

1 Introduction
Cyclodextrins (CDs) are cyclic oligosaccharides composed of α-d-glucopyranose units. The most common naturally occurring CDs are composed of 6, 7 and 8 α-1,4-linked d-glucopyranose and are usually referred to as α-, β- and γ-CD, respectively. They have the property of forming inclusion complexes with various guest molecules with suitable polarity and dimension because of their special molecular structure of the hydrophobic internal cavity and the hydrophilic external surface [1–3].
Tolfenamic acid, N-(3-chloro-2-methyl-phenyl) anthranilic acid (TA), is a non-steroidal inflammatory drug with associated gastrointestinal side effects and problems with bioavailability. Furthermore, it is extremely insoluble in water. β-CD can increase the solubility of TA by forming inclusion complex [4–8]. In 2013, Floareet al. reported the experimental study on the inclusion complex of β-CD and TA [4]. The results showed that the stoichiometry of the formed complex was determinated as 1:1, and confirms the existence of a bimodal binding process of TA into β-CD.
Molecular modelling is one of the most important methods to investigate the formation of cyclodextrin inclusion complexes [9–22]. This study is aimed to give information about the geometries and the driving forces of the formation of inclusion complex of TA and β-CD by semi empirical and ONIOM2 methods.
2 Calculation procedure
The structure of TA was constructed using Hyperchem 7.5 molecular modelling package [23]. The starting geometry of β-CD was taken from Chem-Office 3D ultra (version 10, Cambridge software). Then, the two structures TA and β-CD were optimized by MPW1PW91/6-31G(d) and PM3MM, respectively (see Fig. 1).

(Color online.) Geometrical structures of TA and β-CD optimized at MPW1PW91/6-31G(d) and PM3MM methods.
All calculations were performed using Gaussian 09 program [24]. The following initial steric conditions were applied for docking of TA into the β-CD cavity. The glycosidic oxygen atoms of the cyclodextrin molecule were placed onto the XY plane and their centre was defined as the centre of the coordination system [25–27]. Two possible orientations of the guest molecule in complex were considered. For simplicity, the orientation in which the COOH group of TA points toward wider rim of β-CD was called the “A orientation”, and the other orientation, in which the carboxylic group of the guest points toward narrow rim of β-CD was called the “B orientation”, (see Fig. 2). Then, TA was moved into the β-CD cavity along the Z-axis from –8 Å to +8 Å with 1 Å step (reference atom is N154). The generated structures at each step were optimized with PM3MM methods without any restriction [28]. The more stable complexes found by PM3MM calculations were optimized by ONIOM2 methods without imposing any symmetrical restrictions.

(Color online.) The proposed structures of TA/β-CD complex for A and B orientations.
The solvent effects on the conformational equilibrium geometries have been investigated using the PCM model for water (ɛ = 78.39) as a solvent with ONIOM2 method.
Finally, the natural bond orbital analyses (NBO) were applied as a powerful approach for the evaluation of the intermolecular interactions between β-CD and TA molecules.
3 Results and discussion
The most important factor in complex formation is the size of the guest. On one hand, the guest should not be too small, because the intermolecular forces will not form if the distances between the guest and the interior of the cyclodextrin molecule are too great. On the other, if the guest is too large, complex formation cannot take place due to steric hindrance. It must also be considered that it is possible for just parts of a guest molecule to be complexed within the cavity. The size of the cavity in relation to the size of the guest is also critical for complex formation.
The linear distance, in optimized TA, between chlorine atom and hydrogen of benzoic group is about 10.01 Å. A lower value is compared to the height of the β-CD cavity (around 7.9 Å). Thus, the reaction of TA should take place inside the β-CD cavity.
The first part of this study is focused on the localization of global minimum along z-axis for A and B orientation by means PM3MM semi empirical calculation. The generated minimums were used in the next parts for ONIOM2 optimization in vacuo and taking in the consideration the effect of water solvent.
The Ecomplexation upon complexation between TA and β-CD is defined in Eq. (1):
| (1) |
where Ecomplex, ETA and Eβ−CD represent the relatives energies of complex, free TA and free β-CD, respectively.
The deformation energy of the guest or the host molecule can be obtained by Eqs. (2) and (3):
| (2) |
| (3) |
where Edeformation (guest) stands for the deformation energy of the guest, is the single point energy of the guest using its geometry in the optimized complex, and is the energy of the optimized geometry of the guest [28].
For A orientation, the lowest energy was found at z = –3 Å indicating that the TA was embedded in the cavity increasing the interactions between the two molecules. In the case of B orientation the most favored complex was located at z = 2 Å then, the energy increases with the departure of TA from the β-CD cavity (Fig. 3).
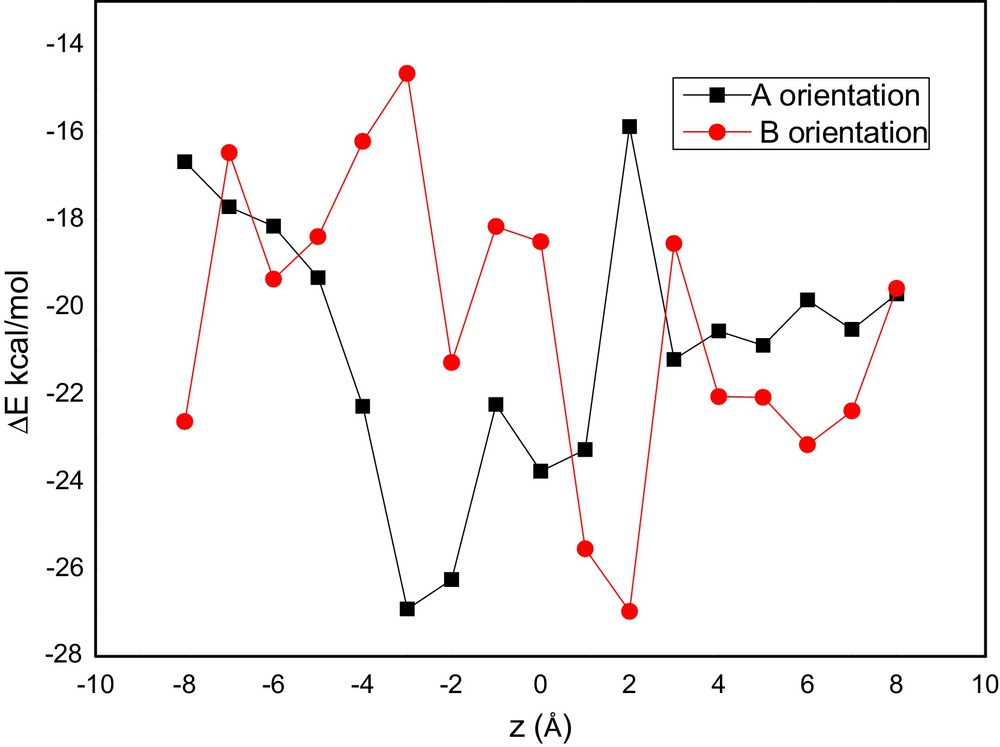
(Color online.) Complexation energies of the inclusion complexation of TA into β-CD at different positions, PM3MM calculations.
The proposed favorable structures are graphically presented in Fig. 4, in all case, TA is embedded in the β-cyclodextrin cavity. In A orientation, the methyl-phenyl ring is totally embedded in the cavity, the chlorine atom is pointed toward secondary hydroxyls and the reference atom N154 shifts downwards by 2.27 Å from the centre of the coordination system. But, in B orientation, benzoic group is encapsulated in the cavity, the chlorine atom is closed to primary hydroxyls and the N154 displaces upwards by 1.85 Å from the centre of the coordination system.
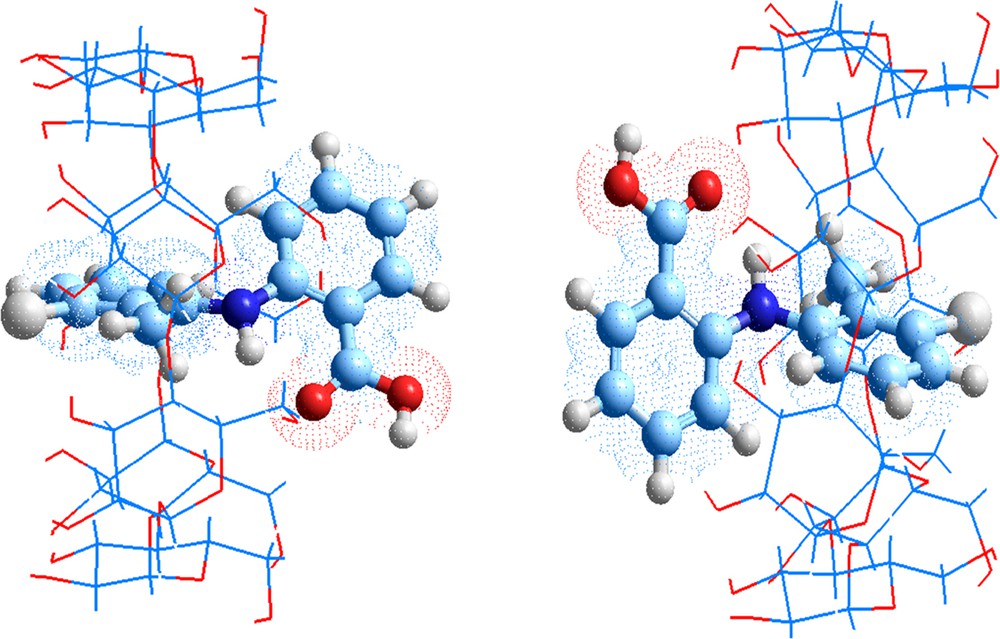
(Color online.) Geometrics structures of TA/β-CD complexes optimized by PM3MM method.
Table 1 showed the calculated complexation energy for TA/β-CD with A and B orientations. The corresponding complexation energies were –26,93 kcal/mol and –26,97 kcal/mol A and B orientations, respectively; the energetic gap is of 0.04 kcal/mol. Thus, indicating the coexistence of these two orientations, which is observed experimentally [4].
Complexation energies (kcal/mol), deformation energies (kcal/mol), HOMO and LUMO energies (eV) and dipole moments (Debye) at PM3MM calculations.
| A orientation | B orientation | Δ(A–B) | |
| Ecomplexation (kcal/mol) | –26,93 | –26,97 | 0.04 |
| Edeformation (TA) (kcal/mol) | 9.03 | 8.68 | 0.35 |
| Edeformation (β–CD) (kcal/mol) | 4.17 | 5.21 | 1.04 |
| HOMO (eV) | –8.68 | –8.65 | 0.03 |
| LUMO (eV) | –0.78 | –0.73 | 0.05 |
| Δ(HOMO–LUMO) | –7.90 | –7.92 | 0.02 |
| (Debye) | 10.22 | 6.87 | 3.35 |
The deformation of host and guest molecules, the difference between energies of frontier orbital's HOMO–LUMO and dipole moment are important factor involving the stability of inclusion complexes with cyclodextrins.
It is clearly showed in Table 1 that TA is more deformed than β-CD in A and B orientation, the HOMO–LUMO gap is of –7.90 and –7.92 eV for A and B orientation respectively indicating a partial charge transfer between HOMO of one component and LUMO of another. The variation of dipole moment of β-CD after complexation indicate that polarity of β-CD is changed and the dipole moment play an important role to stabilize the inclusion complex, it is of 10.22 Debye for A orientation and 6.87 Debye for B one. These indicate that the dipole moment of the complex have a closed relation with the polarity of the guest molecule.
To give more information about the most stable orientation, an ONIOM2 optimization was performed on the most stable complexes obtained by PM3MM calculation. The effect of the water solvent was also taken in consideration with PCM model (ɛ = 78.39).
The ONIOM2 energies for TA/β-CD for A and B orientations with and without solvents are mentioned in Table 2. The calculated energies in vacuo were found equal to –758078.48 kcal/mol for A orientation and to –758085.58 kcal/mol for B orientation; corresponding to a difference between the two orientations equal to 7.10 kcal/mol in favor to B orientation.
Energies (kcal/mol) at ONIOM2 calculations.
| E (kcal/mol) | A complex | B complex | Δ(A–B) |
| Vacuum | –758078.48 | –758085.58 | 7.10 |
| Water | –758122.75 | –758119.23 | –3.52 |
Generally, the formation of the inclusion complexes involving cyclodextrins takes place in aqueous solution; so, the binding behavior of β-CD and TA in solution seems to be more important than they are in vacuo.
The results of calculations in the presence of water solvent are listed in Table 2 and do not support those obtained in vacuo and the energy difference in H2O between the two orientations is 3.52 kcal/mol in favor to A orientation. This difference is smaller than that between the two orientations of the TA/β-CD complex in the vacuum. Solvation effects lead to the inversion of the complexation orientation from the solution to the vacuum.
4 NBO analysis
In this section, we investigate firstly the geometries and mutual interactions between the two molecules in vacuo. Secondly, the effect of solvent was considered by the PCM model.
4.1 In vacuo
The NBO analysis was performed by MPW1PW916-31G level of theory in vacuo at the optimized geometries of ONIOM2 calculations. The Table 3 illustrates only the interactions with stabilization E(2) ≥ 1 kcal/mol.
Donor–acceptor interactions and stabilization energies E(2) (kcal/mol) in vacuo.
| Donor | Acceptor | E(2) (kcal/mol) MPW1PW91/6-31G |
| TA/β-CD (A) | ||
| β-CD proton donor and TA proton acceptor | ||
| σ C 3–H 81 | σ* C 160–H 173 | 2.13 |
| σ C 15–H 93 | σ* C 162–H 175 | 1.44 |
| σ C 17–H 95 | σ* C 162–H 174 | 2.31 |
| σ C 21–H 100 | σ* C 162–H 176 | 2.15 |
| σ C 39–H 121 | σ* C 153–H 168 | 2.36 |
| σ C 41–H 123 | σ* C 152–H 167 | 2.84 |
| TA proton donor and β-CD proton acceptor | ||
| σ C 152–H 167 | σ* 41–H 123 | 2.54 |
| σ C 153–H 168 | σ* C 39–H 121 | 2.55 |
| σ C 160–H 173 | σ* C 3–H 81 | 2.48 |
| σ C 162–H 174 | σ* C 17–H 95 | 2.13 |
| σ C 162–H 175 | σ* C 15–H 93 | 2.60 |
| σ C 162–H 176 | σ* C 21–H 100 | 2.43 |
| LP (1) O 164 | σ* O 59–H 135 | 1.30 |
| TA/β-CD (B) | ||
| β-CD proton donor and TA proton acceptor | ||
| σ C 3–H 81 | σ* C 152–H H 167 | 2.72 |
| σ C 5–H 83 | σ* C 153–H 168 | 2.33 |
| σ C 11–H 90 | σ* C 160–H 173 | 1.83 |
| σ C 12–H 147 | σ* C 159–H 172 | 2.91 |
| σ C 21–H 100 | σ* C 162–H 174 | 2.32 |
| σ C 23–H 102 | σ* C 162–H 176 | 1.72 |
| TA proton donor and β-CD proton acceptor | ||
| σ C152–H 167 | σ* C 3–H 81 | 2.72 |
| σ C153–H 168 | σ* C 5–H 83 | 2.49 |
| σ C159–H 172 | σ* C 12–H 147 | 1.58 |
| σ C160–H 173 | σ* C 11–H 90 | 2.44 |
| σ C162–H 174 | σ* C 21–H 100 | 2.48 |
| σ C162–H 176 | σ* C 23–H 102 | 2.97 |
As can be seen from Table 3 for A orientation, the following occupied orbital's of β-CD: σ C 3–H 81, σ C 15–H 93, σ C 17–H 95, σ C 21–H 100, σ C 39–H 121 and σ C 41–H 123 play an important role to donate proton to vacant orbital's σ* C 160–H 173, σ* C 162–H 175, σ* C 162–H 174, σ* C 162–H 176, σ* C 153–H 168 and σ* C 152–H 167, respectively of TA molecule.
When TA acts as proton donation the following orbital's: σ C 152–H 167, σ C 153–H 168, σ C 160–H 173, σ C 162–H 17, σ C 162–H 175 and σ C 162–H 176 participate to proton donation to: σ* C 41–H 123, σ*C 39–H 121, σ*C 3–H 81, σ*C 17–H 95 and σ*C 15–H 93, respectively. Additionally, the formation of H-bond was observed between O164 of TA and H135 with stabilization energy equal to1.30 kcal/mol.
In the case of B orientation, the occupied orbital's of β-CD donate proton to unoccupied orbital of TA with stabilization energies comprised between1.72 kcal/mol and 2.91 kcal/mol. In the other hand, the unoccupied orbital's of β-CD accept proton from TA with stabilization energies ranges from 1.58 to 2.97 kcal/mol.
From Fig. 5, we can see that TA is included in the β-CD cavity; for A orientation, the methyl-phenyl ring is totally embedded in the cavity but in B orientation benzoic group is encapsulated in the cavity.

(Color online.) Geometrics structures of TA/β-CD complexes optimized by ONIOM2 method in vacuo.
The intermolecular distances in A orientation between occupied and unoccupied orbital's given in NBO analysis of the two molecules are: (i) C3–C160: 3.74 Å; (ii) C15–C162: 3.62 Å; (iii) C17–C162:3.69 Å; (iv) C21–C162: 3.66 Å; (v) C39–C153: 3.81 Å; (vi) C41–C152: 3.82 Å; (vii) O164–H135: 1.79 Å. Thus, the methyl-phenyl ring is closed to internal behavior of β-CD, which explains the augmentation of stabilization energies.
For B orientation, the COOH group is directed toward primary hydroxyls, the Cl atom is oriented toward secondary hydroxyls, while the methyl-phenyl ring is totally tramped in the cavity. The intermolecular distances between occupied and unoccupied orbital's given in NBO analysis of the two molecules are: (i) C3–C152: 3.77 Å; (ii) C5–C153: 3.81 Å; (iii) C11–C160: 3.66 Å; (iv) C12–C159: 3.48 Å; (v) C21–C162: 3.72 Å; (vi) C23–C162: 3.64 Å.
It is important to note that NH group of TA do not participate in intermolecular interaction with β-CD because it is engaged on intramolecular H-bond with O164 of COOH group; the two lone pairs of O164 donate proton to H169 linked to N154 with stabilization energies 7 and 9.11 kcal/mol. The same remark is observed with Cl atom, lone pair of Cl interact with vacant orbital σ C148–C150 of TA (E(2) = 9.80 kcal/mol).
4.2 In water
In the following, are given the results of NBO analysis of optimized geometries of ONIOM2 calculation in water, NBO analysis were also done by MPW1PW91/6-31G level of theory in water (Table 4).
Donor–acceptor interactions and stabilization energies E(2) (kcal/mol) in water.
| Donor | Acceptor | E(2) (kcal/mol) MPW1PW91/6-31G |
| TA/β-CD (A) | ||
| β-CD proton donor and TA proton acceptor | ||
| σC 3–H 81 | σ*C 160–H 173 | 2.20 |
| σC 15–H 93 | σ*C 162–H 175 | 1.38 |
| σC 17–H 95 | σ*C 162–H 174 | 2.23 |
| σC 21–H 100 | σ*C 162–H 176 | 2.05 |
| σC 39–H 121 | σ*C 153–H 168 | 2.62 |
| σC 41–H 123 | σ*C 152–H 167 | 2.48 |
| TA proton donor and β-CD proton acceptor | ||
| σC 151–C 152 | σ*C 5–H 83 | 1.38 |
| σC 152–H 167 | σ*C 41–H 123 | 2.50 |
| σC 153–H 168 | σ*C 39–H 121 | 2.60 |
| σC 159–C 160 | σ*O 74–H 144 | 1.04 |
| σC 160–H 173 | σ*C 3–H 81 | 2.28 |
| σC 162–H 174 | σ*C 17–H 95 | 2.33 |
| σC 162–H 175 | σ*C 15–H 93 | 2.66 |
| σC 162–H 176 | σ*C 21–H 100 | 2.51 |
| LP (1) O 164 | σ*O 59–H 135 | 12.54 |
| LP (2) O 164 | σ*O 59–H 135 | 7.47 |
| TA/β-CD (B) | ||
| β-CD proton donor and TA proton acceptor | ||
| σC 3–H 81 | σ* C 152–H 167 | 2.41 |
| σC 5–H 83 | σ* C 153–H 168 | 2.31 |
| σC 11–H 90 | σ* C 160–H 173 | 1.64 |
| σC 12–H 147 | σ* C 159–H 172 | 2.68 |
| σC 21–H 100 | σ* C 162–H 174 | 2.20 |
| σC 23–H 102 | σ* C 162–H 176 | 1.57 |
| TA proton donor and β-CD proton acceptor | ||
| σC 152–H 167 | σ* H 81 | 1.03 |
| σC 152–H 167 | σ* C 3–H 81 | 2.66 |
| σC 153–H 168 | σ* C 5–H 83 | 2.34 |
| σC 160–H 173 | σ* C 11–H 90 | 2.26 |
| σC 162–H 174 | σ* C 21–H 100 | 2.59 |
| σC 162–H 176 | σ* H 102 | 1.21 |
The given stabilization energies for A orientation between donor orbital's of β-CD and acceptor one of TA are ranged between 1.38 and 2.62 kcal/mol. When TA acts as donor the stabilization energies are between 1.04 and 12.54 kcal/mol. Additionally, the two lone pairs of O164 of TA are engaged in the formation of H-bond and contribute largely in the stabilization of the complex.
In the case of B orientation, charge transfer is occurred between donor and acceptor orbital's of both TA and β-CD, but no intermolecular H-bond was formed.
The geometries of the formed complexes in water were illustrated if Fig. 6 indicating that TA is embedded in the cavity. The intermolecular distances between occupied and unoccupied orbital's given in NBO analysis of the two molecules are ranged between 3.51 and 4.03 Å for A orientation and comprised between 3.48 and 3.82 Å for B one.
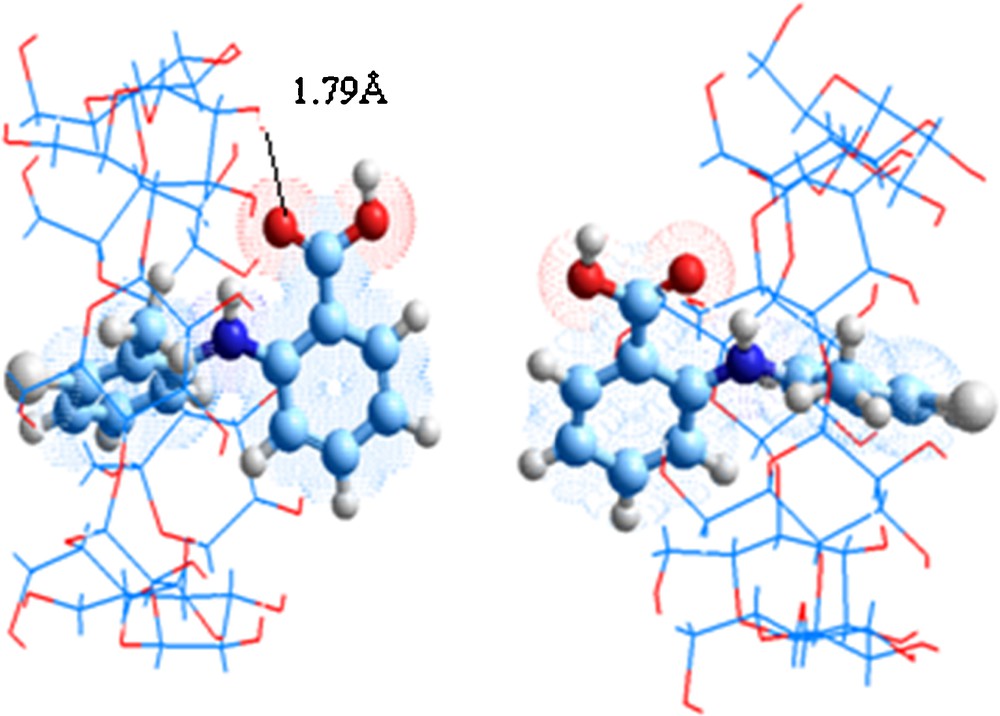
Geometrics structures of TA/β-CD complexes optimized by ONIOM2 method in water.
5 Conclusion
The PM3MM semi empirical and ONIOM2 calculations were applied to study the complexation of TA with native β-CD. PM3MM calculation shows that the formed complex is stable. Also, ONIOM2 calculation support PM3MM results and give that TA/β-CD is stable with and without solvent.
Finally, NBO analysis reveals that charge transfer between occupied and unoccupied orbital's of both TA and β-CD is the major contribution of the stabilization of the complex.
Acknowledgement
This study was supported by the Algerian Ministry of Higher Education and Scientific Research and General Direction of Scientific Research as part of projects E01520090010, E01520120013 and E01520100004.