

1 Introduction
Hydrogen production, through the reduction of water in electrolyzers, is currently one of the most convenient ways to store energy durably, provided that electrical energy is initially obtained from renewable resources. While electrolysis is a mature and robust technology, the most promising devices, based on proton exchange membranes, rely on the use of platinum as an electrocatalyst to accelerate both hydrogen evolution and water oxidation reactions. This rare and expensive metal is not itself a renewable resource, so the viability of a hydrogen economy depends on the design of new efficient and robust electrocatalytic materials based on Earth-abundant elements. A competitive alternative to platinum could be found in living micro-organisms that metabolize hydrogen using hydrogenases [1,2]. Catalysis in hydrogenases requires only base-metal centers (nickel and iron), and the structures of their active sites have inspired the design of new synthetic catalysts based on these metals [3–8], cobalt [8,9], or other Earth-abundant elements such as molybdenum [10] and manganese [11]. Probably one of the most successful examples of such a bio-inspired approach is the series of nickel bisdiphosphine complexes designed by D. L. DuBois (Scheme 1) [12,13]. While structurally dissimilar to hydrogenase active sites (Scheme 1), these compounds combine features of both [NiFe]- and [FeFe]-hydrogenases, borrowing the Ni ion from the former, and from the latter the pendant amine groups [14,15], which act as a proton relay in the enzymatic mechanism. Depending on the nature of the substituents on the N and P atoms, these compounds have been shown to display remarkable catalytic properties for hydrogen evolution, and currently stand as the only series capable of catalytic hydrogen oxidation, with some compounds being able to achieve bidirectional catalysis [16,17]. Immobilization of such synthetic catalysts on multiwall carbon nanotubes (CNTs), either covalently or through π–π stacking interactions, has yielded catalytic nanomaterials that when interfaced with a Nafion membrane show bidirectional and reversible catalytic activity for hydrogen evolution and oxidation at the thermodynamic equilibrium, prolonged stability under turnover conditions, and resistance to CO poisoning [2,18,19]. However, the reported preparation of such nickel bisdiphosphine compounds requires the use of anhydrous nickel salts in the form of the hexakisacetonitrilenickel(II) complex, which must be synthesized by the oxidation of metallic nickel by nitrosonium cations in acetonitrile. We report herein that the same nickel bisdiphosphine complexes can be prepared starting from hydrated nickel salts. Additionally, in the course of this study, we identified that chloride anions can bind to the nickel center in nickel bisdiphosphine complexes, which significantly affects their electrochemical behavior and strongly diminishes their catalytic activity for hydrogen evolution.
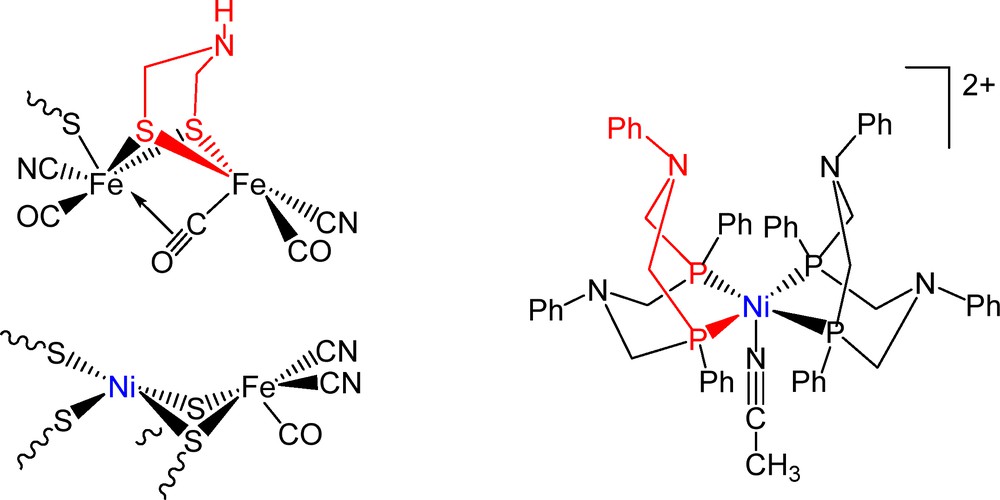
(Color online.) Representation of the [Ni(PPh2NPh2)2(CH3CN)]2+ cation (right) and of the active sites of the [FeFe]-hydrogenase in the reduced Hred state (top left), and the [NiFe]-hydrogenase in the Ni-SI state (bottom left).
2 Results and discussion
The reaction of [Ni(H2O)6](BF4)2 or [Ni(H2O)6](ClO4)2 with two equivalents of the PPh2NPh2 diphosphine ligand (Scheme 1) in acetonitrile yields the corresponding nickel complexes [Ni(PPh2NPh2)2(CH3CN)](BF4)2 (1) and [Ni(PPh2NPh2)2(CH3CN)](ClO4)2 (2) in good yields. These complexes show 1H and 31P NMR spectra similar to those of the complex synthesized from the hexakisacetonitrilenickel(II) precursor as prepared in our laboratory and previously reported by DuBois and coworkers [12]. The cyclic voltammograms of complexes 1 and 2 (Fig. 1), recorded in acetonitrile at a glassy carbon electrode, both display a reversible reduction at −0.84 V vs Fc+/0 (peak-to-peak separation of 70 mV), followed by a quasi-reversible reduction at −1.02 V vs Fc+/0 (which becomes irreversible at scan rates below 100 mVs−1). These data compare well to the one-electron reductions at −0.84 V and −1.02 V vs Fc+/0 reported by DuBois [12] and also to the values of −0.82 V and −1.03 V vs Fc+/0 obtained from a sample prepared in our laboratory (Fig. 1). The same procedure also allowed the preparation of the [Ni(P2PhN2Bz)2](ClO4)2 compound, again with similar characteristics as those previously reported for a complex prepared from [Ni(CH3CN)6](BF4)2.

(Color online.) Cyclic voltammograms of compounds 1 (blue trace), 2 (red trace) and [Ni(PPh2NPh2)2](BF4)2 (black trace) prepared from [Ni(CH3CN)6](BF4)2 (1 mmol.L−1, GC working electrode, 100 mV.s−1, 0.1 mol.L−1 n-Bu4NBF4 in MeCN, potentials are quoted versus the Fc+/0 couple).
The addition of increasing amounts of the acid [DMFH]OTf:DMF results in the appearance of a catalytic wave with half-wave potential of −0.92 V and −0.89 V vs Fc+/0 for compounds 1 and 2, respectively. These values compare well with the half-wave potential of −0.86 V vs Fc+/0 as reported by DuBois [20,21]. An ic/ip plot for 1 (Fig. 2, inset) shows the catalytic current upon addition of acid, normalized to the current of the first reduction peak in the absence of acid, as a function of acid concentration [22]. Overlaying this plot with that obtained for the complex prepared from the hexakisacetonitrilenickel(II) precursor shows identical catalytic activities.
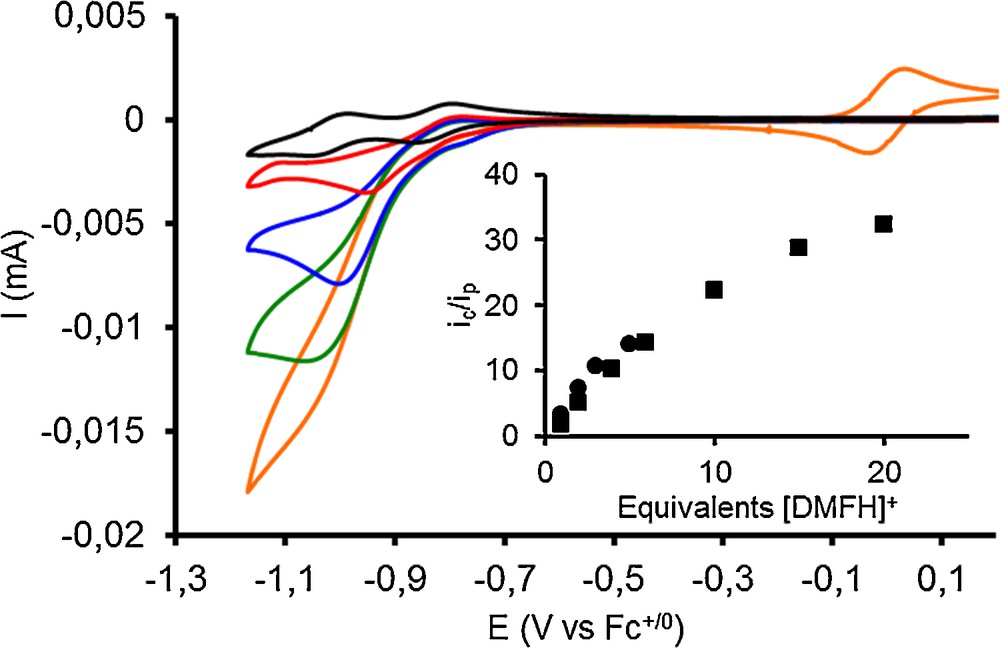
(Color online.) Cyclic voltammograms of compound 1 (1 mmol.L−1) in CH3CN in the presence of 0, 1, 2, 3, and 5 equiv of [DMFH]OTf:DMF. (GC working electrode, 100 mV.s−1, 0.1 mol.L−1 n-Bu4NBF4 in MeCN, potentials are quoted versus the Fc+/0 couple added in the solution for the last measurement shown with the orange trace). Inset: trace of the ratio ic/ip (where ic is the peak current of the catalytic wave and ip is the peak current of a monoelectronic Ni-centered wave) towards the equivalents of [DMFH]OTf:DMF for 1 (circles) and [Ni(PPh2NPh2)2](BF4)2 prepared from [Ni(CH3CN)6](BF4)2 (squares).
Compounds 1 and 2 were crystallized through the diffusion of diethyl ether into solutions of the complexes in acetonitrile. The structures obtained by X-ray crystallography (Fig. 3) show the same coordination environment and geometry as that reported by DuBois and coworkers for [Ni(PPh2NPh2)2](BF4)2 [12]. Both structures comprise a nickel(II) center in a distorted trigonal bipyramidal environment, in which the axial positions are occupied by a single phosphorus donor from each of the two PPh2NPh2 ligands, and the three equatorial positions are occupied by the two other phosphorus atoms and a coordinated acetonitrile molecule. Each PPh2NPh2 ligand forms two six-membered rings through coordination with the nickel(II) center, of which one is in the chair conformation and the other is in the boat conformation. The bond lengths and bond angles of both complexes closely match those of the structure reported by DuBois, with Ni–P1 distances of 2.207(2) Å, 2.2063(15) Å and 2.212(2) Å, Ni–P2 distances of 2.187(2) Å, 2.1865(16) Å and 2.183(2) Å, P1–Ni–P3 angles of 136.29(7)°, 136.50(6)° and 136.13(8)° and P2–Ni–P4 angles of 177.86(8)°, 177.76(6)° and 177.73(9)° for the complex reported by DuBois [12], compound 1 and compound 2, respectively.

(Color online.) Asymmetric units for compounds 1·0.5 H2O (left) and 2 (right) (50% probability thermal ellipsoids). Hydrogen atoms and disorder present in a phenyl ring and one of the tetrafluoroborate anions in compound 1 have been omitted for clarity.
Attempts to synthesize the complex starting from [Ni(H2O)6]Cl2 as the precursor, however, resulted in a purple product (compound 3) with different NMR spectra and electrochemical properties. The cyclic voltammogram shows only a single reduction event at −1.08 V vs Fc+/0, the reversibility of which increases with increasing scan rate. This could be interpreted as being a two-electron event, comprising a one-electron reduction with the simultaneous loss of the chloride ligand, and the instantaneous reduction of the resulting complex, which should have a more positive reduction potential than the chloride species. Addition of the acid [DMFH]OTf:DMF results in the appearance of a catalytic wave (Fig. 4), however with a half-wave potential of −1.06 V, a detrimental increase of 200 mV in overpotential. The activity of complex 3 is thus significantly lower than that of complex 1 or of the compound prepared from the hexakisacetonitrilenickel(II) precursor, as can be seen by the lower slope in the ic/ip plot of complex 3 as compared to that of [Ni(PPh2NPh2)2](BF4)2 prepared from [Ni(CH3CN)6](BF4)2 (Fig. 4).

Cyclic voltammograms of compound 3 (1 mmol.L−1) in CH3CN in the presence of 0, 0.5, 1.5, 2.5, 5, 7.5, 10 and 15 equiv of [DMFH]OTf:DMF. (GC working electrode, 500 mV.s−1, 0.1 mol.L−1 n-Bu4NBF4 in MeCN, potentials are quoted versus the Fc+/0 couple added in the solution for the last measurement shown with the brown trace). Inset: trace of the ratio ic/ip (where ic is the peak current of the catalytic wave and ip is the peak current of a monoelectronic Ni-centered wave) towards the equivalents of [DMFH]OTf:DMF for 3 (diamonds) and [Ni(PPh2NPh2)2](BF4)2 prepared from [Ni(CH3CN)6](BF4)2 (triangles).
Electrospray ionization-mass spectrometry indicates that a chloride ligand is strongly attached to the nickel bisdiphosphine moiety. This is confirmed by crystallographic analysis (Fig. 5), which shows that the complex again adopts the same coordination geometry as the previously reported complex, comprising a nickel(II) center in a distorted trigonal bipyramidal environment, in which the axial positions are occupied by a single phosphorus donor from each one of the two PPh2NPh2 ligands, and the three equatorial positions are occupied by the two other phosphorus atoms, but with a coordinated chloride ligand in place of the acetonitrile found in 1, 2 and the structure reported by DuBois [12]. The bond lengths and bond angles are once again quite similar to those of the structure reported by DuBois, with a Ni–P1 distance of 2.1918(12) Å, a Ni–P2 distance of 2.2028(12) Å, a P1–Ni–P3 angle of 175.74(5)° and a P2–Ni–P4 angle of 136.50(5)°. The formula of compound 3 thus stands as [NiCl(PPh2NPh2)2]Cl.

(Color online.) Crystal structure of compound 3.4CH2Cl2 (50% probability thermal ellipsoids). Hydrogen atoms and crystallized solvent molecules have been omitted for clarity.
The treatment of a solution of 1 in CD2Cl2 with 0.5 and 1 equiv of NEt4Cl in CD3CN causes an upfield shift and broadening of the singlet in the 31P NMR spectrum, suggesting a dynamic replacement of the coordinated acetonitrile molecule with chloride. We therefore note that the formation of 3 or related compounds may spontaneously occur during electrochemical measurements if there is some leakage from a KCl-based reference electrode and thus recommend the use of non-aqueous reference electrodes, as in this study, for measurements on DuBois catalysts.
Conversely, the treatment of 3 with two equivalents of silver triflate (AgOTf) in CH2Cl2 resulted in a red compound (4), of which the NMR spectrum and electrochemical behavior again resemble those of the compound prepared in the manner described by DuBois. The cyclic voltammogram shows a reversible wave at −0.83 V and a quasi-reversible wave −1.02 V (both potentials vs Fc+/0), comparable to the potentials of the waves found in the complex obtained from [Ni(CH3CN)6](BF4)2.
3 Conclusion
DuBois nickel bisdiphosphine compounds currently stand as electrocatalysts with high potential for technological implementation as a substitute for platinum in hydrogen fuel cells [23], hydrogen producing electrolyzers and photochemical cells. This, however, requires that the preparation of such compounds can be scaled-up and achieved at low cost. The synthesis of the diphosphine ligands is straightforward and easily tunable to generate a range of variously substituted compounds. There was, however, still a need to develop a synthetic protocol for the nickel complex using commercially available and easy-to-handle metal salts as precursors. In particular, the hexakisacetonitrilenickel(II) precursor used in the previously reported synthetic procedure is moisture-sensitive, which is not the case of final complexes, some of which can operate as electrocatalysts even in fully aqueous media. We show herein that the synthesis of DuBois complexes can be achieved from commercially available hydrated nickel salts in acetonitrile, taking advantage of the stronger binding properties of acetonitrile compared to water. Besides allowing for a much simpler synthetic protocol, this study now allows the preparation of a variety of compounds with various counter anions, with the obvious advantage of being able to tune the solubility of the compound. In the course of the study, we have found that chloride, introduced as one of these counter-anions, binds to the nickel bisdiphosphine complex, modifying its electrochemical behavior to the detriment of its catalytic activity. From this compound, the chloride ligand can be exchanged through a reaction with silver salts, affording another method for easy anion metathesis and modification of solubility properties of the final complexes. These new synthetic methodologies hold promise for simplifying the preparation of molecular-engineered electrode materials initially developed in our group.
4 Experimental
4.1 Methods
NMR spectra were recorded at room temperature in 5 mm tubes on a Bruker AC 300 spectrometer equipped with a QNP probehead, operating at 300.0 MHz for 1H, and 121.5 MHz for 31P. Solvent peaks were used as internal references relative to Me4Si for 1H chemical shifts; H3PO4 (85%) was used as external reference for 31P NMR spectra. All 31P NMR spectra were proton decoupled. ESI mass spectra were recorded with a Finnigan LCQ thermoquest ion-trap.
Electrochemical experiments utilized a Bio-logic SP300 potentiostat, and were carried out in a three-electrode electrochemical cell with a glassy carbon working electrode (1 mm or 1.6 mm in diameter). The auxiliary electrode was platinum, titanium or graphite, and the reference electrode was a non-aqueous Ag/(AgNO3 10−2 M in CH3CN) electrode. All the potentials given in this work are with respect to the Fc+/0 couple. The voltammograms were referenced to ferrocene by addition of an internal standard after the final experiment. The experiments were conducted in anhydrous acetonitrile (water content < 50 ppm) with tetrabutylammonium tetrafluoroborate (n-Bu4NBF4) as the supporting electrolyte, degassed by bubbling with solvent-saturated argon, and kept under a blanket of argon during experiments. Additions of [DMFH](OTf):DMF were made with a syringe, using a freshly prepared 2:1 (mol:mol) mixture of dimethylformamide (4.9 mL) and triflic acid (2.8 mL) with a density of 1.28 and a [DMFH]+ concentration of 4.1 M.
4.2 Synthesis
The ligand 1,3,5,7-tetraphenyl-1,5-diaza-3,7-diphosphacyclooctane (PPh2NPh2), [12] the complex [Ni(CH3CN)6](BF4)2 [24], and the complex [Ni(PPh2NPh2)2(CH3CN)](BF4)2 [12] starting from [Ni(CH3CN)6](BF4)2 were all synthesized using published procedures.
4.2.1 [Ni(PPh2NPh2)2(CH3CN)](BF4)2 (1)
The ligand PPh2NPh2 (29 mg, 0.064 mmol) and [Ni(H2O)6](BF4)2 (11 mg, 0.032 mmol) were suspended in 5 mL of acetonitrile and stirred at room temperature overnight. The red solution was filtered and the solvent removed under vacuum to give 30 mg of a red powder (0.026 mmol, 81%).
1H NMR (CD2Cl2): δ 7.25–7.47 (m, 40 H, Ph), 4.60 (d, J = 14 Hz, 8 H, CH2), 4.04 (d, J = 13.5 Hz, 8 H, CH2) 2.02 (s, 3 H, CH3CN); 31P NMR (CD2Cl2): δ 4.96.
4.2.2 [Ni(PPh2NPh2)2(CH3CN)](ClO4)2 (2)
The ligand PPh2NPh2 (39.5 mg, 0.087 mmol) and [Ni(H2O)6](ClO4)2 (16.5 mg, 0.045 mmol) were suspended in 5 mL of acetonitrile and stirred at room temperature overnight. The red solution was filtered and the solvent removed under vacuum to give 36 mg of a red powder (0.03 mmol, 69%).
1H NMR (CD2Cl2): δ 7.20–7.43 (m, 40 H, Ph), 4.51 (d, J = 14 Hz, 8 H, CH2), 4.01 (d, J = 14 Hz, 8 H, CH2), 2.06 (s, 4 H, CH3CN); 31P NMR (CD2Cl2): δ 4.60.
4.2.3 [NiCl(PPh2NPh2)2]Cl (3)
The ligand PPh2NPh2 (78.8 mg, 0.173 mmol) and [Ni(H2O)6]Cl2 (19.8 mg, 0.083 mmol) were suspended in 10 mL of ethanol and refluxed for two hours. The solvent was evaporated, and the residue added to 10 mL dichloromethane. The purple solution was filtered and the solvent removed under vacuum to give 75 mg of a purple powder (0.073 mmol, 88%).
1H NMR (CD2Cl2): δ 7.11–7.50 (m, 40 H, Ph), 4.68 (d, J = 14 Hz, 4 H, CH2), 4.27 (d, J = 13 Hz, 4 H, CH2), 3.80 (t, J = 15 Hz, 8 H, CH2); 31P NMR (CD2Cl2): δ 3.40 (br); ESI-MS: m/z = 1001.4 ([Ni(PPh2NPh2)2Cl]+).
4.2.4 [Ni(PPh2NPh2)2](OTf)2 (4)
Complex 3 was dissolved in 10 mL of distilled CH2Cl2 and the purple solution transferred into a suspension of AgOTf (30 mg, 0.116 mmol) in 10 mL CH2Cl2 at −40 °C. This suspension was stirred at −40 °C for one hour and then allowed to warm to room temperature and filtered. The solvent was evaporated under reduced pressure giving a red powder (90%).
1H NMR (CD2Cl2): δ 7.21–7.48 (m, 40 H, Ph), 4.59 (d, J = 14 Hz, 4 H, CH2), 4.02 (d, J = 14 Hz, 4 H, CH2); 31P NMR (CD2Cl2): δ 4.33.
4.2.5 Crystal structure analysis
Crystallographic data are summarized in Table 1. Data collection was performed at 150 K with an Oxford-diffraction XCalibur S diffractometer with a CCD area detector, with graphite monochromated Mo Kα radiation (λ = 0.71073 Å). An empirical absorption correction using Abspack software was performed. Molecular structures were solved by the charge-flipping method [25] and refined on F2 by full-matrix least-squares techniques with the SHELXL [26] and Olex2 package [27], with anisotropic thermal parameters. All non-hydrogen atoms were refined anisotropically. Hydrogen atoms were fixed on ideal positions and refined as riding atoms for compounds 2 and 3·4 CH2Cl2; for compound 1·0.5 H2O they were found by Fourier transformation and refined with individual isotropic displacement parameters, except for the disordered phenyl group, where hydrogen atoms were fixed on ideal positions as for 2 and 3·4 CH2Cl2.
Crystal data and structural refinement details for complexes 1·0.5 H2O, 2 and 3·4 CH2Cl2.
| Compound | 1·0.5 H2O | 2 | 3·4 CH2Cl2 |
| Formula | C58H60B2F8N5NiO0.50P4 | C58H59Cl2N5NiO8P4 | C60H64Cl10N4NiP4 |
| Molecular mass | 1191.32 | 1207.59 | 1378.24 |
| Color | Orange | Red | Purple |
| Crystal size (mm) | 0.544 × 0.059 × 0.046 | 0.217 × 0.084 × 0.039 | 0.430 × 0.224 × 0.072 |
| Crystal system | Orthorhombic | Orthorhombic | Monoclinic |
| Space group | P212121 | P212121 | P21/n |
| a [Å] | 12.9812 (5) | 13.0088 (6) | 21.0453 (8) |
| b [Å] | 18.2673 (11) | 18.2801 (7) | 12.9418 (4) |
| c [Å] | 23.0389 (11) | 23.2098 (11) | 22.8815 (10) |
| α [°] | 90 | 90 | 90 |
| β [°] | 90 | 90 | 90.678 (4) |
| γ [°] | 90 | 90 | 90 |
| V [Å]3 | 5463.2 (5) | 5519.4 (4) | 6231.6 (4) |
| Z | 4 | 4 | 4 |
| ρcalcd [g.cm−3] | 1.448 | 1.453 | 1.469 |
| μ [cm−1] | 5.46 | 6.25 | 8.86 |
| Reflections collected | 16727 | 18710 | 34426 |
| Unique reflections (Rint) | 12,668 (0.0502) | 11,230 (0.0617) | 15,434 (0.0624) |
| Observed reflections [I > 2σ(I)] | 5517 | 5158 | 10217 |
| Refined parameters | 930 | 704 | 750 |
| Flack parameter | –0.009 (13) | 0.007(14) | |
| Final R indices (observed reflections) | R1 = 0.0431, wR2 = 0.0396 | R1 = 0.0513, wR2 = 0.0680 | R1 = 0.0740, wR2 = 0.1743 |
| Goodness of fit S | 0.530 | 0.714 | 1.061 |
| Δρ (max/min) [e.Å−3] | 0.684 and −0.440 | 0.899 and −0.697 | 1.884 and −1.205 |
X-ray suitable crystals of compounds 1 and 2 were grown by vapor diffusion of diethyl ether into a filtered concentrated solution of each complex in acetonitrile, whereas crystals of compound 3 were grown by vapor diffusion of diisopropyl ether into a filtered concentrated solution of the complex in dichloromethane. Compound 1·0.5 H2O crystallized in the space group P212121. The asymmetric unit contains one [Ni(PPh2NPh2)2]2+ cation, of which one phenyl ring is disordered over two positions with relative occupancy of 0.63(2)/0.37(2), two BF4 anions, the fluorine atoms of one of which are disordered over two positions with relative occupancy of 0.69(2)/0.31(2), and a water molecule with an occupancy of 0.5. The compound 2 also crystallized in the P212121 space group, and the asymmetric unit contains one [Ni(PPh2NPh2)2]2+ cation and two ClO4 anions. Compound 3·4 CH2Cl2 crystallized in the space group P21/n. The asymmetric unit contains a [NiCl(PPh2NPh2)2]+ cation, a chloride anion and four dichloromethane solvent molecules, of which two are disordered over two positions with relative occupancy of 0.55(2)/0.45(2) and 0.57(2)/0.43(2).
The atomic coordinates have been deposited in the Cambridge crystallographic database (CCDC code 1037913 for compound 1·0.5 H2O, 1,037,914 for 2 and 1,037,912 for 3·4 CH2Cl2).
Acknowledgements
This work was supported by ANR (EnzHyd project, ANR-08-PANH-008, Caroucell project, ANR-13-BIME-003 and Labex program ARCANE, ANR-11-LABX-0003-01) and the FCH Joint Undertaking (Nano-Cat project, grant No. 325239).