


 CC-BY 4.0
CC-BY 4.0
1. Introduction
The photo-synthesis of reduced forms of carbon from CO2 holds the promise to set us on track to mitigate the alarming anthropogenic amount of CO2 we are dumping into our biosphere from the massive use of fossil fuels [1, 2]. Blueprints of this strategy are directly provided by photosynthesis and cellular respiration that have enabled the development of energy infrastructures for solar energy harnessing and its storage in chemical compounds [3]. The efficient capture of sunlight, its conversion into a chemical potential and coupled to oxidative and reductive catalytic processess are the elemental steps that must be synchronized and optimized.
Since the early reports in the 80s on homogeneous photocatalytic systems for CO2 reduction based on transition metals [4, 5], a multitude of electron donor (ED)/photosensitizer (PS)/catalyst (Cat) combinations have been reported to reduce CO2 in organic and aqueous media [6, 7, 8, 9, 10, 11]. In the scheme of photocatalytic CO2 reduction, the PS mediates the transfer of electrons from the ED to the Cat, but utilizes photons to drive the otherwise endergonic electron transfer steps. In a simple multicomponent approach, the three components can be simply mixed in solution and the kinetics of the electron transfer steps are expected to be diffusion-limited without kinetic advantage for a particular interaction. An alternative aproach to overcome this diffusion limit and favor productive reaction steps consists of covalently attaching the PS and the Cat in a PS–Cat dyad to promote fast electron transfer from the PS to the Cat [12, 13, 14, 15].
We and others have demonstrated that iron-porphyrins are one of the best performing homogeneous catalysts for the electrocatalytic reduction of CO2 [16, 17, 18, 19]. Their electrocatalytic properties such as overpotential, turnover number (TON) and turnover frequency (TOF) can be further improved by introducing substituents on the porphyrin macrocycle such as electron withdrawing or donating groups, proton relays, hydrogen bond donors or electrostatic groups [10, 11, 20, 21, 22, 23, 24]. When associated with a PS in a multicomponent system, iron-porphyrins were also recently shown to catalyze the photoreduction of CO2 to CO [25, 26, 27], and even further to methane in some conditions [28, 29]. However, to the best of our knowledge, there are no reported supramolecular dyads consisting of an iron porphyrin catalyst and any photosensitizer [30]. This is in part due to the fact that the high absorption coefficient of the porphyrin catalyst across the visible region, overlaps and sometimes overwhelms the light-absorption role of the photosensitizer, leading to short-lived, and therefore non-productive, excited states. As such, for most multicomponent systems, there is a common trend of employing huge differences in the concentration of the iron porphyrin catalyst (μM range) and the photosensitizer (mM range). This piqued our curiosity to examine if a photoredox-iron-porphyrin catalyst design may lead to a new reactivity pattern.
Structures and abbreviation of the porFe-bpyRu supramolecular complex and the corresponding model complexes porFe and bpyRu and the electron donors BIH and Asc.
In this work, we report the synthesis and characterization of a molecular assembly porFe-bpyRu composed of an iron porphyrin as catalyst covalently attached to a ruthenium trisbipyridine, the photosensitizer, through an amide linker (Scheme 1). Methoxy groups were introduced on the porphyrin as electron donating groups to enhance the nucleophilic character of the iron center and promote CO2 reduction [16]. The electrochemical and photophysical properties of the porFe-bpyRu dyad were systematically compared with iron porphyrin (porFe) and ruthenium trisbipyridine (bpyRu) reference compounds. The electrochemical data indicate that the amide linkage does not promote strong electronic communication between the Cat and the PS, keeping the independent redox properties of the constitutive modules intact. Laser flash photolysis studies demonstated that upon excitation of the photosensitizer in the dyad, an efficient energy transfer occurs towards the porphyrin catalyst, leading to a very fast quenching (<20 ns) due to the presence of the Fe metal, thereby shutting off the possibility for the light activation of the catalytic unit. In the presence of an electron donor, an immediate dark electron transfer is observed in the catalyst (porFe-bpyRu or porFe) forming Fe(II) species from Fe(III). For an independent mixture of the PS/PS–Cat and a reversible electron donor, a light induced charge shift leading to the reduced Fe(I) species was evidenced. Accordingly, upon exciting an exogenous bpyRu added to the porFe-bpyRu/electron donor mixture, a photo-induced electron transfer was observed from the exogenous Ru(I) photoreductant to the Fe(II) species, consequently forming the two-electron reduced form Fe(I). More interestingly, under continuous irradiation in the presence of dimethylphenylbenzimidazoline (BIH) as sacrifical electron donor, the porFe-bpyRu dyad displayed a surprisingly higher photocatalytic activity and CO2-to-CO selectivity compared to the referenced iron porphyrin (porFe). Comprehensive photophysical and electrochemical studies pinpoint to the fact that despite the extinction of the photophysical properties of the photosensitizer in the supramolecular dyad, its appendage to the porFe was beneficial inasmuch as it acts as an electron reservoir next to the catalytic unit that promotes the formation of the catalytically active species at minimal thermodynamic cost.
2. Experimental section
2.1. General procedure
Proton and carbon nuclear magnetic resonance (1H and 13C NMR) spectra were recorded at room temperature on Bruker Advance spectrometers. The electrospray ionization mass spectrometry (ESI-HRMS) experiments were performed on TSQ (Thermo Scientific. 2009) with an ESI+ method. Ground state absorption spectra were measured on a Specord spectrophotometer (Analytik Jena). EPR spectra were recorded at 40 K on a Bruker ELEXSYS 500 spectrometer equipped with a Bruker ER 4116DM X band resonator, an Oxford Instrument continuous flow ESR 900 cryostat, and an Oxford ITC 503 temperature control system. Cyclic voltammetry measurements were performed in an electrochemical cell composed of a glassy carbon (3 mm diameter) working electrode, Ag/AgNO3 (10−2 M) reference electrode, and a platinum wire counter electrode with tetra-N-butylammonium hexafluorophosphate (TBAP) as supporting electrolyte. Scan rate was chosen at 100 mV/s and a CH Instruments potentiostat workstation was utilized to control the applied voltages and to measure resulting current. Ferrocene was used as an internal reference and the potentials are converted to NHE [31]. Spectroelectrochemical measurements were performed in a thin-layer quartz cuvette cell using a platinum honeycomb electrode (PINE Research) and a Pt wire reference electrode. Nanosecond transient absorption measurements were performed on a home-built set-up which has been described in detail previously [32] and a commercial Edinburgh Instruments LP920 Laser Flash Photolysis Spectrometer system. Photocatalytic experiments were performed in a 41.5 mL vial containing 6.5 mL CO2-saturated DMF/H2O (9:1 v/v) + 2 μM catalyst + 50 μM photosensitizer + 50 mM electron donor. A SugarCUBE high intensity LED fiber optic illuminator was utilized with a blue light output centered at 463 nm with a full width at half maximum (FWHM) characteristic of about 50 nm, adjusted to produce 9.5 mW⋅cm−2. The reaction vessel is connected in line with a micro gas chromatography Inficon system to analyze the products of the reaction.
2.2. Synthesis and characterization
2.2.1. Synthesis of 1,3-dimethoxybenzene (1)
Potassium bicarbonate, K2CO3 (6.28 g, 45 mmol) and methyl iodide CH3I (2.6 mL, 41.4 mmol) are added simultaneously and dropwise to a stirred solution of resorcinol (2.00 g, 18 mmol) in anhydrous acetone, at 0 °C under Ar atmosphere. The reaction mixture is stirred at room temperature for 12 h. Once reaction reached completion, the volatiles are evaporated under reduced pressure and 30 mL of H2O are added to the residue. The crude product mixture is extracted with ethyl acetate (3 × 30 mL) and the combined organic layers are dried over Na2SO4. After evaporation of the solvent, the obtained residue is purified via silica column chromatography (hexane/ethyl acetate (5:1 v/v)) to give 1,3-dimethoxybenzene as a light-yellow oil (1.92 g, 13.9 mmol, 77%). 1H NMR (500 MHz, CDCl3): 𝛿 = 7.19 (t, J = 8.2 Hz, 1H), 6.52 (d, J = 2.4 Hz, 1H), 6.51 (d, J = 2.4 Hz, 1H), 6.47 (t, J = 2.4 Hz, 1H), 3.8 (s, 6H).
2.2.2. Synthesis of 2,6 dimethoxybenzaldehyde (2)
1,3-dimethoxybenzene (1.4 mL, 1.51 g, 10.9 mmol) is dissolved in 50 mL of dry THF, under N2 and the resulting solution is purged with N2 for at least 20 min. Afterwards, under vigorous stirring, n-BuLi (8.2 mL, 86.8 mmol) is added dropwise at 0 °C and once the addition is completed, the reaction mixture is stirred at room temperature for 2 h under N2 atmosphere. Then, dry DMF (2.1 mL, 27 mmol) is added to the solution and the mixture is stirred for an additional 2 h. After 2 h, 20 mL of H2O are added and extractions with ethyl acetate (3 × 30 mL) are carried out. The collected organic layers are dried over Na2SO4 and evaporated under vacuum to obtain a yellowish oily residue. The desired product was recrystallized from hexane (15 mL) and collected as a light brown solid (1.26 g, 7.6 mmol, 70%). 1H NMR (500 MHz, CDCl3): 𝛿 = 10.51 (s, 1H), 7.45 (t, J = 8.5 Hz, 1H), 6.58 (d, J = 8.5 Hz, 2H) 3.90 (s, 6H).
2.2.3. Synthesis of 5-(4-nitrophenyl)-10,15,20-tris-(2,6-dimethoxyphenyl)-21H, 23H porphyrin (3)
2,6-dimethoxybenzaldehyde (0.75 g, 4.5 mmol) and 4-nitrobenzaldehyde (0.23 g, 1.5 mmol) are dissolved in 600 mL of CHCl3 and the resulting solution is subsequently purged with N2 for at least 30 min under vigorous stirring. Then, pyrrole (0.42 mL, 6 mmol) is added dropwise in the absence of light and the reaction mixture is purged with N2 for an additional period of 10 min, before BF3OEt2 (233 μL, 1.88 mmol) is added. After the addition of BF3OEt2, the reaction mixture is stirred in the dark at room temperature for 3 h under N2 atmosphere. Then, DDQ (1.36 g, 6 mmol) is added to the solution and it stirred for 2 h at room temperature. The resulting crude product mixture is subsequently filtered through silica pad and then is purified via silica column chromatography (CH2Cl2/hexane 4:1 (v/v)). However, only a small percentage of the desired porphyrin can be obtained clean. Given that, without any further purification, the 148 mg of the crude mixture was then used for the synthesis of 5-(4-aminophenyl)-10,15,20-tris-(2,6-dimethoxyphenyl)-21H, 23H porphyrin (4). 1H NMR (500 MHz, CDCl3): 𝛿 = 8.77 (d, J = 4.7 Hz, 2H), 8.72 (s, 4H), 8.64 (d, J = 4.7 Hz, 2H), 8.59 (d, J = 8.7 Hz, 2H), 8.39 (d, J = 8.7 Hz, 2H), 7.71 (m, 3H), 6.99 (m, 6H), 3.53 (s, 6H), 3.51 (s, 12H), −2.57 (s, 2H). 13C NMR (75 MHz, CDCl3): 𝛿 = 160.7, 150.1, 147.5, 135.3, 130.3, 121.7, 120.0, 119.8, 115.53, 112.2, 111.9, 104.3, 104.3, 56.2. UV/Vis (CH2Cl2): 𝜆max (𝜀, mM−1⋅cm−1) = 419 (262.5), 514 (16.6), 550 (5.5), 590 (5.3), 644 (2.5).
2.2.4. Synthesis of 5-(4-aminophenyl)-10,15,20-tris-(2,6-dimethoxyphenyl)-21H, 23H porphyrin (4)
To a stirred solution of the isomeric mixture of porphyrin derivative (3) (148 mg) in DCM (15 mL), 2.5 mL of conc. HCl and SnCl2⋅2H2O (0.2 g, 0.89 mmol) are added at 0 °C and the mixture is heated to 70 °C for at least 20 h. The progress of the reaction is monitored by thin layer chromatography (TLC, SiO2, CH2Cl2/MeOH (99:1 v/v)). Upon reaction completion, 50 mL of DCM are transferred to the solution and it is left stirring for 15 min. Afterwards, the organic phase is collected, quenched with sat. aqueous solution of NaHCO3 and washed thoroughly with H2O. Then, the obtained organic layer is dried over Na2SO4 and evaporated until dryness. Porphyrin (4) is obtained as a purple solid after silica column chromatography (DCM/EtOH (98:2 v/v)) purification (67.4 mg, 0.083 mmol, overall yield of two steps 5.5%). 1H NMR (500 MHz, CDCl3): 𝛿 = 8.77 (d, J = 4.7 Hz, 2H), 8.72 (s, 4H), 8.64 (d, J = 4.7 Hz, 2H), 8.59 (d, J = 8.7 Hz, 2H), 8.39 (d, J = 8.7 Hz, 2H), 7.71 (m, 3H), 6.99 (m, 6H), 3.53 (s, 6H), 3.51 (s, 12H), −2.57 (s, 2H). 13C NMR (75 MHz, CDCl3): 𝛿 = 160.7, 150.1, 147.5, 135.3, 130.3, 121.7, 120.0, 119.8, 115.53, 112.2, 111.9, 104.3, 104.3, 56.2. UV/Vis (CH2Cl2): 𝜆max (𝜀, mM−1⋅cm−1) = 419 (262.5), 514 (16.6), 550 (5.5), 590 (5.3), 644 (2.5).
2.2.5. Synthesis of 4′-methyl-4-carboxy-2,2′- bipyridine (5)
4,4-dimethyl-2,2′-bipyridine (1.1 g, 6 mmol) is dissolved in 65 mL of 1,4-dioxane and SeO2 (800 mg, 7.2 mmol) is added. The resulting solution is stirred and heated to reflux for 24 h. After 24 h, the hot solution is passed through a short pad of celite to remove the elemental Se that is formed during the reaction. The filtrate once it is cooled down to room temperature, it is evaporated under reduced pressure. The obtained yellow residue is dissolved in 40 mL of EtOH and under vigorous stirring, 10 mL of aqueous solution of AgNO3 (1.1 g, 6.6 mmol) is added in the absence of light. Then, over a period of 30 min, 25 mL of aqueous solution of 1 M NaOH are added dropwise and the reaction mixture is stirred for 15 h at room temperature. Afterwards, the solution is concentrated under vacuum and the formed precipitate is filtered and washed first with 1.3 M NaOH (2 × 15 mL) and then with 15 mL of H2O. The filtrate is collected and extracted with DCM (4 × 30 mL). Next, the pH of the collected basic aqueous layer is adjusted to pH = 3.5 by the addition of 4.0 N HCl/CH3COOH (1:1 v/v). Upon acidification, the precipitation of a pink solid is observed and the mixture is kept at −10 °C for an additional period of 20 h for further precipitation. The precipitate is filtered and dried at 90 °C for 12 h. Finally, the crude mixture is purified via a Soxhlet extractor apparatus using anhydrous acetone as the eluent for 3 days. 4′-methyl-4-carboxy-2,2′-bipyridine is obtained as a light-yellow solid (255 mg, 1.19 mmol, 20%). 1H NMR (500 MHz, DMSO-d6): 𝛿 = 13.89 (br s, 1H), 8.85 (br d, J = 4.9 Hz, 1H), 8.82 (br s, 1H), 8.57 (br d, J = 4.9 Hz, 1H), 8.27 (br s, 1H), 7.85 (dd, J = 4.8 Hz, J = 1.4 Hz, 1H), 7.33 (br d, J = 4.8 Hz, 1H), 2.43 (s, 3H).
2.2.6. Synthesis of cis–bis(2,2′-bipyridine)dichloro- ruthenium(II) (Ru(bpy)2Cl2) (6)
2,2′-bipyridine (0.331 g, 2.1 mmol), 10 mL of dry DMF, RuCl3 (0.200 g, 0.96 mmol) and LiCl (0.290 g, 6.75 mmol) are transferred into a two-neck round bottom flask and the resulting dark purple solution is heated to reflux overnight. After cooling down to room temperature, the formed orange precipitate is filtered and washed thoroughly first with acetone and then with H2O. Finally, it is washed with diethyl ether and hexane. Ru(bpy)2Cl2 is obtained as a dark purple solid (0.160 g, 0.33 mmol, 34%). 1H NMR (500 MHz, DMSO-d6): 𝛿 = 9.97 (br d, J = 4.6 Hz, 2H), 8.64 (br d, J = 7.9 Hz, 2H), 8.49 (br d, J = 7.8 Hz, 2H); 8.06 (m, 2H), 7.76 (m, 2H), 7.68 (m, 2H), 7.51 (br d, J = 4.9 Hz, 2H), 7.10 (m, 2H).
2.2.7. Synthesis of compound 7
In a schlenk tube, compound 5 (48 mg, 0.224 mmol) is dissolved in 9 mL of SOCl2 and the resulting stirring solution is heated up to 78 °C under N2 for 2 h. In the next step, SOCl2 is evaporated under reduced pressure and to the remaining residue 15 mL of dry THF is added. Then, porphyrin (4) (60 mg, 0.074 mmol) and anhydrous Et3N are transferred to the solution under N2. The reaction mixture is heated up to 50 °C for 20 h. Once the reaction reaches completion, the volatiles are removed under vacuum and the crude solid is purified via column chromatography (SiO2, DCM/MeOH (99:1 v/v)) affording derivative (7) as a purple solid (73 mg, 0.073 mmol, 98%). 1H NMR (500 MHz, CDCl3): 𝛿 = 8.94 (s, 1H), 8.93 (s, 1H), 8.78 (d, J = 4.7 Hz, 2H), 8.76 (s, 1H), 8.74 (d, J = 4.7 Hz, 2H), 8.71 (m, 4H), 8.61 (m, 1H), 8.36 (m, 1H), 8.21 (d, J = 8.5 Hz, 2H), 8.07 (d, J = 8.3 Hz, 2H), 8.01 (dd, J1 = 4.8 Hz, J2 = 1.9 Hz, 1H), 7.70 (t, J = 8.5 Hz, 3H), 7.23 (m, 1H), 6.99 (m, 6H), 3.52 (s, 6H), 3.50 (s, 12H), 2.53 (s, 3H), −2.59 (s, 2H). 13C NMR (75 MHz, CDCl3): 𝛿 = 164.2, 160.7, 157.4, 155.1, 150.6, 149.2, 148.8, 143.3, 139.5, 137.1, 135.2, 131.9, 130.2, 125.6, 122.5, 122.2, 120.1, 119.9, 118.6, 118.1, 117.5, 111.7, 111.0, 104.3, 56.2, 21.4. UV/Vis (CH2Cl2): 𝜆max (𝜀, mM−1⋅cm−1) = 419 (382.9), 513 (16.5), 546 (5.4), 589 (4.9), 645 (2.5).
2.2.8. Synthesis of compound 8
To a stirring solution of porphyrin derivate (4) (45 mg, 0.06 mmol) in dry THF, benzoyl chloride (50 μL, 0.38 mmol) and 70 μL of dry Et3N are added. The mixture is heated up to 70 °C for 12 h under N2 atm. Afterwards, THF and Et3N are evaporated under reduced pressure and the reaction product mixture is purified by column chromatography (SiO2, CH2Cl2/EtOH (99.7/0.3 v/v)). The desired product is afforded as a purple solid (40 mg, 0.043 mmol, 73%). 1H NMR (500 MHz, CDCl3): 𝛿 = 8.76 (d, J = 4.7 Hz, 2H), 8.72 (m, 6H), 8.14 (d, J = 8.4 Hz, 2H), 8.12 (s, 1H), 7.98 (m, 2H), 7.92 (d, J = 8.4 Hz, 2H), 7.69 (m, 3H), 7.58 (m, 1H), 7.52 (m, 2H), 6.98 (m, 6H), 3.51 (s, 6H), 3.49 (s, 12H), −2.53 (br s, 2H). 13C NMR (75 MHz, CDCl3): 𝛿 = 166.1, 160.7, 139.0, 137.4, 135.1, 132.0, 130.1, 129.0, 127.3, 126.4, 120.3, 120.0, 118.2, 111.6, 110.9, 104.3, 56.2. UV/Vis (CH3CN): 𝜆max (𝜀, mM−1⋅cm−1) = 415 (401.1), 511 (17.3), 544 (6.0), 588 (5.1), 644 (2.8).
2.2.9. Synthesis of compound 9
In a two-neck round bottom flask, porphyrin (7) (60 mg, 0.06 mmol) is dissolved in 38 mL of CH3COOH and the stirring solution is purged with N2 for 15 min at room temperature. After degassing, Ru(bpy)2Cl2 (57.6 mg, 0.12 mmol) is transferred to the flask and the reaction mixture is heated to reflux for 12 h under N2. Then CH3COOH is evaporated under reduced pressure and the obtained orange reddish residue is washed with H2O (5 × 20 mL). Then, the crude residue is purified by column chromatography (SiO2, CH3CN∕H2O∕KNO3(sat) (30:2:1 v/v)). Afterwards, the counter anions of RuII are exchanged with anions. More specifically, the dyad is dissolved in the minimum amount of CH3CN and to the resulting solution, a saturated solution of NH4PF6 in MeOH is added. Then, H2O was added slowly. Upon addition of H2O, the precipitation of a brown-purple solid is observed. The precipitate is subsequently filtered and washed thoroughly with H2O to remove the excess of the NH4PF6 salt giving porphyrin derivative (9) as an orange-purple solid (21 mg, 0.012 mmol, 20%). 1H NMR (500 MHz, (CD3)2CO): 𝛿 = 10.50 (br s, 1H), 9.43 (s, 1H), 8.98 (s, 1H), 8.76 (s, 1H), 8.76 (s, 4H), 8.73 (s, 4H), 8.64 (br s, 2H), 8.39 (br s, 2H), 8.26 (d, J = 8.2 Hz, 2H), 8.16–8.09 (m, 2H), 8.03–7.95 (m, 5H), 7.84 (br d, J = 5.6 Hz, 1H), 7.79 (t, J = 8.6 Hz, 3H), 7.57–7.49 (m, 4H), 7.45 (d, J = 5.4 Hz, 1H), 7.14 (d, J = 8.6 Hz, 6H), 3.52 (s, 18H), 2.61 (s, 3H), −2.50 (s, 2H). 13C NMR (75 MHz, (CD3)2CO): 𝛿 = 163.3, 161.3, 159.0, 157.8, 157.1, 153.2, 152.5, 152.4, 151.7, 151.6, 144.2, 139.3, 138.9, 138.7, 135.5, 131.4, 129.9, 128.6, 126.7, 126.3, 125.1, 125.0, 122.9, 120.0, 199.7, 119.4, 118.9, 113.0, 112.6, 104.9, 56.1, 21.2. UV/Vis (CH3CN): 𝜆max (𝜀, mM−1⋅cm−1) = 288 (87.8), 415 (413.5), 476 (22.9), 510 (24.2), 544 (9.3), 587 (6.7), 642 (4.1).
2.2.10. Synthesis of porFe-bpyRu
FeBr2 (0.088 g, 0.41 mmol) and derivate (9) (0.023 g, 0.014 mmol) are dissolved in 5 mL of dry and degassed THF. The reaction mixture is kept under stirring and heated at 50 °C for 12 h under Ar. The progress of the reaction is monitored by absorption spectroscopy. Once the reaction is completed, THF is evaporated under reduced pressure. The remaining solid is dissolved in 20 mL of DCM and treated with 4N HCl (3 × 15 mL). The organic layers are combined, washed with H2O (3 × 15 mL) and dried over Na2SO4. The crude product is purified via silica column chromatography (CH3CN∕H2O∕KNO3(sat) (30:2:1 v/v)). Then, the counter anions of RuII were exchanged with anions, as described previously for the synthesis of compound 9. The final compound is given as an orange-purple solid (18 mg, 0.010 mmol, 71%). UV/Vis (CH3CN): 𝜆max (𝜀, mM−1⋅cm−1) = 288 (83.0), 377 (59.4), 417 (95.8), 465 (27.7), 505 (17.9), 576 (5.3), 655 (4.1), 689 (4.3). ESI-HRMS: m/z calculated for a chemical formula C82H65ClFeN11O7Ru2+ [M]2+ = 754.1543, found 754.1580.
2.2.11. Synthesis of porFe
To a solution of FeBr2 (0.272 g, 1.27 mmol) in 5 mL of dry and degassed THF, porphyrin (7) (0.037 g, 0.041 mmol) is added and the mixture of the reaction and heated at 55 °C for 12 h under Ar. After reaction reaches completion, THF is evaporated and the residue is dissolved in 20 mL of DCM. Then, 15 mL of 4N HCl is added. The organic phase is collected and extracted for additional two times with 4N HCl (2 × 15 mL). Finally, the organic layers are collected, washed with H2O (3 × 15 mL) and dried over Na2SO4. PorFe is afforded as an orange-purple solid after column chromatography (SiO2, CH2Cl2/MeOH (99:1 v/v)) purification (24 mg, 0.024 mmol, 58%). UV/Vis (CH2 Cl2): 𝜆max (𝜀, mM−1⋅cm−1) = 274 (31.6), 341 (32.0), 417 (87.2), 499 (11.2), 506 (11.0), 577 (5.4), 640 (3.0). ESI-HRMS: m/z calculated for a chemical formula C57H45FeN5O7 [M–Cl]+ = 967.2619, found 967.2664.
Synthesis of the porFe-bpyRu supramolecular complex and the corresponding reference complex porFe.
3. Results and discussion
3.1. Synthesis
As shown in Scheme 2, dyad porFe-bpyRu and iron-porphyrin porFe share the same porphyrin platform 4. This synthetic intermediate was prepared in an acid-catalyzed condensation of four pyrroles, one para-nitrobenzaldehyde and three di-ortho-methoxybenzaldehyde (2) followed by an oxidation of the obtained macrocyle using dichloro-5,6-dicyano-p-benzoquinone (DDQ), then by a reduction of the nitro group into amino using tin chloride in acidic conditions. Aldehyde 2 was obtained in two steps from resorcinol by first methylating the hydroxy groups and then a lithium-mediated formylation reaction of the aromatic carbon in position 2 takes places. The coupling between porphyrin 4 and one of the bipyridines of the bpyRu moiety was performed using modified bipyridine 5 bearing a carboxyl group in position 4 that was prepared by a SeO2 oxidation of one of the methyl groups in the 4,4′-dimethyl-2,2′-bipyridyl. After an activation of the carboxyl group using thionyl chloride, 5 reacts with the aniline group in porphyrin 4 to yield the amide-bridged porphyrin-bipyridine ligand 7. The bipyridine in 7 can displace the two chlorides of the dichlorobis(bipyridine)ruthenium 6 to form the porphyrin-bpyRu complex (9) that leads, after complexation of the iron, to the aimed dyad porFe-bpyRu. Reference iron-porphyrin porFe was prepared using a similar synthetic scheme and by replacing the bipyridine 5 with benzoyl chloride.
Cyclic voltammograms (CV) of the supramolecular and reference complexes at concentrations of 0.5 mM in dimethylformamde with 100 mM tetra-N-butylammonium hexafluorophosphate (a) under Ar and (b) under CO2 and 5.5 mM H2O. Inset in (b) shows magnification of the onset of catalysis.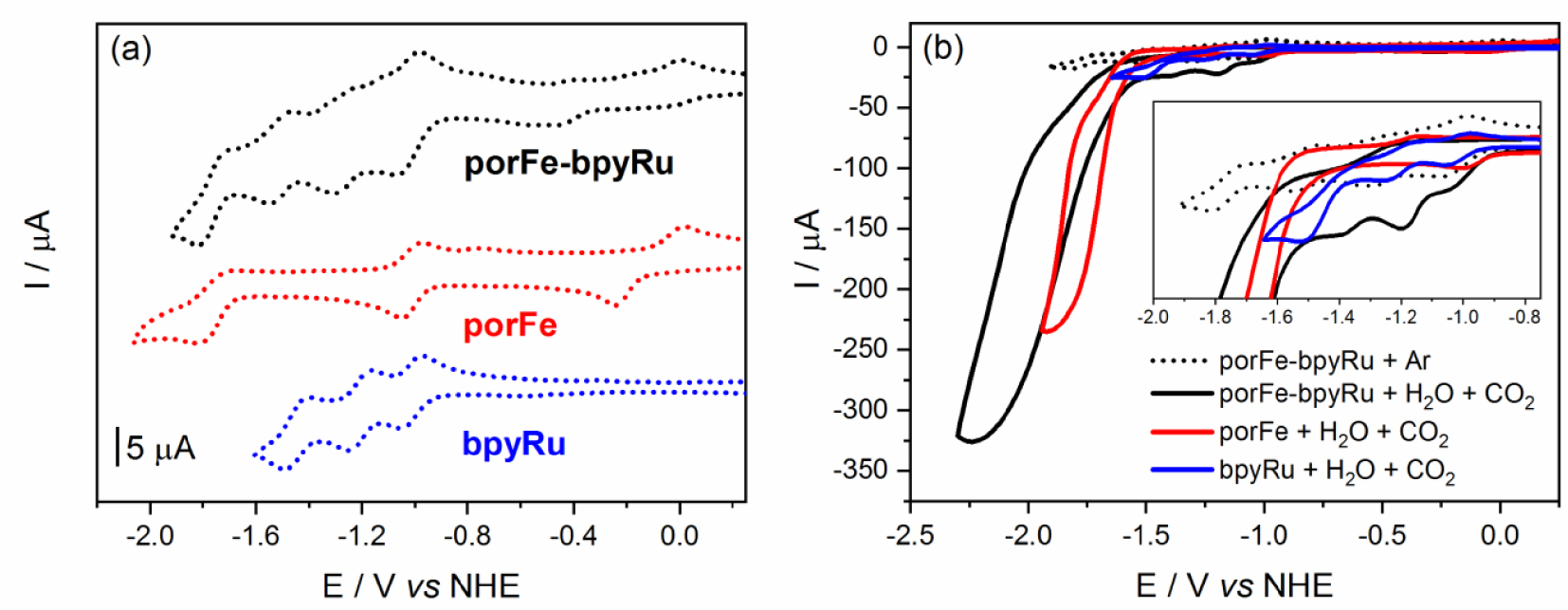
Electrochemical potentials of the formal redox couples involved in the supramolecular and reference complexes, referenced vs NHE in dimethylformamide with 100 mM tetra-N-butylammonium hexafluorophosphate
| Complex | Fe(III/II) | Fe(II/I) | Fe(I/0) | Ru(II/I) | Ru(I/0) | Ru(0/-I) |
|---|---|---|---|---|---|---|
| porFe-bpyRu | −0.23 | −1.03 | −1.77 | −1.03 | −1.27 | −1.51 |
| porFe | −0.12 | −1.02 | −1.76 | — | — | — |
| bpyRu | — | — | — | −1.01 | −1.21 | −1.45 |
3.2. Electrochemical characterization
Cyclic voltammetry (CV) of model compound porFe in argon-degassed dimethylformamide (DMF) containing 100 mM of tetra-N-butylammonium hexafluorophosphate (TBAP) shows three reversible redox waves corresponding to the formal Fe(III/II), Fe(II/I), and Fe(I/0) couples (Figure 1(a), Table 1). As expected, due to the presence of the methoxyl groups, these three waves are shifted to more negative potentials compared to those of the previously reported non-functionalized iron-tetraphenyl-porphyrin (FeTPP) [18]. Three successive reduction waves are also observed for bpyRu complex which were previously reported to be mainly centered on the bipyridine ligands [33]. The CV of porFe-bpyRu displays a combination of the porFe and bpyRu reduction waves. A noticeable anodic shift of about 100 mV is observed for the Fe(III/II) couple in the dyad compared to the reference complex but no significant shifts are observed for the Fe(II/I) and Fe(I/0) redox couples. Similarly, the bpy-based reduction waves of the Ru moiety in the dyad showed minimal anodic shifts of around 5 mV. These are all indicative that the amide linker does not establish a strong electronic communication between the two moieties. The redox couples Fe(II/I) and Ru(II/I) overlap in the dyad as a two-electron reduction wave, which is indicative that the one-electron reduced form of the PS can reduce both the Fe(II) and Fe(III) states of the catalyst in the dyad with thermodynamic driving force of 0 and −800 meV, respectively. However, this same photoreductant would have an uphill penalty of +760 meV to reduce the iron-porphyrin moiety to its catalytically active Fe(0) form.
(a) Photocatalytic curves for the production of CO (solid lines) and H2 (dotted lines) during the irradiation of the following in CO2-saturated DMF/H2O (9:1 v/v): 2 μM porFe-bpyRu + 50 μM bpyRu + 50 mM BIH (black); 2 μM porFe + 50 μM bpyRu + 50 mM BIH (red). (b) Comparison of the photocatalytic performance in terms of selectivity towards CO production (columns) and turnover numbers (TON).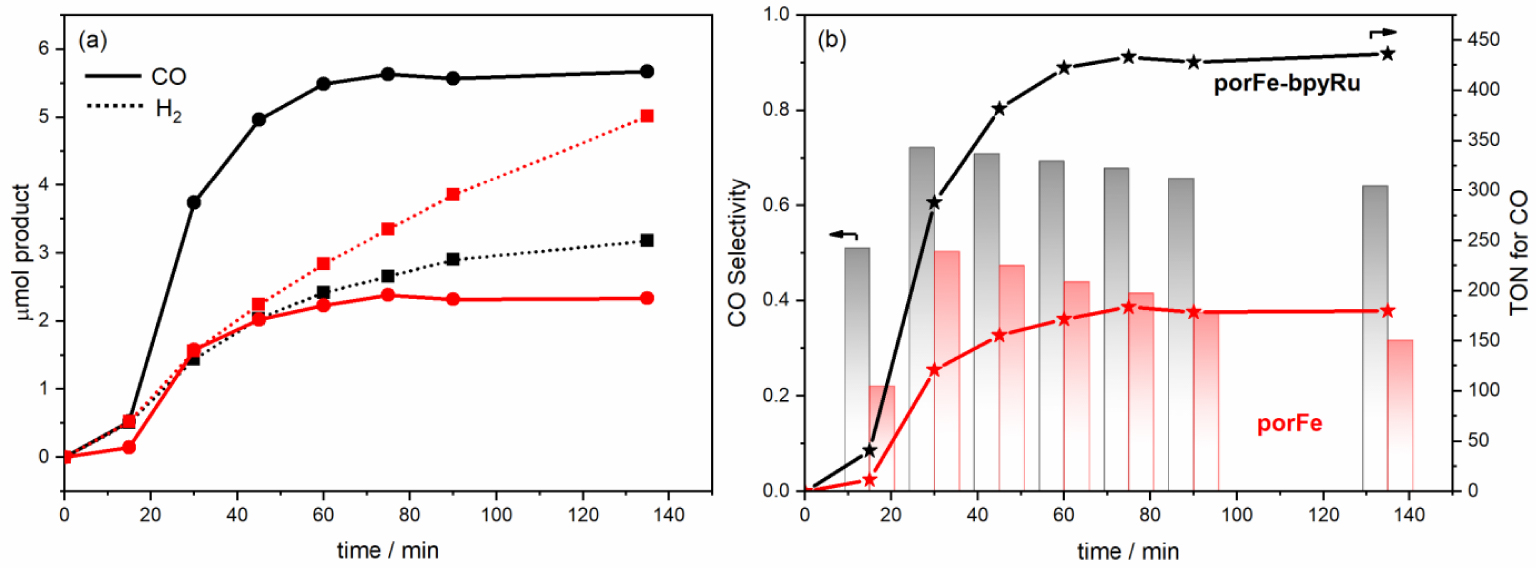
Summary of photocatalytic activities showing the production of CO and H2 after irradiation for 135 min of a CO2-saturated DMF/H2O (9:1 v/v) solution containing the photosensitzer–catalyst system investigqted in this study
| Photosensitizer | Catalyst | μmol CO | μmol H2 | TON CO | TON H2 | SelectivityCO |
|---|---|---|---|---|---|---|
| 2 μM porFe-bpyRu | n.d. | n.d. | — | — | — | |
| 2 μM porFe-bpyRu | 50 μM bpyRu | 5.7 | 3.2 | 436 | 244 | 64% |
| 2 μM porFe | 50 μM bpyRu | 2.3 | 5.0 | 180 | 386 | 32% |
| 2 μM porFe | — | n.d. | n.d. | — | — | — |
| — | 50 μM bpyRu | 1.4 | 0.5 | 4.5 | 1.6 | 74% |
n.d. = not detectable.
When the CVs were performed under a CO2 atmosphere and in presence of water as a proton source (Figure 1(b)), porFe and porFe-bpyRu displayed a typical catalytic current corresponding to CO2 reduction at the last reduction wave (Fe(I/0) redox couple) with a significantly higher current intensity in the case of porFe-bpyRu. More interestingly, the onset potential of catalysis started at a significantly more positive potential in the dyad (Figure 1(b) inset), almost at the Fe(II/I) and Ru(II/I) waves with a smaller current intensity increase preceding a much higher current observed after −1.5 V. A control experiment with bpyRu in the same conditions shows minimal current intensity increase and only at the third bpy-reduction wave, confirming that the iron-porphyrin catalyst is responsible for the observed catalytic wave in the porFe-bpyRu dyad. The early onset potential for the first catalytic regime of the dyad takes place right after the electro-generation of the Fe(I) species that is accompanied concomitantly with addition of one electron on the bipyridine holding the PS and the Cat. This proposal is based on the fact that the bridging bipyridine is chemically modified with a C-terminated amido group and is under the influence of two metallic cations, rendering it the privilege locus for the addition of an electron as compare to the other two bipyridine ligands. This species can therefore be looked at as an Fe(I) coupled to an anion radical species that can trigger the catalytic activity.
(a) Absorption spectra of of the supramoleclar porFe-bpyRu dyad in comparison with the reference complexes in ACN/H2O (6:4 v/v). Inset shows magnification of the Q bands region. (b) Transient emission spectra of porFe-bpyRu in DMF upon laser excitation at 460 nm. Inset shows kinetics at 650 nm revealing emission lifetime of 18 ns. Similar transient emission was recorded in ACN/H2O (Figure S2).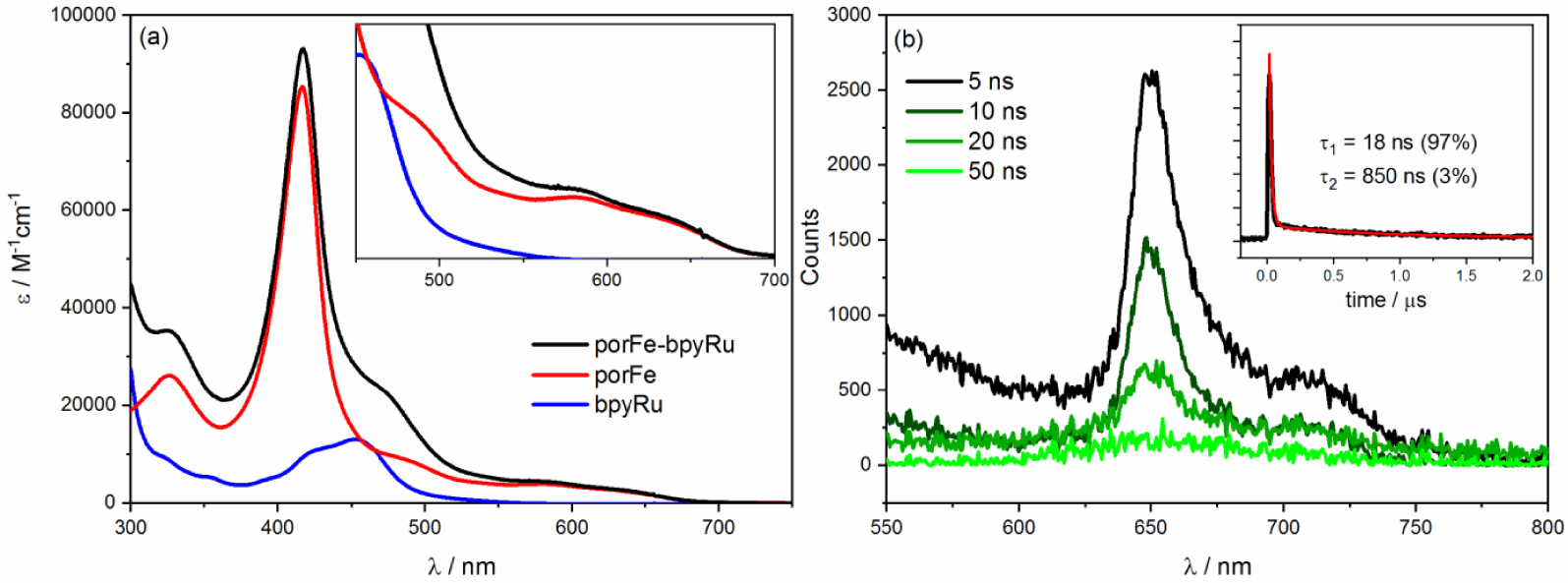
3.3. Photocatalytic evaluation
Motivated by the promising results from the electrochemical measurements, we undertook the evaluation of the photocatalytic performance of the supramolecular dyad toward CO2 reduction. An initial experiment was performed by irradiating with a blue LED source a solution containing 2 μM porFe-bpyRu as photo-catalyst and 50 mM BIH as sacrificial electron donor in DMF/H2O (9:1 v/v). Unfortunately, the gas chromatography analysis of the reaction vessel headspace was not able to detect any gas product. Similarly, no detectable products were observed for a solution containing only 2 μM porFe and 50 mM BIH. In the same conditions but in a bimolecular configuration using 2 μM porFe as catalyst and 50 μM bpyRu as PS, the reaction produces 5.0 μmol of H2 as the major product and 2.3 μmol of CO (Figure 2(a), Table 2). However, in a hybrid configuration where exogenous bpyRu (50 μM) in combination with 2 μM porFe-bpyRu were used, 5.7 μmol CO and 3.2 μmol H2 were produced after 135 min of irradiation (Figure 2(a), Table 2). To ascertain that the photocatalytic activity comes mainly from the dyad, a control experiment was performed with 50 μM of bpyRu and in absence of the dyad. Under these conditions, much smaller amounts of CO (1.4 μmol) and H2 (0.5 μmol) are produced (Figure S1), possibly due the minor CO2 reduction activity of the Ru bisbipyridyl, a degradation product of bpyRu that was shown to form in absence of the electron acceptor, in this case the Cat, and exhibit some CO2 reduction activity [34, 35, 36]. In the same catalytic conditions, the porFe-bpyRu dyad displays a higher selectivity ( ∼64%) for CO production (Figure 2(b)) during the course of photocatalysis, while in the case of porFe the selectivity was below 40%. This improved selectivity exhibited by the supramolecular porFe-bpyRu dyad also translates in a significantly higher TON of ∼440 compared to that obtained with the porFe as catalyst (TON = 180). Even though the porFe-bpyRu dyad didn’t fulfill the initially intended photo-catalyst role, it surprisingly improves the selectivity and TON when used as a catalyst in presence of exogenous PS.
UV-Vis spectral changes when Asc is added to a solution of (a) porFe-bpyRu or (b) porFe in Ar-saturated ACN/H2O (6:4 v/v) and its similarity to (c) the spectral evolution during the 1st reduction FeIII→FeII of porFe in ACN using spectroelectrochemistry, SEC (more details in Figure S3).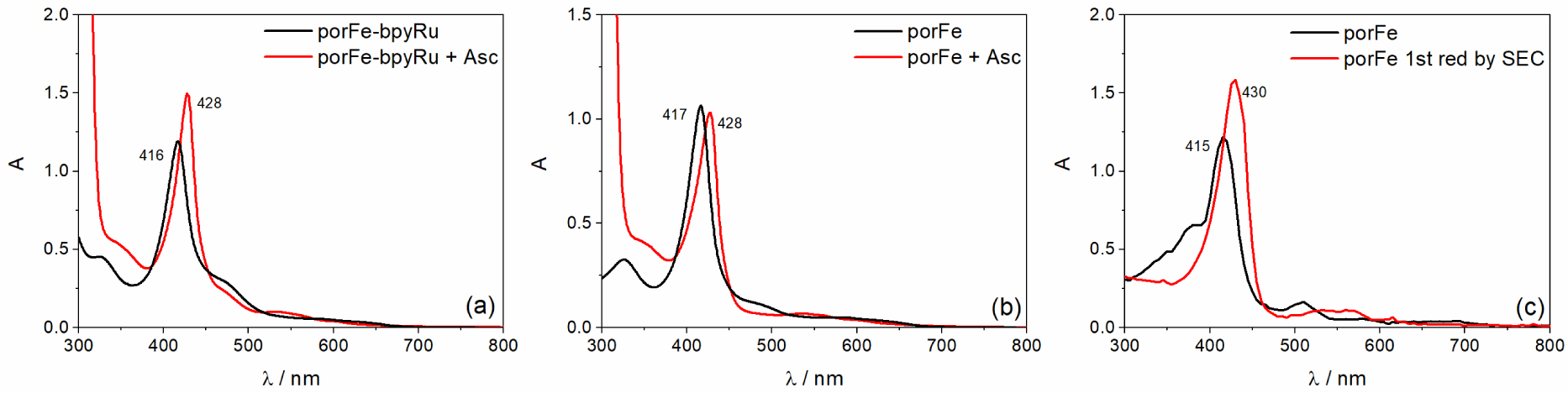
3.4. Photophysical and mechanistic analyses
The intriguing and peculiar photocatalytic performance of the porFe-bpyRu dyad merited further photophysical investigation. The absorption spectrum of the dyad (Figure 3(a)) consists of combination of spectral features of the mononuclear complexes porFe and bpyRu: a Soret band at 417 nm and Q bands at 490, 585, and 635 nm characteristic of the iron porphyrin catalyst and the 3MLCT band of Ru PS as broad shoulder at 460 nm. The absence of significant spectral changes between the porFe-bpyRu dyad and the mixture of porFe and bpyRu confirm the previous observation that the amide linkage does not alter the electronic properties of the individual components in the ground state. Upon excitation at 460 nm to excite mainly the Ru PS (a similar excitation domain of the blue LED lamp employed in the photocatalytic experiments), only emission from the porFe could be detected (Figure 3(b); narrow band at 650 nm with a shoulder at 700 nm). The absence of emission from the Ru excited state, expected as a broad band at 610 nm, is indicative of ultrafast quenching of the Ru excited state. As expected, the lifetime of the porFe fluorescence is shorter than the time resolution (20 ns) of the ns laser flash photolysis apparatus. No transient absorption peaks were observed (even at the shortest time scale of the experiment) indicating the absence of any long-lived exited triplet Ru species. These observations are attributed to (a) an efficient energy transfer from the excited singlet Ru species to singlet porphyrin, which is then significantly quenched due to the presence of the heavy Fe metal center, and (b) competing photon absorption ( ∼50%) between the PS and the Cat.
Upon addition of sodium ascorbate (Asc) as a reversible electron donor for the photophysical measurements, a noticeable red shift in the Soret band (416 nm → 428 nm) of the ground state spectrum of porFe-bpyRu was observed in Ar-saturated ACN/H2O solutions (Figure 4(a)). A similar shift was observed for the reference porFe catalyst (Figure 4(b)), indicating that there is a dark chemical reaction occurring between the Asc and the catalyst. Spectroelectrochemical measurement on the reference complex porFe shows a similar characteristic red-shift of the Soret band for the Fe(II) species (Figures 4(c) and S3–S4) which indicates that the observed red shift of the Soret band is due to the reduction of Fe(III) to Fe(II). A similar reduction of Fe(III) is occurring when BIH was used as sacrificial electron donor. The electron paramagnetic resonance (EPR) spectra of porFe-bpyRu (Figure S5) shows the disappearance of the high spin Fe(III) signal upon addition of the electron donor in the dark. We also investigated the photophysical properties of the singly reduced Fe(II) state of porFe-bpyRu upon excitation at 460 nm. Here too, no transient absorption peaks were observed. Hence, revealing again an efficient quenching of any excited states. Accordingly, the absence of long-lived excited species and the lack of light-induced electron transfer processes leading to reduced species for redox reactions, explain why the porFe-bpyRu dyad in the presence of BIH did not show any photocatalytic CO2 reduction activity under continuous illumination. Therefore, these results highlight that linking the bpyRu PS and porFe catalyst through the single amido function was disadvantageous in the scenario of eliminating the diffusion control limits in the photo-driven electron transfer processes and catalysis. Reasons behind this in our particular case probably originate from the competition for photon absorption between the catalyst and the photosensitizer, a mismatch of emission and absorption properties of both constitutive chromophores and finally the short distance between the two modules which promote an efficient energy transfer and quenching of the excited states.
(a–c) Optical transient absorption (OTA) spectra in three time domains showing the spectral evolution of each species during single flash photolysis of a solution containing 13 μM porFe-bpyRu dyad, 31 μM bpyRu, and 100 mM Asc in ACN/H2O (6:4 v/v) (laser excitation at 460 nm, 10 mJ per pulse). (d) Globally-fitted transient kinetics at some wavelengths with fitting parameters used to plot the (e) decay-associated difference spectra. (f) Comparison of the OTA decay spectra corresponding to (Fe(I)–Fe(II)) with that obtained from spectroelectrochemistry (SEC) in Figure S3 (in the absence of H2O).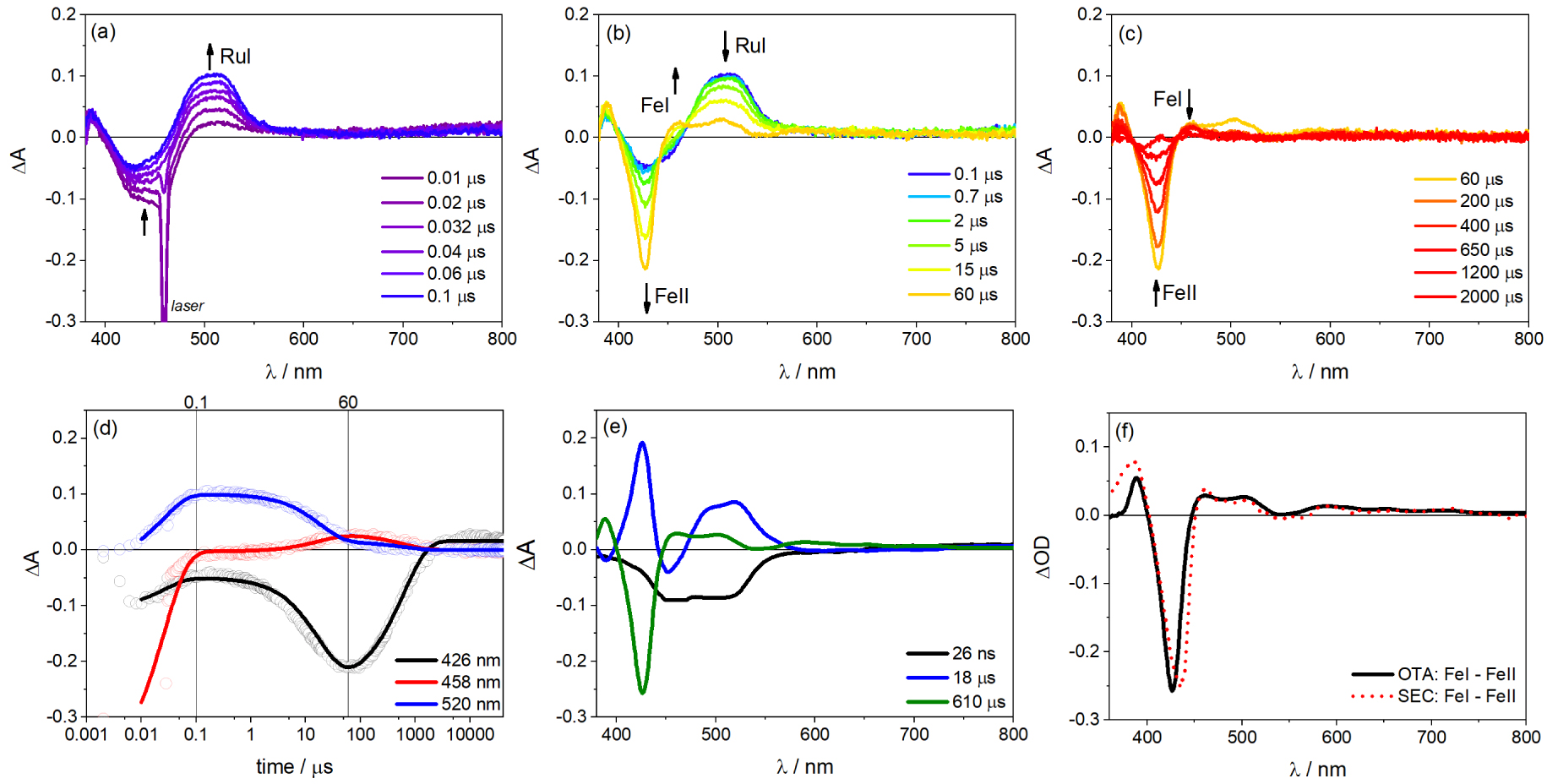
As it turns out, the photoredox-catalyst molecular dyad did not lead to the expected photo-induced charge separation and charge accumulation towards the catalyst. We then pursue the photophysical investigation by the addition of an exogenous ruthenium (II) trisbipyridine photosensitizer (bpyRu) together with Asc a reversible electron donor considering the porFe-bpyRu as basically a dyad with an extinct photosensitizer unit but with a functioning porFe catalyst unit. In this configuration, photo-induced electron transfer steps are observed as summarized in Figures 5(a)–(c). At short time domain (0–100 ns in Figure 5(a)), upon laser excitation at 460 nm and formation of the excited state Ru∗ (bleaching of Ru(II) MLCT at 450 nm), there is a simultaneous formation of the one-electron reduced form of the PS, formal Ru(I) species, characterized by an absorption at 520 nm, and oxidized Asc⋅+ (absorbing at 360 nm). Global fitting of the kinetic traces with a triexponential function [a1 exp(−t∕𝜏1) + a2 exp(−t∕𝜏2) + a3 exp(−t∕𝜏3) + c] gave satisfactory fits (Figure 5(d)). This first photo-induced electron transfer event (Ru(II) + Asc → Ru(I) + Asc⋅+) occurs with an apparent time constant of 26 ns with 100 mM Asc. In the proceeding time scale (100 ns to 60 μs in Figure 5(b)), the Ru(I) species decays to give a new spectral feature at 458 nm, corresponding to the reduction from the Fe(II) to Fe(I) oxidation state of the porphyrin catalyst moiety of the porFe-bpyRu dyad. This is confirmed by two control experiments: (a) similar transient absorption changes are observed for the solution containing the reference complexes porFe, bpyRu and Asc in the similar time window (Figure S6) and (b) a similar differential spectrum is observed during spectroelectrochemical measurement of the reference complex porFe when going from the Fe(II) to the Fe(I) oxidation states (Figures 5(e), (f) and S3). This second electron transfer event (Ru(I) + Fe(II) → Ru(II) + Fe(I)) occurs with a diffusion-limited second order rate constant of 7.9 × 109 M−1⋅s−1 estimated from the apparent time constant of 18 μs and formation of 7.1 μM Fe(I). Finally, at longer time scale (>60 μs in Figure 5(c)), this Fe(I) species decays back to the Fe(II) state after charge recombination with oxidized Asc⋅+ (Fe(I) + Asc⋅+→ Fe(II) + Asc) with a time constant of 610 μs.
These photophysical investigations have shown that when the porFe-bpyRu dyad is assimilated as only the catalyst in the presence of exogenous ruthenium (II) trisbipyridine photosensitizer and a sacrificial electron donor, three electrons can be accumulated on the catalyst to form the Fe(I)–Ru(II) species: one electron coming from a dark reaction with the electron donor and two electrons coming from the photo-induced Ru(I) reductant. These first three electron transfer events are similarly envisioned as the initial steps in the proposed photocatalytic cycle in Figure 6. However, from the CV of porFe catalyst (Figure 1(b)) it is clear that the Fe(0) oxidation state must be reached before the catalytic reduction of CO2 can proceed. With this thermodynamic constraint, the Ru(I) photo-reductant faces an uphill thermodynamic penalty of +760 meV to proceed. An alternative route may come from the non-innocent reducing radical BI⋅ resulting from the first electron donor step of BIH (E = −1.39 V vs NHE [37] ensuing a less positive 𝛥G ∼ +370 meV for this reaction), as we and others have previously reported [38, 39, 40]. However, an interesting feature of the porFe-bpyRu dyad is the possibility for the appended ruthenium (II) trisbipyridine to act as an electron reservoir, as suggested by the earlier onset of catalysis (Figure 1(b)) occurring at a potential corresponding to the Fe(II/I) and Ru(II/I) couples. Since there is only a minimal driving force for the exogenous Ru(I) photo-reductant to reduce the appended Ru moiety in the dyad (𝛥G ∼ +20 meV), we hypothesize that this occurs to form a Fe(I)–Ru(I) state of the dyad: Fe(I)–Ru(II) + Ru(I) → Fe(I)–Ru(I) + Ru(II). The formal Fe(I)–Ru(I) state can then be regarded as an Fe(I) species backed up by an additional electron with the same reducing power probably delocalized on the bipyridine extending onto the porphyrin macrocycle. Such a reduced species may then start the two-electron activation of CO2 at a lower overpotential than the classic Fe(0) active form. The lower thermodynamic penalty along this route can account for the higher performances of the porFe-bpyRu dyad, in terms of TON and CO selectivity, than the porFe catalyst in the presence of the same amount of exogenous ruthenium (II) trisbipyridine as photosensitizer. As a consequence, even though the bpyRu photoredox unit in the porFe-bpyRu dyad is shut down due to fast deleterious energy transfer processes, it assists in lowering the overpotential for CO2 reduction by acting as a reservoir for providing an extra reducing power to ignite the catalysis at the formal Fe(I) state of the catalyst.
Proposed photocatalytic cycle involving the supramolecular porFe-bpyRu dyad and exogenous bpyRu PS with BIH as sacrificial electron donor. Dashed arrows show competing minor pathways.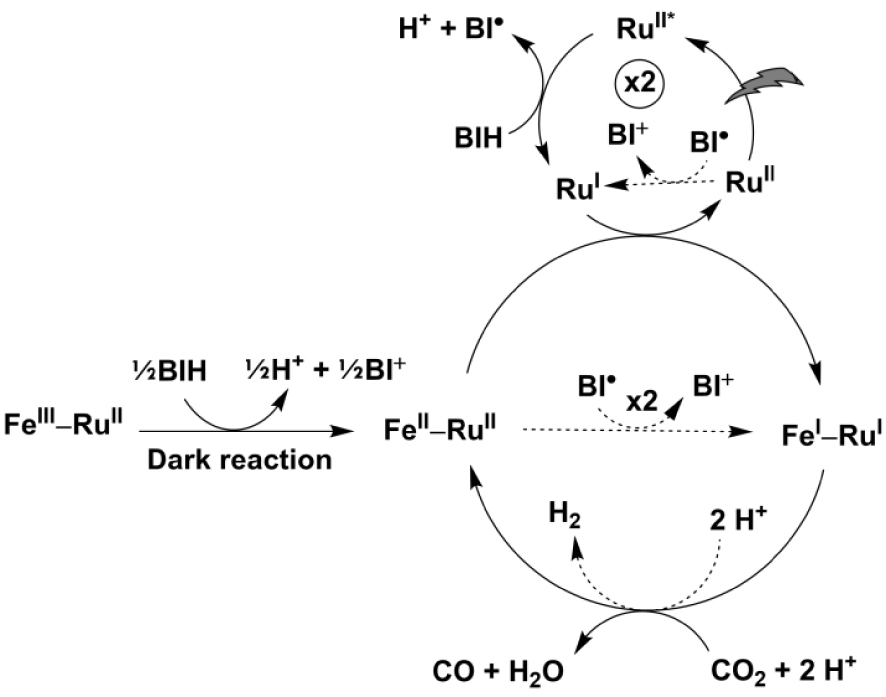
4. Conclusion
We have synthetized a new photoredox-catalyst couple, the porFe-bpyRu, where a ruthenium (II) trisbipyridine photosensitizer bpyRu is covalently attached to an iron porphyrin catalyst porFe through an amide linker. We found that this molecular dyad did not lead to the expected photo-induced charge separation and charge accumulation towards the catalyst due to a non-productive quenching of the excited state of the photosensitizer by energy transfer to the catalyst. A comparative electrochemical study points to a change in the electrocatalytic pattern of the porFe-bpyRu dyad compared to the porFe catalyst. Indeed, for the porFe, the catalytic wave is observed at the Fe(0) state while for the porFe-bpyRu dyad, a catalytic wave starts at the formal Ru(I)–Fe(I) species at some 700 mV lower overpotential than the porFe catalyst. This species can be best described as a formal Fe(I) species at the catalyst in interaction with a radical anion on the bipyridine ligand holding the porFe. Interestingly though, in presence of exogenous bpyRu photosensitizer, the porFe-bpyRu dyad presents a significant enhancement of the turnover number and CO2-to-CO selectivity of the catalysis compared to the porFe catalyst analogue under the same photocatalytic conditions. Reasons behind this probably come from the role of the bpyRu unit in the porFe-bpyRu dyad that acts an electron reservoir to power the photocatalytic activity. DFT calculations are underway to provide more insights in the functioning of such dyad.
Acknowledgements
This work has been supported by the French National Research Agency (ANR-19-CE05-0020-02, LOCO). We thank CNRS, CEA Saclay, LABEX CHARMMMAT, ICMMO and University Paris-Saclay for the financial support. This research was also funded by the General Secretariat for Research and Technology (GSRT) and Hellenic Foundation for Research and Innovation (HFRI; project code: 508). In addition, this research has been co-financed by the European Commission’s Seventh Framework Program (FP7/2007-2013) under grant agreement no. 229927 (FP7-REGPOT-2008-1, Project BIO-SOLENUTI).
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.104 or from the author.
It contains transient emission, transient absorption, EPR, spectroelectrochemistry, NMR and ESI-HRMS spectra.