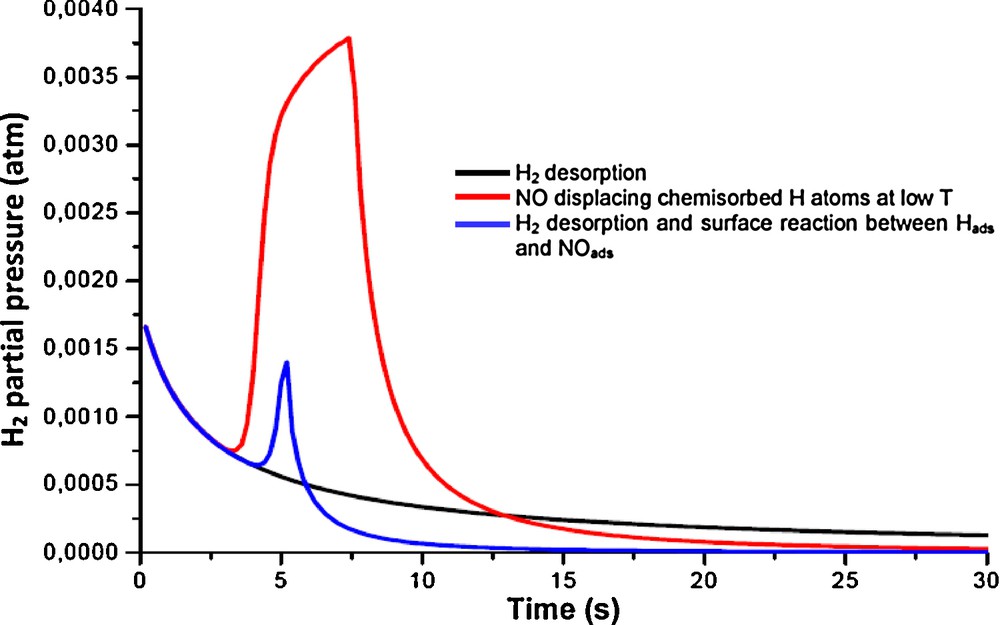
(Color online.) Predicted desorption profiles after NO admission on a hydrogen pre-adsorbed Pd surface and competitive surface reactions taking place at 90 °C.

Temporal response after NO exposure of H2-pre-adsorbed Pd/Al2O3 at 70 °C.
2.2 Monometallic noble metal based catalysts
2.2.1 Discussion on the nature of the rate determining step
Initially, Hecker and Bell [49] proposed a reaction mechanism for depicting the NO/H2 reaction over pre-reduced and pre-oxidized Rh/SiO2 catalysts (Scheme 1). A positive effect of the partial pressure of hydrogen on the turn-over frequencies is noticeable irrespective of the nature of the pretreatment. In parallel, they found a selectivity behavior towards N2O and N2 formation insensitive to the partial pressure of NO which suggests a selectivity independent of NO coverage, as previously demonstrated over Pd(111) under UHV conditions [42]. Hecker and Bell rationalized their results by suggesting a reaction mechanism involving H-assisted NO dissociation step as rate determining. They also neglected the recombination of two adjacent chemisorbed nitrogen atoms for N2 formation (step (4) reported in the Scheme 1), in favor of step (5). This mechanism, and related assumptions, successfully described the kinetics of the NO/H2 reaction in numerous investigations. In particular, Harold et al. [54,55] have determined an activation energy of 107.8 kJ·mol−1 for step (3) in Scheme 1 and attributed a value of 60 kJ·mol−1 for step (3′) (Tables 1 and 2). Those data are consistent with those obtained from theoretical calculations by applying the Unity Bond Undex-Quadratic Exponential Potential method (UBI-QEP) developed by Shustorovitch et al. [60,61]. Calculations performed on model Pd(111), Pt(111) surfaces [8] and recently extended on Rh(111) [50] led to the same tendencies as exemplified in Table 3. The deviations observed by comparing theoretical and experimental values can be simply related to the fact that theoretical calculations account for coordinated chemisorbed nitrosyl species via a two-fold bridged site. At higher coverage, the stabilization of on-top ad-NO species seems more realistic which would correspond to higher activation barriers towards NO dissociation. While hydrogen appears to promote the dissociation of NO, the participation of step (3′) involving a nearest-neighbor-vacant site for NO dissociation cannot be completely ruled out. This seems particular true in the presence of oxygen in the feed or at low temperature, typically during the cold start engine when the reaction is controlled by oxygen scavenging. By way of illustration Frank et al. [62] modeled their kinetic data on Pt-Mo-Co/α-Al2O3 via step (3′) in the temperature range 142–160 °C while they also observed a beneficial effect of hydrogen. However, they found their results more consistent with previous explanations given by Pirug and Bonzel [63] who suggested that co-adsorbed hydrogen can displace molecularly adsorbed NO releasing vacant sites requested for NO dissociation. As a matter of fact, such an explanation seems in contradiction by examining the calculated heat of adsorption for H2 and NO provided by those authors. Indeed, the numerical values of respectively –59 and –77 kJ·mol−1 still suggest that NO would displace chemisorbed H atoms on Pt-Mo-Co/α-Al2O3. Figs. 4 and 5 bring experimental evidences on palladium catalyst which seems more in agreement with this latter statement.

Reaction mechanism earlier suggested for depicting the NO/H2 reaction over supported noble metal based catalysts [8,49,50,55,62].
Rate parameters for the NO/H2 reaction on Pt/Al2O3.
| Reaction | Ai,f (mol·m–3·s–1) or S0 | Ei,f (kJ·mol–1) | Ai,r (mol·m–3·s–1) or S0 | Ei,r (kJ·mol–1) |
| H2 + 2Pt ⇆ 2 H-Pt | 0.046 | 4.02 × 1014 | 73.0 | |
| H-Pt + O-Pt ⇆ OH-Pt + Pt | 4.02 × 1014 | 11.5 | 4.02 × 1014 | 74.9 |
| OH-Pt + H-Pt → H2O + 2Pt | 4.02 × 1014 | 17.4 | ||
| NO-Pt + H-Pt ⇆ N-Pt + OH-Pt | 4.02 × 1012 | 60.0 | 4.60 × 1014 | 143.7 |
| N-Pt + 3 H-Pt ⇆ NH3-Pt + 3Pt | 8.00 × 1014 | 117.0 | 2.00 × 1017 | 146.5 |
| NO-Pt + 3 H-Pt ⇆ NH3-Pt + O-Pt + 2Pt | 8.00 × 1014 | 60.0 | 4.00 × 1015 | 109.8 |
| NH3-Pt ⇆ NH3 + Pt | 8.50 × 1015 | 48.5 | 1.00 | |
| NH3-Pt + 3 O-Pt → N-Pt + 3 OH-Pt | 1.00 × 1017 | 80.0 | ||
| NH3-Pt + 3 NO-Pt → 4 N-Pt + 3 OH-Pt | 4.00 × 1014 | 80.0 |
Rate parameters for the NO decomposition on Pt. Underlined parameters were fixed.
| Reaction | Ai,f (mol·m–3·s–1) or S0 | Ei,f (kJ·mol–1) | Ai,r (mol·m–3·s–1) or S0 | Ei,r(kJ·mol–1) |
| NO + Pt ⇆ NO-Pt | (2.38 ± 0.19) × 108(*) | (2.45 ± 0.9) × 1016 | ||
| NO-Pt + Pt ⇆ N-Pt + O-Pt | (8.76 ± 12.56) × 1013 | 107.8 | (6.42 ± 10.54) × 1014 | 100.0 |
| 2 N-Pt + H-Pt → N2 + 2Pt | (2.71 ± 1.17) × 1016 | 130.0 | ||
| NO-Pt + N-Pt ⇆ N2O + 2 Pt | (3.69 ± 0.2) × 109 | 81.5 ± 4.5 | (1.18 ± 65.90) × 103(*) | 36.2 |
| 2 NO-Pt → N2O + O-Pt + Pt | (6.45 ± 0.6) × 1015 | 152.1 ± 1.4 |
Enthalpies (ΔH) and activation energies (E) in kJ·mol–1 of suggested elementary steps for the NO/H2 reaction on Pd(111) and Rh(111) from UBI-QEP calculations.
| Reaction | Rh(111)a | Pd(111)b | Pd(111)b | |||
| ΔH | E | ΔH | E | ΔH | E | |
| NOg + * → NOads | –109 | 0 | –134 | 0 | –109 | 0 |
| H2g + 2* → 2Hads | –76 | 12 | –84 | 9 | –75 | 13 |
| O2g + 2* → 2Oads | –357 | 0 | –232 | 0 | –213 | 0 |
| NOads + * → Nads + Oads | –172 | 27 | –143 | 38 | –100 | 53 |
| NOads + Hads → Nads + OHads | –130 | 9 | –113 | 7 | –78 | 22 |
| NOads + Hads → NHads + Oads | –40 | 39 | 1 | 89 | 32 | 96 |
| Nads + Nads → N2g + 2* | 27 | 112 | 142 | 179 | 25 | 111 |
| NOads + Nads → N2g + Oads + * | –145 | 0 | –1 | 53 | –75 | 7 |
| NOads + Nads → N2Og + 2* | 114 | 114 | 197 | 197 | 113 | 113 |
| NOads + NOads → N2Og + Oads + * | –58 | 0 | 53 | 60 | 13 | 34 |
| Oads + Hads → OHads + * | 42 | 101 | 30 | 91 | 22 | 85 |
| OHads + Hads → H2Og + 2* | –24 | 52 | –67 | 0 | –77 | 0 |
An interesting kinetic feature, also reported by Frank et al., is related to the promoting effect of oxygen. This promoting effect is currently observed for the reduction of NOx by hydrocarbons via the formation of NO2 as intermediates [7]. However, in the case of the NO/H2 reaction, those authors proposed an alternative explanation based on the fact that H2 would strongly adsorb at the surface of noble metals further inducing a significant inhibiting effect on the rate of NO conversion to nitrogen. Based on this statement, they predominantly associated this beneficial effect with a faster removal of abundant hydrogen atoms by reaction with oxygen.
2.2.2 Operando spectroscopic investigation of the NO/H2 reaction
Based on previous observations, it seems obvious that hydrogen assists the NO bond scission corresponding to a significant lowering of the activation energy values for the step related to NO dissociation. As previously reported by Hecker and Bell [49], further comparisons of the reactivity of pre-reduced and pre-oxidized Rh/SiO2 led to similar turn-over frequencies with no significant impact of oxygen coverage. It was also noticeable that no significant deviation on the apparent activation energy is discernible on Rh/SiO2 which cannot strictly rule out a classical promotional effect simply due to the release of free vacant sites even if these authors privileged step (3) in Scheme 1 for representing the promotional effect of hydrogen. Hence, an indirect beneficial effect of hydrogen should be taken into account especially when oxygen is co-fed with NO and H2 in a large extent. In order to check the validity of this assumption, we have investigated this reaction by operando IR spectroscopy [64,65]. Conversion and selectivity can be compared to the nature of nitrosyl species accumulated at the surface and the evolution of their concentration during a rise in temperature (Fig. 6). Let us notice that these experiments were not realized with a large excess of oxygen but near stoichiometric conditions. Positively charged Rh(NO)δ+, neutral Rh(NO)0 and negatively charged Rh(NO)δ–species can be clearly distinguished. At low temperature, an increase in intensity of IR bands related to Rh(NO)δ+, and neutral Rh(NO)0 species is discernible and likely due to oxygen accumulation from NO dissociation. Subsequent withdrawing effects of oxygen would lower the electron back-donation into the π* anti-bonding molecular orbitals of NO. In those typical conditions, Rh surface is likely quasi-completely covered by nitrosyl species which favor a predominant formation of nitrous oxide (N2O). At more elevated temperature a sharp decrease takes place in the concentration of Rh(NO)δ–species recognized as the most reactive species towards NO dissociation. Meantime, when the H2/O2 reaction becomes predominant, a sharp decrease in O coverage would lead to the restoration of Rh(NO)δ–species at the expense of Rh(NO)0 and Rh(NO)δ+ less reactive towards NO dissociation. This corresponds to a significant decrease in N2O production and in parallel to a more accentuated N2 and ammonia formation. Hence, those observations emphasize the fact that the H2/O2 reaction may interfere by changing the concentration of nitrosyl species more reactive towards dissociation parallel to the releasing of nearest-neighbor-vacant sites requested for NO dissociation.
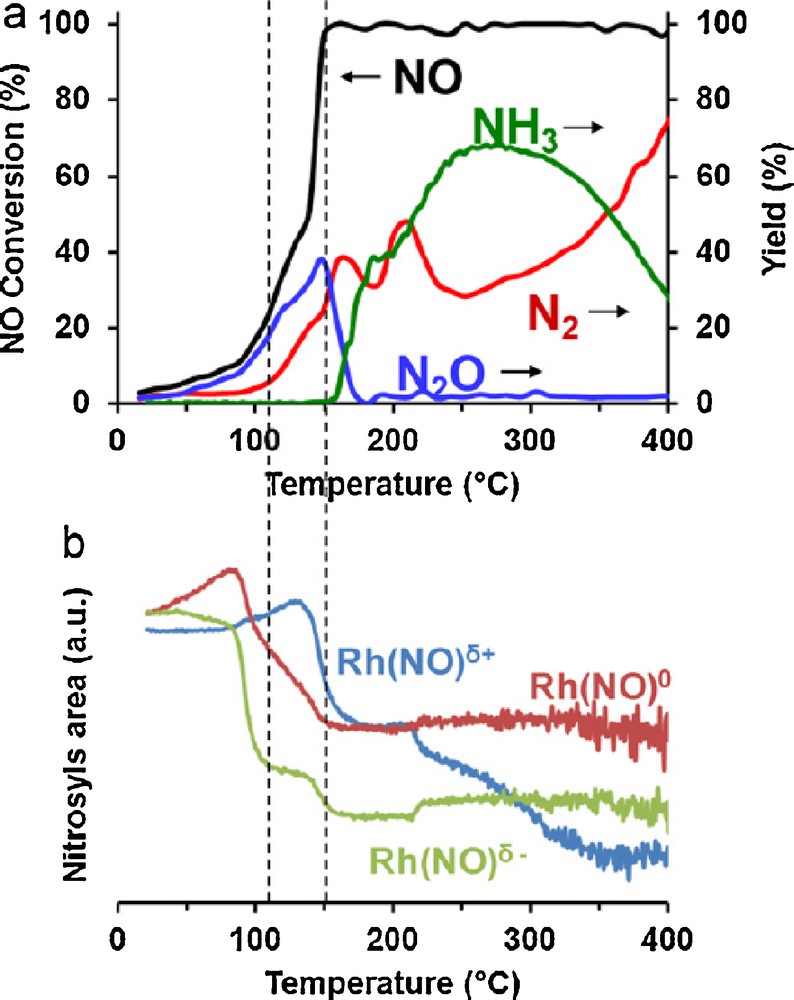
(Color online.) Temperature-programmed reaction on Rh/Al2O3 in stoichiometric conditions with 0.095 vol.% NO, 0.3 vol.% H2, 0.1025 vol.% O2 (GHSV = 10,000 h−1; dT/dt = 2.2 °C/min). Corresponding conversion and yield curves vs. temperature (a) changes in the integral IR band related to nitrosyl species (b).
Reproduced with permission from reference [64].
Operando spectroscopic experiments are also useful to investigate macroscopic evolutions of the selectivity and to relate them to changes in adsorbate compositions. This is particularly demonstrative in the case of the reduction of NO since different selectivity behaviors can be identified depending on the temperature range of the study as reported elsewhere [64,65]. From those observations, the predominance of steps (4) or (5) (Scheme 1) for the production of nitrogen can be discussed. Such kinetic features can be illustrated for the NO/H2 reaction on Rh for which the selectivity to N2 and N2O formation can be insensitive to changes in NO partial pressure at low temperature and then becoming sensitive with a rise in temperature. This is nicely illustrated in Fig. 7 based on transient experiments in which Rh/Al2O3 is periodically exposed to H2 and NO atmospheres [64]. At low temperature, the production of N2 and N2O does not vary significantly with changes in NO concentration occurring during the successive switches. On the other hand, drastic changes take place at more elevated temperature with a maximum in N2O formation coinciding with the highest NO partial pressure. Qualitatively, the low temperature behavior means that N2 and N2O formation occurs via the same intermediates (reaction between NOads and Nads). In other terms, only steps (5) and (6) would prevail as compared to the recombination of two adjacent chemisorbed N atoms which would become significant at much higher temperature. We will come back later on this aspect on bimetallic Pd–Rh/Al2O3 catalysts which behave similarly.

(Color online.) Transient experiments on Rh/Al2O3 with sequential NO (2 min) and H2 (40 s) exposures at 150 °C. Comparison between IR spectra and relative outlet gas composition from mass spectrometry analysis: 1st cycle (a) and 9th cycle (b).
Reproduced with permission from reference [64].
2.3 Steady-state kinetics of the NO/H2/O2 on bimetallic Pd–Rh catalysts
Intense discussions in the past have been dedicated to the reduction of NO over bimetallic catalysts with the aim to elucidate if their activity is governed by structural or ligand effects [66–68]. In fact, this is not an easy task especially on supported polycrystalline catalysts since the reduction of NO is considered as structure-sensitive.
2.3.1 Activity behavior of Pd–Rh/Al2O3
Previous investigations on bimetallic Pt-Rh catalysts [10,69], more particularly for the NO/CO reaction, led to more complex kinetic features compared to those characteristics of monometallic Pt and Rh-based catalysts. Most of those results agree with a much stronger NO adsorption on Rh compared to Pt. Hence, the remaining question was related to the preservation of their peculiar properties or their alteration after Rh incorporation to Pt producing alloyed particles with average adsorptive properties different than those characterizing Pt and Rh alone. A first response was provided by Ng et al. [69] on Pt10Rh90(111) who observed lower activities compared to Rh(111) ascribed to a dilution effect of platinum. Clearly, this explanation was in favor of structural effects rather than electronic ones. Later on, Granger et al. [10] demonstrated on Pt-Rh/Al2O3 the occurrence of preferential NO and CO adsorptions on Rh and Pt respectively based on the comparison of the equilibrium adsorption constants estimated on the bimetallic catalyst compared with those previously calculated on the monometallic ones. This statement suggests differences concerning step (3) related to the composition of the nearest-neighbor-vacant site. Better predictions were achieved considering a higher probability to find vacant Pt sites at the vicinity of chemisorbed NO molecules rather than Rh ones. This configuration is in rather good agreement with a random distribution of Pt and Rh atoms at the surface. All these conclusions provided correct interpretations to describe the weak NO partial pressure dependency of the selectivity towards N2 and N2O production especially on Rh-based catalysts with steps for N2 and N2O involving only Rh sites.
Returning to the NO/H2 reaction over NGV Pd–Rh/Al2O3 catalyst, we have to consider that the reducing agent is different which leads to different reactivities since both Pt and Pd become in that case much more active than Rh for the NO/H2 reaction [56] (Table 4). Such an observation can be explained by a stronger NO adsorption on Rh which hinders the dissociative adsorption of hydrogen [50]. None competitive adsorptions would prevail on Pd–Rh/Al2O3 with preferential adsorptions of NO on Rh and hydrogen on Pd. Hence, the reaction mechanism can be slightly modified replacing adsorption steps (1) and (2) by the following steps (12) and (13), and including a dissociation step (14) which accounts for N-O bond scission assisted by chemisorbed hydrogen or more conventionally taking place on a nearest-neighbor Pd site (15). Let us note that those proposals can correctly predict the beneficial effect of hydrogen according to the above-mentioned statements. Indeed, the predominance of step (15) could be related to the release of Pd sites due to a fast H2/O2 reaction.
- (12) NO + *’ ⇆ NOads
- (13) H2 + 2* ⇆ 2Hads
- (14) NOads + Hads → Nads + OHads
- (15) NOads + * → Nads + Oads
Optimised kinetic and thermodynamic parameters calculated for the NO/H2 reaction on supported Pd- and Rh-based catalysts.
| Catalyst | T (reaction) (°C) | k14a | k15a | KNO (atm–1) | (atm–1) | (atm–1) |
| 2.5Pd/Al2O3 | 40 | 0.53 | 0.25 | 4 | 25 | 200 |
| 0.18Rh/Al2O3 | 210 | 8.5 × 10–3 | ≅ 0 | 700 | 15 | 75 |
| 2.5Pd–0.18Rh/Al2O3 | 40 | 5.3 × 10–3 | 5.8 × 10–4 | 1500 | 20 | 93 |
a mol·h−1·g−1.
The different kinetic and thermodynamic constants associated with NO and hydrogen adsorptions have been adjusted from steady-state measurements [50] and then compared to the corresponding optimized ones on monometallic catalysts. As seen in Table 4, the examination of those numerical solutions, especially the rate constant associated with steps (14) and (15), suggests that the dissociation assisted by H atoms is much faster. However, one cannot completely rule out step (15). The order of magnitude of the equilibrium constant for NO adsorption (KNO) on Pd–Rh/Al2O3 seems also in agreement with a preferential adsorption of NO on Rh. Indeed, the calculated value on this latter catalyst should converge to the same order of magnitude as that obtained on Rh/Al2O3 if we take into account the difference in temperature. On the other hand, it is worthwhile to note that the comparison of the rate constant k14 estimated on Pd–Rh/Al2O3 is much lower than that for Pd/Al2O3. This could reflect the structure sensitivity of NO dissociation which suggests that a higher proportion of Pd atoms in bimetallic Pd–Rh particles would be located on terraces rather than edges, corners or others defective sites where NO dissociates more readily.
2.3.2 Selectivity behavior towards N2O formation
The formation of N2O on NGV catalysts is a key factor during the cold start engine especially on Pd which behaves like Rh where NO strongly adsorbs and displaces chemisorbed H atoms. The formation of N2O must be prevented because of its high global warming potential of approximately 300 times higher than that of CO2. As previously illustrated in Fig. 8, the formation of N2O on Rh/Al2O3 predominates at low temperature and conversion and is insensitive to change in NO partial pressure. In such conditions, the surface is essentially covered of chemisorbed NO molecules. As previously reported [50], it is noteworthy that Pd–Rh/Al2O3, mimics the selectivity behavior of Rh/Al2O3 which means that steps for the formation of N2 and N2O would only involve Rh sites in the bimetallic catalyst (Fig. 8). Such features are consistent with non competitive adsorptions of NO and H2 related to a strong NO adsorption on Rh as previously argued. Previous calculations demonstrated that the selectivity is related to according to Eq. (16). By applying the steady-state approximation to chemisorbed N atoms θN/θNO can be calculated which leads to Eq. (17). The weak sensibility of N2O selectivity to partial pressure conditions implies a ratio much lower than 1 which requires: (i) a very high equilibrium constant for NO adsorption, (ii) high values for the rate constants k5 and k6, (iii) very low values for the rate constants for NO dissociation and Nads recombination, respectively k3 and k4, which likely differentiate Pd from Rh. Accordingly, on Rh and Pd–Rh/Al2O3.
| (16) |
| (17) |

Selectivity behaviour of 2.5Pd/Al2O3 (); 0.18Rh/Al2O3 (□); 2.5Pd-0.18Rh/Al2O3 () reflected by changes in the relative rate vs. under steady-state conditions according to the following conditions with constant inlet partial pressures of H2 and O2 of respectively 3.0 × 10−3 and 1.0 × 10−3 atm and NO NO partial pressure varying in the range (0.7–1.2) × 10−3 atm.
Reproduced with permission from reference [50].
3 Development of molecular kinetic scheme for methane activation over NGV catalysts
3.1 Versatile kinetic behavior of NGV catalyst depending on the richness
Methane is a relatively environment-friendly combustible because of its high ratio H/C = 4, producing less CO2 per kJ than other typical combustibles: 5.5 × 10−5 kg/kJ, as compared with 6.5 × 10−5 for n-propane, 7 × 10−5 for n-octane and 10 × 10−5 for anthracite. On the other hand, the emissions of unburned methane, particularly from NGVs, lead to environmental damages, because of its strong greenhouse gas behavior, and high chemical stability in atmospheric conditions. Therefore the catalytic converters for NGV must deal not only with NOx, but also with unburned methane [70].
Catalytic oxidative reactions involving methane have been largely studied in the past, in particular reforming, selective oxidation and total oxidation (combustion). In the case of NGV, where combustion is desired, supported palladium seems to be best choice, since it is able to convert methane both in lean and slightly rich conditions, although with worse performances in this later case as illustrated in Fig. 9 on Pd–Ce/Al2O3 [71]. On another catalyst Rh/Al2O3, Wang et al. [72] investigated the adsorption of methane on differently pretreated surfaces in a TAP reactor. The highest conversion was obtained on a catalyst fully oxidized at 600 °C (fraction adsorbed 90% at 450 °C), while on the catalyst completely reduced only 1% methane was adsorbed. Activation energies obtained on fully oxidized surface and on a surface partially reduced (conversion 40%) were identical at 45 kJ·mol−1. Under those operating conditions, more complex reaction network should be taken into account, including methane combustion and parallel reforming and partial oxidation reactions responsible for syngas formation, the water-gas-shift reaction and consecutive reactions involving H2 and CO as reducing agent as well [73]. Kinetic models for the partial oxidation of methane on 1.4-wt% Pt/Pr0.3Ce0.35Zr0.35Ox catalyst coated on the surface of a triangular corundum channel were previously compared [74] accounting for an oxygen-assisted methane activation or methane dissociation without oxygen assistance. Those authors obtained a reasonable good description of the experimental data, suggesting that the initial activation of methane is of less importance than the subsequent oxidation of carbonaceous intermediates. The active role the ceria for this latter step was pointed out. All these changes can be also intimately related to surface reconstructions during lean/rich switches with related open questions concerning the exact nature of oxygen species.

Stationary lambda sweep at 500 °C over monolith 2 (solid line, Pd = 6.71 and Ce = 16.77 g/dm3) and monolith 1 (broken line, Pd = 6.11 and Ce = 0.81 g/dm3), GHSV = 66,500 h−1 (10 °C/min) (■): NO; (): CH4; (●): CO.
Reproduced with permission from reference [71].
3.2 General assessments on the kinetics of methane conversion and related surface structure
Up to now, methane adsorption has been preferentially investigated over Ni, Pt, Rh and Ru and more recently on Pd based catalysts with the emergence of Natural Gas-fuelled Vehicle catalysts (NGV) indicating a complex surface chemistry with sometimes controversial assessments as illustrated in Scheme 2. As shown various parallel pathways reveal the importance of oxygen coverage in the hydrogen abstraction from gaseous CH4 or chemisorbed CHx species [75–78] and also point out the experimental fact that OH groups would be a more efficient oxidizer than chemisorbed O atoms [79]. The emergence of theoretical calculations based of the Density Functional Theory provided outstanding information but essentially on model surfaces [80,81] allowing the clarification of the mechanism of CH4 oxidation with a lower activation barrier on PdxCe1–xO2(111) [82] than on Pd(111) and pre-covered PdO(111) explained by the oxygen storage capacity of ceria usually enhanced when noble metals interact with CeO2. Regarding conventional alumina supports, its role of oxygen reservoir has been clearly established by supplying oxygen from spill-over process of OH groups to activate methane over noble metal sites [72,75,78]. However, up to now, no detailed quantitative information concerning this process is given whereas the influence of the support as oxygen supplier is often mentioned [75,77,78,83]. Also, some controversies have arisen in the past two decades regarding the influence of oxygen chemisorbed over transition metal with sometimes a beneficial effect of oxygen on Pd in methane conversion found by Wang and Liu [84]. On the contrary, Valden et al. [85,86] observed the reverse trend on Pd(110) and Pd(111) with an absolute dissociation probability to find two neighbor-vacant sites decreasing with a raise in oxygen pre-coverage in agreement with a first-order kinetics with one oxygen atom capable of blocking two adjacent vacant adsorption sites. As a matter of fact, theoretical and DFT calculations with further comparisons with UHV investigations on model catalysts are useful for evaluating the catalyst properties. Unfortunately, they cannot directly take into account the effect of surface reconstructions on supported catalysts during steady-state kinetic measurements and the impact of adsorbate concentration on the rate of surface reactions [87]. For illustration, complex interactions, which take place when Pd interacts with oxygen [88], have been explained by a model that distinguishes three different chemical environments for oxygen: chemisorbed oxygen on metallic palladium, surface palladium oxide and sub-surface palladium oxide. Time-resolved in situ XANES spectroscopy measurements during methane oxidation on Pt/Al2O3 catalyst and DFT calculations also led to the conclusion that an intermediate O/Pt ratio is needed to get an optimal activity, an oxygen-rich surface hindering the dissociative adsorption of methane [89]. Returning to TAP observations, it was shown that the ageing process can be followed using the TAP technique. On Pd and Pd–Rh catalysts, aged as previously described [90], the adsorption of methane was strongly reduced, particularly at low temperature and on Pd-only catalyst. On the contrary, a reductive or oxidative pretreatment of the catalysts had no significant impact [90]. This observation and the changes in the outlet flow curves of reaction products, in particular of H2, suggest important modifications of the metal-support interaction during thermal ageing.

Suggested elementary steps on pre-reduced and pre-oxidized noble-metal-based catalysts during methane single-pulse experiments [90] on the basis of earlier mechanistic proposals [75–79]. S1 stands for the Pd adsorption site, OHs represents hydroxyl groups on alumina which would spill-over further interacting with S1 to form OHads (step (22)) and/or reacting with Cads at the metal/support interface.
3.3 TAP as a powerful technique to investigate the role of the metal/support interface
Temporal Analysis of Product (TAP) reactor represents a unique tool for in situ characterization that allows the elucidation of adsorption kinetics, surface reactions, and diffusion processes on a wide variety of catalysts, especially on polycrystalline supported catalysts, for which the metal/support interface might play a key role [30,91–93]. TAP, experiments consist in exposing the catalyst sample in an evacuated fixed-bed microreactor to series of pulses of gas mixtures, and analyzing the effluent gases by means of one or more QMS analyzers. The process is performed at a sufficiently low pressure to be governed by Knudsen diffusion laws. Single-Pulse (SP) TAP experiments on real catalysts come near to surface science ultra-high-vacuum (UHV) investigations usually restricted to well-defined single crystals with different characteristics than those currently observed on heterogeneous catalysts. Hence, TAP experiments under high vacuum with small amounts of reactive molecules injected compared to the number of accessible active sites at the surface can bridge the gap between surface and industrial catalysts as argued elsewhere [30,91–93]. This suppresses the influence of diffusion on observed reaction rates and allows the use of simple laws of Knudsen domain to account for the diffusion in bed void spaces. Thus, considering the surface characteristics as constant in typical experiments (single-pulse TAP experiments), it makes possible the determination of kinetic constants of elementary steps with characteristic times of order of milliseconds and, in so-called Thin-Zone TAP reactor, to determine directly reaction rates without prior assumptions on the nature of reaction mechanisms [94]. Small amounts of reacting gases, of the order of 1 nanomol used in such experiments, suppress the influence of thermal effects of reactions studied [95] which may distort the results in classical kinetic experiments [96]. In this sense, the strategies associated with the utilization of TAP reactors can be considered as an intermediate between classical kinetic studies and surface science approach with pressure ranges higher than those currently encountered in this latter case [92,97,98].
It is obvious that this features make TAP reactors as outstanding instruments for characterizing surface processes including adsorption, surface reactions and chemical processes taking place at the metal/support interface, qualitatively and even quantitatively, by determining kinetic parameters representative of spill-over processes toward the support or a storage component [99]. Such studies with supported Pt-group metals have been carried out in the past, by TPD of hydrogen pre-adsorbed on alumina support and desorbing after reverse spill-over to metal [100] or by isotopic exchange [101–103]. These previous studies concerning mostly alumina-supported catalysts, led to quantitative models of exchange of hydrogen between the metal and the support and its diffusion on the support [100,103]. Other well known effects include modifications of the electronic properties of metals, strong metal–support interactions and activation of reacting species at the metal–support boundary.
3.4 Quantitative study of the effect of ageing
Up to now, only few papers can be found in which the involvement of the metal/support interface in the catalytic processes is discussed on the basis of quantitative and accurate determination of kinetic parameters. Most of those investigations report essentially qualitative information formulated from the profile of the outlet response of reactants and products that can be useful for the elucidation of reaction mechanisms [75–78,97] while this technique can answer to a formidable challenge in establishing direct structure/activity relationship under reactive mixture. In this context, TAP analysis is a powerful technique which can allow the identification of kinetically significant steps based on the calculation of rate constants and activation barriers. In a recent paper [90] kinetic parameters of methane adsorption on Pd/Al2O3 and Pd–Rh/Al2O3 have been determined on fresh and aged catalysts as well as the respective outlet flow curves of products. As illustrated in Figs. 10 and 11, different product distributions can be observed according to the nature of the pretreatment in reductive or oxidative conditions. On reduced Pd/Al2O3 and Pd–Rh/Al2O3 catalysts, CO and H2 are mainly produced, whereas oxygen pre-coverage essentially leads to the formation of CO2. The lack of observation of peaks of H2O may be attributed to a very slow desorption and/or to a re-adsorption of this species on the support, as observed for γ-alumina-supported Rh [75] and Ru [72], but not on bulk Rh or supported on α-alumina [83]. Interesting information can be obtained from the comparative examination of the shapes of the outlet curves of H2 and CO on reduced samples. On fresh catalysts, the former presents a broad signal at 400 °C accompanied at higher temperatures by another sharp one, increasing with temperature, suggesting the existence of two parallel pathways for H2 production and/or important contribution of slow processes associated with hydrogen spill-over. The initial fast and well-resolved process is not correlated to a simultaneous CO formation. Afterwards, a slower H2 production takes place accompanied with a low production of CO for which in the case of reduced catalysts indicates a contribution of alumina as mentioned in earlier papers [104,105]. On pre-oxidized surface, the lack of H2 formation is likely due to its fast reaction with chemisorbed oxygen over palladium sites. As expected, CO2 predominantly forms. A standard comparison [91] of the effluent curves of CH4 and Ar indicates an irreversible adsorption of methane in agreement with a direct dissociation mechanism rather than a precursor-mediated mechanism involving a molecular precursor state as proposed elsewhere [85,86] on Pt(111), Pt(110) and Pd(110). Such a statement seems in rather good agreement with the fact that a large fraction of methane, above 84%, is converted into reaction products (Table 5).

MS responses during single-pulse TAP experiments at 400 °C (a) and 550 °C (b) on fresh-2.5 wt.% Pd/Al2O3 pre-reduced in 72 mbar hydrogen at 550 °C for 30 minutes. MS responses recorded at 400 °C (c) and 550 °C (d) on pre-activated samples in pure oxygen at 450 °C for 50 minutes. Corrected data from QMS tuned to m/z = 15 (CH4), 44 (CO2), 28 (CO, CO2) and 2 (H2, CH4).

MS responses during single-pulse TAP experiments at 400 °C (a) and 550 °C (b) on aged-2.5 wt.% Pd/Al2O3 pre-reduced in 72 mbar H2 at 550 °C for 30 minutes. MS responses recorded at 400 °C (c) and 550 °C (d) on pre-activated samples in pure oxygen at 450 °C for 50 minutes. Corrected data from QMS tuned to m/z = 15 (CH4), 44 (CO2), 28 (CO, CO2) and 2 (H2, CH4).
Surface characterization of freshly-prepared and aged supported Pd catalyst and related diffusion and kinetic constant from modeling single-pulse TAP experiments.
| Catalysts | Dispersiona | dPdb | Pretreatment | SP Reaction temp. | kadsc | E | |
| (%) | (nm) | 400 ̊C | 550 ̊C | (kJ·mol–1) | |||
| 2.5 wt.% Pd/Al2O3 | 0.26 | 4.3 | Reduced | 0.84 | 0.86 | 0.48 | 7.0 |
| Oxidized | 0.88 | 0.89 | 0.55 | 5.0 | |||
| Aged-2.5 wt.% Pd/Al2O3d | 0.13 | 8.8 | Reduced | 0.01 | 0.21 | 0.01 | 45.0 |
| Oxidized | 0.58 | 0.84 | 0.41 | 27.0 |
a Calculated from H2 titration at 100 °C assuming atomic H/Pd = 1.
b Estimated from the Pd dispersion assuming hemispherical particle.
c Calculated at 417 °C expressed in m3gas·s–1·kgcata–1.
d After exposure to 10 vol.% H2O diluted in air at 980 °C for 4 h.
Thermal ageing in wet atmosphere (10 vol.% H2O diluted in air) was implemented to speed up surface reconstructions that usually occur on NGV catalysts after long term running conditions. Changes in catalysis properties were expected based on previous investigations emphasizing the role of the metal/support interface and also the influence of particle size with low coordination sites controlling the overall rate of methane reforming [106]. Hence, the very low fraction of methane adsorbed on pre-reduced aged-Pd/Al2O3 at lower temperature (400 °C and 450 °C) can be easily correlated to those surface alterations (Table 5). It is remarkable that a quasi-complete loss of conversion on reduced surface at 400 °C is then partly restores with a raise in temperature. The bimetallic catalyst Pd–Rh/Al2O3 retains some activity upon thermal ageing even at the lowest temperature indicating some protective properties of Rh preventing particle sintering of Pd as observed in Fig. 12. Aged bimetallic catalysts, pre-exposed to oxidative atmosphere prior to SP experiments, preserve a significant capacity to adsorb and convert methane showing a comparable behavior at 550 °C as that previously reported on fresh catalysts. However, the most significant observation is provided by the examination of the product effluent curves in Fig. 11 especially the signal related to hydrogen since the slow part of the signal, observed in the case of pre-reduced catalysts, is suppressed.
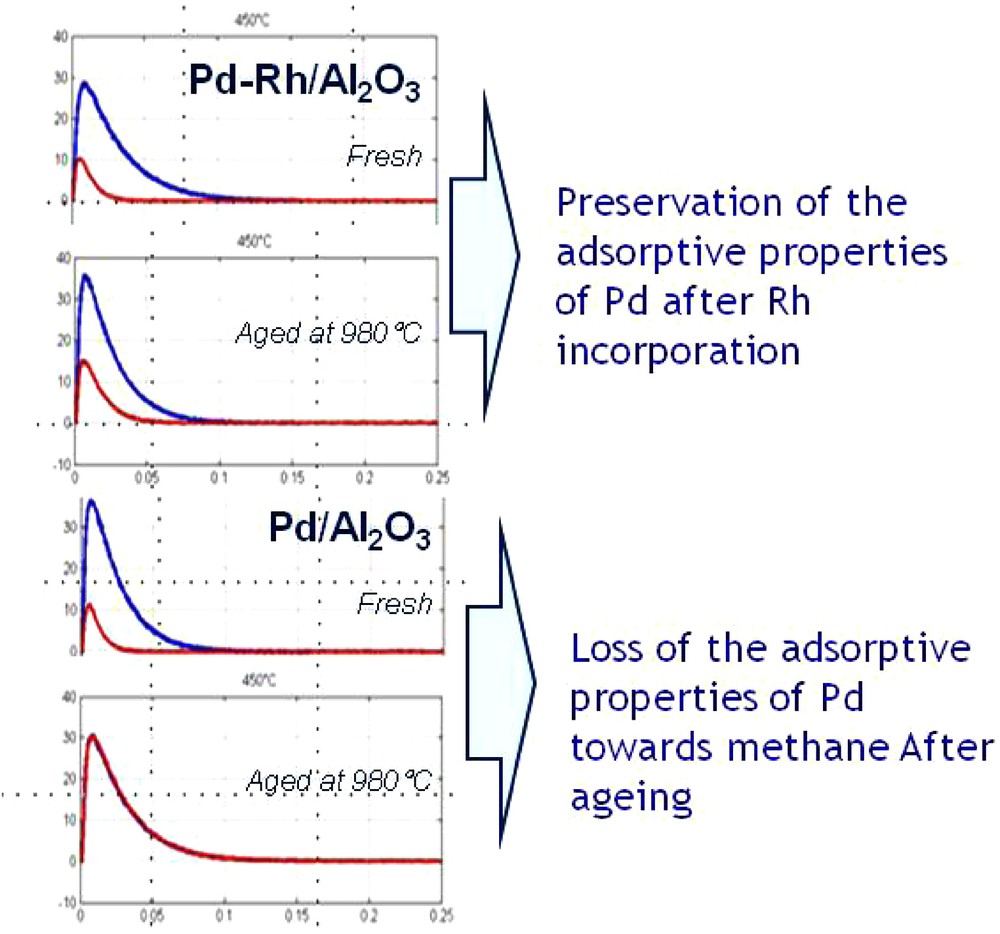
(Color online.) Comparison of normalized CH4 and Ar outlet flow curves FiMi1/2 vs. t/Mi1/2 recorded on pre-reduced Pd/Al2O3 Pd–Rh/Al2O3 at 450 °C before and after ageing where Fi stands for the pulse intensity normalized flow multiplied by Mi1/2 where Mi stands for molar weight of the component i–Freshly-prepared sample (a); aged sample (b). The ageing procedure consisted in exposing the catalysts to wet atmosphere (air in the presence of 10 vol.% H2O) at 980 °C for 4 hours.
Rate constants for methane adsorption collected in Table 5 have been computed using a standard numerical model of TAP microreactor described elsewhere [90]. Activation energies have been estimated from the Arrhenius plots using rate constant values calculated in the temperature range 400–550 °C on fresh and aged-Pd/Al2O3 pre-activated in hydrogen or oxygen. Data collected in Table 5 reveal comparable rate constants on freshly-prepared catalysts irrespective of the nature of the pre-activation treatment (reduction or oxidation) but abnormally low in comparison with usually reported in the literature. On the contrary, significant deviations are noticeable on aged samples with a very weak adsorption at 400 °C on pre-reduced samples partly restored with a raise in temperature especially when the catalyst is pre-covered by oxygen. The determination of the activation barrier also leads to significant deviations with much higher numerical solutions on aged samples in particular on pre-reduced one. The abnormally high values in this latter case can be partly related a very poor accuracy due to negligible estimates determined for the rate constant at low temperature. However, these overestimated values, can be consistent and would likely reflect the impact of oxygen since methane adsorption is surface sensitive with an oxygen-induced effect on surface reconstructions that would favor methane adsorption [106,107]. Subsequent theoretical calculations by using the UBI-QEP method also support the involvement of OH species as exemplified in Table 6 leading to a substantial decrease in the activation barrier. Hence, this comparison emphasizes the importance of the metal/support associated with the migration of OH groups from alumina to the metallic Pd particle would migrate and react with methane on Pd and/or at the metal/support. One aged samples, methane dissociation involving preferentially metallic sites and/or pre-covered by oxygen coincides with higher activation barrier.
Heat of reaction and activation barrier calculated from the UBI-QEP method for various elementary steps involved in methane adsorption on Pd (111) (estimated margin of error ± 15 kJ/mol).
| Elementary steps | ΔHa (kJ·mol–1) | Ef (kJ·mol–1) | Er (kJ·mol–1) |
| CH4g + 2* ⇆ CH3,a + Ha | 2.9 | 41.6 | 38.7 |
| CH4,a + Oa ⇆ CH3,a + OHa | 33.0 | 59.6 | 26.6 |
| CH4,a + Oa ⇆ CH3Oa + Ha | –11.2 | 47.6 | 21.6 |
| CH4,a + OHa ⇆ CH3,a + H2Oa | –21.6 | 0 | 21.6 |
a Calculations integrate heat of adsorptions in the zero-coverage limit.
4 Conclusion
As a conclusion, this overview highlights the potentialities of steady-state and transient kinetic studies based on the illustration of two different reactions taking place over monometallic and bimetallic Pd–Rh and NGV catalysts: NO reduction by H2 and CH4 interaction with differently pretreated Pd and Pd–Rh-based catalysts. Among the different techniques that can be implemented, this overview had the objective to point out the advantages in coupling these different techniques ranging from UHV to practical conditions in order to get better insight into reaction mechanisms and related surface properties. It is obvious that previous surface science studies provided key information related to the nature elementary steps and allowed to draw relevant interpretation of the structure sensitivity of various reactions especially NO dissociation on noble metals which needs a nearest-neighbor-vacant site. Subsequent, extrapolation from UHV to steady-state experiments under realistic pressure conditions is not an easy task. However, it is remarkable to highlight similar kinetic features which can explain the selectivity behavior related to high NO coverage during the NO/H2 reaction. On the other hand, only few data in UHV conditions are related to oxygen abstraction from chemisorbed NO molecules assisted by chemisorbed H atoms as demonstrated from steady-state approximation. This beneficial effect would take place even in the presence of oxygen. However, in such conditions operando spectroscopic measurements also reveal an indirect effect ascribed to the fast H2/O2 reaction further releasing free vacant sites.
Now regarding methane conversion over NGV catalysts, there is still an outstanding point that needs an answer. Precisely, the low activity of those catalysts under rich conditions is a critical point from a practical view point. From this overview, this behavior can be relied to a wide number of parameters which are often difficult to isolate for further detail investigation. TAP experiments appear to be suitable for the characterization of the impact of the phenomena occurring at the metal/support interface going farther on the existing studies that essentially justify the involvement of the metal/support interface based on qualitative information. Accurate determination of rate constants and diffusivities necessitates however a careful modeling. As observed, the sole examination of the rate constant would not be sufficiently sensitive to draw decisive arguments whereas the activation barrier reflects without any ambiguity the participation of the alumina support. Hence, Temporal Analysis of Products (TAP) reactor offers a possibility to draw direct structure/activity relationships checked on Natural Gas-fuelled Vehicle (NGV) catalysts. The determination of kinetic constants for methane adsorption and subsequent surface reactions provides a straightforward visualization of the loss of the metal/support interface accompanied with qualitative changes of interactions at this interface during thermal ageing.
Acknowledgments
The authors would like to thank, the French agency of energy and environment (Ademe), the North Region through the Institut de Recherche en Environnement Industriel (IRENI), and the Research National Agency (ANR) for supporting this work. Also we would like to thank Umicore company which supplied model NGV catalyst samples.