

 CC-BY 4.0
CC-BY 4.0
1. Introduction
Guanidines, due to their wealth of catalytic functionalities, occupy a prominent position in the repertoire of organocatalysts [1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, 12]. They carry out all four basic chemical functions: free guanidines are capable of acting as Brønsted or Lewis bases, while their corresponding conjugated acids (guanidinium cation) can serve as Brønsted or Lewis acids (Scheme 1). These activation modes enable the triaza (CN3) core to be an efficient tool in organic transformations, such as Michael reaction [13, 14, 15, 16], Mannich reaction [17, 18, 19, 20], aldol reaction [21, 22, 23], ring-opening polymerization [24, 25, 26, 27, 28], epoxidation reaction [29, 30, 31, 32] and many others. Guanidines have long been known as strong organic bases [33].
Schematic representation of functionalities in (a) free guanidine and (b) guanidinium cation.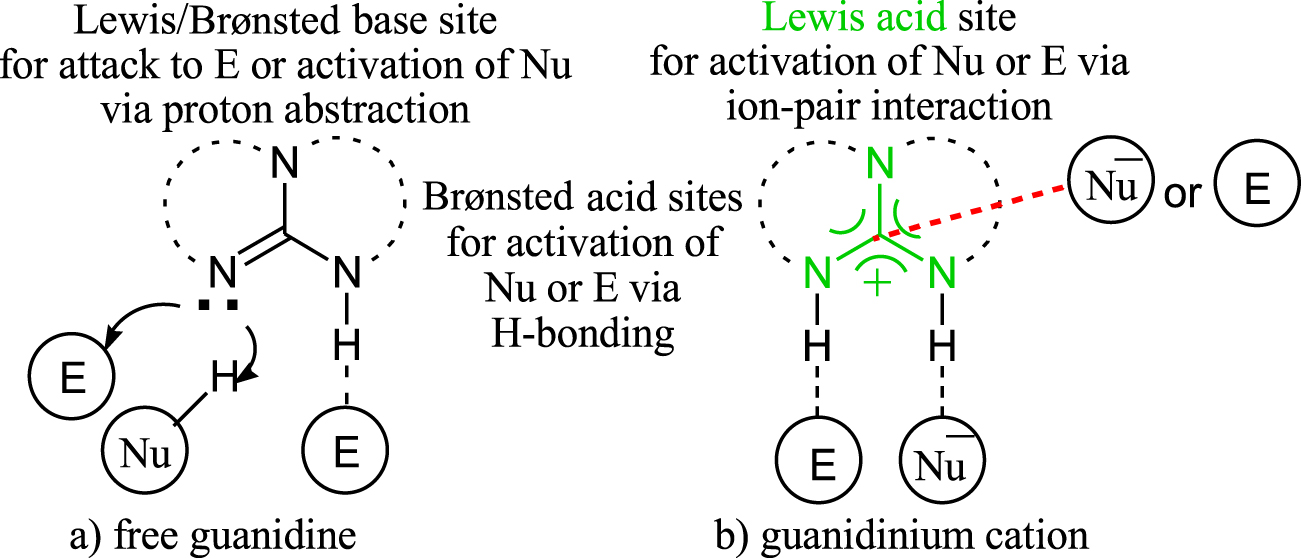
However, knowledge about the Lewis acidity feature of guanidinium cation is scarce. The guanidinium cation is produced when a free guanidine abstracts a proton from a substrate through the Brønsted-base mechanism (Scheme 1b). Through NMR supported studies, Olah and co-workers have demonstrated that the Y-aromaticity of the CN3 system provides a considerable driving force for the deprotonation of guanidine scaffolds. The overall process results in a guanidinium salt, featuring a Lewis base–acid complex formed by electrostatic interaction between the central carbon atom of CN3 (Lewis acid) and the anionic nucleophile or non-bonding electron of an electrophile (Lewis base). The positively charged Lewis acidic center will polarize the ligated partners, which leads to the enhancement of their intrinsic reactivity. This process is referred to as ‘organic Lewis acid catalysis’, and it distinguishes guanidine frameworks from heterocyclic compounds because the term ‘Lewis acid catalysis’ is almost exclusively used for metal salts or metal complexes [34]. Experimental and computational studies demonstrated that the Lewis acid interaction not only prevents deactivation but also improves the nucleophilicity of a Lewis base by increasing the density of the negative charge on it as a result of drawing the electron density from the Lewis acid [35, 37].
It has been claimed that the guanidinium cation serves as a Lewis acid catalyst in a few number of chemical processes such as ring-opening reaction [24, 37], cleavage of phosphodiester and DNA [38] and decomposition of alkyl formate [39, 40]. However, these reports are not supported by theoretical evidence. The density functional theory (DFT) calculation has been recognized as a reliable method to shed light on this mechanism and the quality of activation modes in organocatalyst reactions [41]. However, the computational investigation has been conducted only in a handful of cases where the guanidinium species contribute to reaction through the Lewis acid activating mode (Scheme 2) [16, 42, 43, 44, 45, 46, 47, 48, 49, 50].
This shortcoming in the literature has been attributed to the difficulties involved in establishing theoretical support. Accordingly, in 2012, Wong and co-workers reported a pioneering study on the characterization of the bifunctional Lewis–Brønsted acid activation mode (Scheme 2a) under the catalytic activity of bicyclic guanidine 1,4,6-triazabicyclo[3.3.0]oct-4-ene (TBO) [16].
The calculation revealed the Lewis acid–base complexation between guanidinium and thiolate during the asymmetric thio-Michael reaction. Later, the same group of researchers extended their study to a broader range of substrates [42, 43, 44, 45, 46, 47, 48, 49]. In 2015, the Wong group demonstrated that the unconventional bifunctional Brønsted–Lewis acid activation mode performs a catalytic function in the isomerization reaction of alk-3-ynoate (Scheme 2b) [50].
Herein, we report a one-pot, four-component reaction leading to the synthesis of tricyclic guanidine, containing the unprecedented heterocyclic core, namely pyrido[4,3-d]pyrimido[1,2-a]pyrimidine (Scheme 2c). DFT calculations were conducted to investigate the mechanism and activation modes of the CN3 functional group in bicyclic guanidine (precursor). Among the several activation modes, the role of anion receptor in Lewis acid interactions stands out. The calculation revealed that the Lewis acid–base complexation between the strongly positively charged central carbon atom and the anionic sulfur atom has an important role in thioamidation and N-Michael addition steps.
2. Experimental section
2.1. General information
Melting points were measured on an Electrothermal 9100 apparatus. Infrared (IR) spectra were recorded as KBr pellets on a NICOLET FT-IR 100 spectrometer. 1H NMR (300 and 500 MHz) and 13C NMR (75 and 100 MHz) spectra were obtained using Bruker DRX-300 AVANCE and Bruker DRX-500 AVANCE spectrometers. All nuclear magnetic resonance (NMR) spectra were recorded in DMSO-d6 at room temperature. Chemical shifts are reported in parts per million (δ) downfield from an internal tetramethylsilane reference. Coupling constants (J values) are reported in hertz (Hz), and spin multiplicities are indicated by the following symbols: s (singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Elemental analyses for C, H and N (CHN) were performed using a Heraeus CHN–O–Rapid analyzer. Mass spectra were recorded on a FINNIGAN-MATT 8430 mass spectrometer operating at an ionization potential of 70 eV. The Cartesian coordinates of all optimized structures are separately attached as .xyz files.
Lewis acidity of guanidinium cations.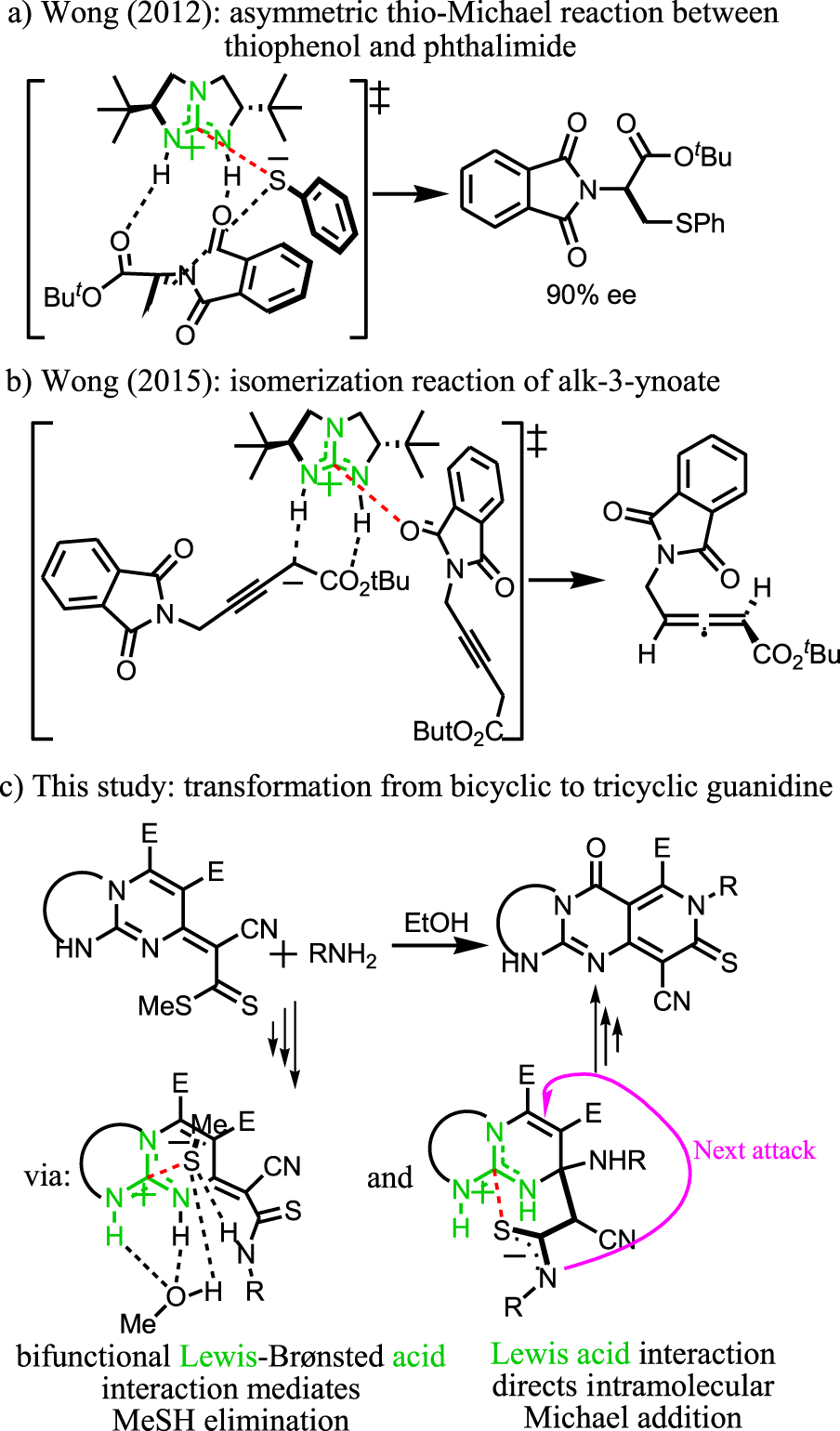
2.2. General procedure for the synthesis of compounds 5a-m
To a solution of the corresponding cyclic thiourea 1 (1 mmol) in DMF (1.0 mL) was added 2-di(methylsulfanyl) methylene malononitrile 2 (0.170 g, 1 mmol). The reaction mixture was stirred for 3 h at 120 °C. Afterward, a solution of dialkyl acetylenedicarboxylates 3 (1 mmol) in EtOH (2.0 mL) was added dropwise to the reaction mixture at room temperature. After completing the addition, the stirring continued for another 20 min. Then, the corresponding N-nucleophiles 4 (1.2 eq) were added to the mixture. After stirring for 1 h at room temperature, the solvent was evaporated by a rotary evaporator and the residue was washed by EtOH to yield the desired adducts 5a-m.
Methyl 8-amino-10-cyano-6-oxo-9-thioxo-1,3,4,6, 8,9-hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5a). Yellow powder, m.p > 300 °C, 0.259 g, yield: 78%. IR (KBr)(νmax, cm−1): 3336 and 3295 (NH2 and NH), 2221 (CN), 1746 (CO2Me), 1693 (C =O), 1614 (C =N), 1578 (C =C), 1295 (C =S), 1084 (C–O). Anal. Calcd. for C13H12N6O3S (332.33): C, 46.98; H, 3.64; N, 25.29%. Found C, 46.97; H, 3.64; N, 25.31. 1H NMR (300 MHz, DMSO-d6): 1.95 (2H, m, CH2), 3.30–3.34 (2H, m, CH2N), 3.81 (2H, t, 3JHH = 4.8 Hz, CH2NH), 3.91 (3H, s, CO2Me), 6.55 (2H, s, NH2), 9.35 (1H, s, NH). 13C{1H} (75.0 MHz, DMSO-d6): 19.00, 39.33, 40.44, 58.83, 102.74, 102.94, 116.99, 147.14, 154.61, 156.57, 158.45, 160.69, 179.73. MS (EI, 70 eV): 332 (M+, 27), 288 (33), 259 (37), 227 (48), 190 (35), 149 (53), 120 (28), 83 (66), 58 (100).
Ethyl 8-amino-10-cyano-6-oxo-9-thioxo-1,3,4,6, 8,9-hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5b). Yellow powder, m.p > 300 °C, 0.266 g, yield: 77%. IR (KBr)(νmax, cm−1): 3290 and 3224 (NH2 and NH), 2216 (CN), 1751 (CO2Et), 1697 (C =O), 1607 (C =N), 1575 (C =C), 1283 (C =S), 1084 (C–O). Anal. Calcd. for C14H14N6O3S (346.36): C, 48.55; H, 4.07; N, 24.26%. Found C, 48.54; H, 4.05; N, 24.27. 1H NMR (300 MHz, DMSO-d6): 1.31 (3H, t, 3JHH = 7.8 Hz, OCH2CH3), 1.92–1.96 (2H, m, CH2), 3.31 (2H, m, CH2N), 3.77–3.81 (2H, m, CH2NH), 4.38 (2H, q, 3JHH = 7.8 Hz, OCH2CH3), 6.55 (2H, s, NH2), 9.32 (1H, s, NH). 13C{1H} (75.0 MHz, DMSO-d6): 14.07, 19.01, 39.33, 40.44, 63.09, 102.63, 102.91, 117.03, 147.30, 154.60, 156.60, 158.44, 160.10, 179.72. MS (EI, 70 eV): 316 (39), 302 (31), 253 (45), 191 (59), 147 (37), 97 (55), 57 (100).
Methyl 10-cyano-6-oxo-9-thioxo-1,3,4,6,8,9- hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5c). Yellow powder, m.p > 300 °C, 0.238 g, yield: 75%. IR (KBr)(νmax, cm−1): 3224 (NH), 2217 (CN), 1748 (CO2M2), 1690 (C =O), 1612 (C =N), 1570 (C =C), 1289 (C =S), 1089 (C–O). Anal. Calcd. for C13H11N5O3S (317.32): C, 49.21; H, 3.49; N, 22.07%. Found C, 49.23; H, 3.52; N, 22.06. 1H NMR (300 MHz, DMSO-d6): 1.99–2.03 (2H, m, CH2), 3.37–3.41 (2H, m, CH2N), 3.47 (3H, s, CO2Me), 4.27–4.31 (2H, m, CH2NH), 8.46 (1H, brs, NH), 9.34 (1H, brs, NH). 13C{1H} (100.0 MHz, DMSO-d6): 18.58, 44.48, 51.48, 88.96, 93.21, 112.64, 147.14, 147.74, 152.32, 161.32, 163.57, 179.17. MS (EI, 70 eV): 279 (18), 251 (35), 222 (44), 185 (53), 149 (100), 139 (19), 104 (55), 69 (57).
Methyl 8-amino-10-cyano-3,3-dimethyl-6-oxo-9- thioxo-1,3,4,6,8,9-hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5d). Yellow powder, m.p > 300 °C, 0.270 g, yield: 75%. IR (KBr)(νmax, cm−1): 3462 and 3281 (NH2 and NH), 2223 (CN), 1762 (CO2Me), 1693 (C =O), 1614 (C =N), 1577 (C =C), 1294 (C =S), 1086 (C–O). Anal. Calcd. for C15H16N6O3S (360.39): C, 49.99; H, 4.47; N, 23.32%. Found C, 50.01; H, 4.45; N, 23.30. 1H NMR (300 MHz, DMSO-d6): 1.01 (6H, s, CMe2), 3.07 (2H, m, CH2N), 3.56 (2H, m, CH2NH), 3.91 (3H, s, CO2Me), 6.57 (2H, s, NH2), 9.39 (1H, s, NH). 13C{1H} (75.0 MHz, DMSO-d6): 23.72, 27.00, 49.98, 50.08, 53.85, 102.72, 103.13, 116.98, 147.15, 153.93, 156.60, 158.74, 160.10, 179.76. MS (EI, 70 eV): 360 (M+, 16), 255 (27), 217 (23), 189 (39), 164 (41), 133 (53), 105 (62), 94 (48), 79 (45), 69 (52), 55 (100).
Ethyl 8-amino-10-cyano-3,3-dimethyl-6-oxo-9- thioxo-1,3,4,6,8,9-hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5e). Yellow powder, m.p > 300 °C, 0.284 g, yield: 76%. IR (KBr)(νmax, cm−1): 3356 and 3293 (NH2 and NH), 2209 (CN), 1752 (CO2Et), 1697 (C =O), 1618 (C =N), 1575 (C =C), 1293 (C =S), 1083 (C–O). Anal. Calcd. for C16H18N6O3S (374.41): C, 51.33; H, 4.85; N, 22.45%. Found C, 51.30; H, 4.88; N, 22.43. 1H NMR (300 MHz, DMSO-d6): 1.03 (6H, s, CMe2), 1.34 (3H, t, 3JHH = 7.2 Hz, OCH2CH3), 3.08 (2H, m, CH2N), 3.58 (2H, m, CH2NH), 4.41 (2H, q, 3JHH = 7.2 Hz, OCH2CH3), 6.59 (2H, s, NH2), 9.39 (1H, s, NH). 13C{1H} (75.0 MHz, DMSO-d6): 14.07, 23.70, 27.03, 49.91, 50.11, 63.12, 102.61, 103.12, 117.02, 147.24, 153.93, 156.64, 158.73, 160.10, 179.75. MS (EI, 70 eV): 374 (M+, 19), 342 (17), 313 (34), 270 (42), 257 (27), 230 (19), 186 (21), 132 (29), 102 (35), 88 (100), 70 (67).
Methyl 10-cyano-3,3-dimethyl-6-oxo-9-thioxo-1, 3,4,6,8,9-hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5f). White powder, m.p > 300 °C, 0.266 g, yield: 77%. IR (KBr)(νmax, cm−1): 3262 (NH), 2212 (CN), 1755 (CO2Me), 1690 (C =O), 1614 (C =N), 1574 (C =C), 1289 (C =S), 1083 (C–O). Anal. Calcd. for C15H15N5O3S (345.37): C, 52.17; H, 4.38; N, 20.28%. Found C, 52.20; H, 4.39; N, 20.30. 1H NMR (500 MHz, DMSO-d6): 1.01 (6H, s, CMe2), 3.13 (2H, m, CH2N), 3.30 (3H, s, CO2Me), 4.06 (2H, m, CH2NH), 8.48 (1H, brs, NH), 9.11 (1H, brs, NH). 13C{1H} (100.0 MHz, DMSO-d6): 23.64, 27.14, 49.88, 51.67, 53.30, 99.73, 104.50, 115.02, 151.66, 152.55, 156.53, 158.09, 167.61, 179.02. MS (EI, 70 eV): 346 (M++1, 37), 314 (29), 275 (36), 233 (17), 191 (100), 153 (64), 125 (49), 111 (58), 58 (77).
Methyl 10-cyano-8-ethyl-3,3-dimethyl-6-oxo-9- thioxo-1,3,4,6,8,9-hexahydro-2H-pyrido[4,3-d] pyrimido[1,2-a]pyrimidine-7-carboxylate (5g). Yellow powder, m.p > 300 °C, 0.295 g, yield: 78%. IR (KBr)(νmax, cm−1): 3289 (NH), 2218 (CN), 1760 (CO2Me), 1687 (C =O), 1610 (C =N), 1571 (C =C), 1278 (C =S), 1089 (C–O). Anal. Calcd. for C17H19N5O3S (378.43): C, 54.68; H, 5.13; N, 18.75%. Found C, 54.67; H, 5.16; N, 18.74. 1H NMR (300 MHz, DMSO-d6): 1.00 (6H, s, CMe2), 1.27–1.28 (3H, m, NCH2CH3), 3.06 (2H, m, CH2N), 3.31 (3H, s, CO2Me), 3.59 (2H, m, CH2NH), 3.99–4.03 (2H, m, NCH2CH3), 8.88 (1H, brs, NH). 13C{1H} (300.0 MHz, DMSO-d6): 14.68, 23.69, 27.00, 49.61, 49.96, 50.00, 51.19, 104.08, 106.86, 117.46, 145.93, 154.00, 154.22, 157.91, 159.78, 179.72. MS (EI, 70 eV): 346 (24), 286 (39), 230 (47), 189 (49), 129 (58), 91 (100), 57 (63), 41 (66).
Methyl 10-cyano-3,3-dimethyl-6-oxo-8-phenyl-9- thioxo-1,3,4,6,8,9-hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5h). Yellow powder, m.p > 300 °C, 0.307 g, yield: 73%. IR (KBr)(νmax, cm−1): 3409 (NH), 2216 (CN), 1744 (CO2Me), 1695 (C =O), 1626 (C =N), 1572 (C =C), 1297 (C =S), 1105 (C–O). Anal. Calcd. for C21H19N5O3S (421.47): C, 59.85; H, 4.54; N, 16.62%. Found C, 59.86; H, 4.57; N, 16.63. 1H NMR (300 MHz, DMSO-d6): 1.02 (6H, s, CMe2), 3.10 (2H, m, CH2N), 3.56 (2H, m, CH2NH), 3.39 (3H, s, CO2Me), 7.20–7.24 (1H, m, CHpara of Ph), 7.46–7.50 (4H, m, 2CHmeta and 2CHortho of Ph), 9.54 (1H, s, NH). 13C{1H} (75.0 MHz, DMSO-d6): 23.69, 27.07, 50.06, 53.54, 103.70, 104.32, 117.14, 129.01, 129.34, 130.09, 139.53, 147.55, 154.13, 157.45, 159.14, 161.28, 181.63. MS (EI, 70 eV): 333 (22), 279 (44), 251 (36), 222 (32), 185 (54), 149 (100), 139 (64), 104 (62), 69 (75).
Methyl 10-cyano-3,3-dimethyl-8-(4- methylphenyl)-6-oxo-9-thioxo-1,3,4,6,8,9-hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5i). Yellow powder, m.p > 300 °C, 0.331 g, yield: 76%. IR (KBr)(νmax, cm-1): 3410 (NH), 2223 (CN), 1746 (CO2Me), 1698 (C =O), 1606 (C =N), 1570 (C =C), 1310 (C =S), 1104 (C–O). Anal. Calcd. for C22H21N5O3S (435.50): C, 60.68; H, 4.86; N, 16.08%. Found C, 60.66; H, 4.85; N, 16.07. 1H NMR (300 MHz, DMSO-d6): 1.05 (6H, s, CMe2), 2.39 (3H, s, Caryl–Me), 3.13 (2H, m, CH2N), 3.48 (2H, m, CH2NH), 3.59 (3H, s, CO2Me), 7.10–7.14 (2H, m, 2CH of Ar), 7.29–7.33 (2H, m, 2CH of Ar), 9.58 (1H, s, NH). 13C{1H} (75.0 MHz, DMSO-d6): 21.29, 23.71, 27.08, 50.10, 53.54, 103.61, 104.34, 117.12, 128.65, 129.79, 137.08, 139.82, 147.73, 154.13, 157.38, 159.13, 161.25, 181.83. MS (EI, 70 eV): 435 (M+, 28), 390 (35), 375 (15), 361 (13), 333 (17), 247 (17), 209 (28), 180 (57), 149 (100), 104 (62), 57 (44).
Methyl 8-(4-chlorophenyl)-10-cyano-3,3- dimethyl-6-oxo-9-thioxo-1,3,4,6,8,9-hexahydro-2H-pyrido[4,3-d]pyrimido[1,2-a]pyrimidine-7-carboxylate (5j). Yellow powder, m.p > 300 °C, 0.328 g, yield: 72%. IR (KBr)(νmax, cm−1): 3210 (NH), 2216 (CN), 1747 (CO2Me), 1690 (C =O), 1605 (C =N), 15777 (C =C), 1295 (C =S), 1080 (C–O). Anal. Calcd. for C21H18ClN5O3S (455.92): C, 55.32; H, 3.98; N, 7.78%. Found C, 55.29; H, 4.01; N, 7.77. 1H NMR (300 MHz, DMSO-d6): 1.03 (6H, s, CMe2), 3.12 (2H, m, CH2N), 3.50 (3H, s, CO2Me), 3.58 (2H, m, CH2NH), 7.13–7.62 (4H, m, 4CH of Ar), 9.60 (1H, brs, NH). 13C{1H} (100.0 MHz, DMSO-d6): 23.69, 27.09, 50.13, 53.73, 103.87, 104.37, 117.01, 129.38, 129.49, 134.91, 138.42, 147.41, 154.19, 157.38, 159.11, 161.25, 181.60. MS (EI, 70 eV): 410 (28), 380 (34), 320 (45), 284 (27), 254 (66), 223 (44), 169 (37), 111 (100), 73 (29).
Methyl 9-amino-11-cyano-7-oxo-10-thioxo-1,2,3, 4,5,7,9,10 octahydropyrido[4’,3’:4,5]pyrimido[1,2-a] [1,3]diazepine-8-carboxylate (5k). Yellow powder, m.p > 300 °C, 0.270 g, yield: 78%. IR (KBr)(νmax, cm−1): 3211 and 3118 (NH2 and NH), 2223 (CN), 1750 (CO2Me), 1692 (C =O), 1610 (C =N), 1571 (C =C), 1295 (C =S), 1064 (C–O). Anal. Calcd. for C14H14N6O3S (346.36): C, 48.55; H, 4.07; N, 24.46%. Found C, 48.56; H, 4.09; N, 24.46. 1H NMR (300 MHz, DMSO-d6): 1.86 (4H, m, 2CH2), 3.43–3.47 (2H, m, CH2N), 3.91 (3H, s, CO2Me), 4.05–4.09 (2H, m, CH2NH), 6.57 (2H, s, NH2), 9.58 (1H, brs, NH). 13C{1H} (75.0 MHz, DMSO-d6): 23.96, 24.57, 42.53, 42.59, 53.83, 103.13, 103.31, 116.90, 147.13, 156.40, 159.68, 159.93, 160.73, 179.76. MS (EI, 70 eV): 345 (M+−1, 15), 316 (22), 279 (34), 241 (31), 216 (29), 191 (42), 149 (40), 111 (57), 73 (100), 44 (89). Crystal data for 5k C14H14N6O3S, C2H3N1 (CCDC 1551358): MW = 387.44, monoclinic, P21/n, 11.5060(13) Å, b = 9.5212(12) Å, c = 16.697(2) Å, 𝛼 = 90.00, 𝛽 = 100.160(10), 𝛾 = 90.00, V = 1800.5(4) Å3, Z = 4, Dc = 1.429 mg/m3, F(000) = 808, MoKα (λ = 0.71073 Å). Intensity data were collected at 298(2) K with a STOE IPDS-II diffractometer with graphite monochromator, and employing the 𝜔∕2𝜃 scanning technique in the range of − 15⩽h⩽15, − 12⩽k⩽13, − 22⩽l⩽22. The structure was solved by a direct method; all non-hydrogen atoms were positioned and anisotropic thermal parameters were refined from 4881 observed reflections with R (int) = 0.2409 by a full-matrix least-squares technique converged to R = 0.0679 and wR2 = 0.0913 [I >2sigma(I)].
Ethyl 9-amino-11-cyano-7-oxo-10-thioxo-1,2,3,4, 5,7,9,10 octahydropyrido[4’,3’:4,5]pyrimido[1,2-a] [1,3]diazepine-8-carboxylate (5l). Yellow powder, m.p > 300 °C, 0.277 g, yield: 77%. IR (KBr)(νmax, cm−1): 3211 and 3118 (NH2 and NH), 2223 (CN), 1750 (CO2Me), 1692 (C =O), 1610 (C =N), 1571 (C =C), 1295 (C =S), 1088 (C–O). Anal. Calcd. for C15H16N6O3S (360.40): C, 49.99; H, 4.47; N, 23.32%. Found C, 50.02; H, 4.44; N, 23.30. 1H NMR (300 MHz, DMSO-d6): 1.32–1.36 (3H, m, OCH2CH3), 1.89–1.91 (4H, m, 2CH2), 3.43–3.47 (2H, m, CH2N), 4.08–4.12 (2H, m, CH2NH), 4.40–4.42 (2H, m, OCH2CH3), 6.60 (2H, s, NH2), 8.84 (1H, brs, NH). 13C{1H} (75.0 MHz, DMSO-d6): 14.08, 23.98, 24.58, 42.58, 63.11, 103.01, 103.26, 116.95, 147.72, 156.41, 159.67, 159.92, 160.14, 179.70. MS (EI, 70 eV): 360 (M+, 31), 328 (42), 299 (100), 256 (32), 227 (49), 186 (38), 132 (22), 97 (64), 54 (62).
Methyl 11-cyano-7-oxo-9-phenyl-10-thioxo-1,2,3, 4,5,7,9,10 octahydropyrido[4’,3’:4,5]pyrimido[1,2-a] [1,3]diazepine-8-carboxylate (5m). Yellow powder, m.p >300 °C, 0.301 g, yield: 74%. IR (KBr)(νmax, cm−1): 3105 (NH), 2220 (CN), 1752 (CO2Me), 1687 (C =O), 1610 (C =N), 1574 (C =C), 1299 (C =S), 1088 (C–O). Anal. Calcd. for C20H17N5O3S (407.44): C, 58.96; H, 4.21; N, 17.19%. Found C, 58.98; H, 4.23; N, 17.20. 1H NMR (300 MHz, DMSO-d6): 1.88–1.92 (4H, m, 2CH2), 3.26 (3H, s, CO2Me), 3.88–3.92 (2H, m, CH2N), 4.07–4.11 (2H, m, CH2NH), 7.05–7.51 (5H, m, 5CH of Ar), 8.98 (1H, s, NH), 9.58 (1H, brs, NH). 13C{1H} (100.0 MHz, DMSO-d6): 18.72, 19.36, 37.36, 37.41, 48.27, 98.81, 99.25, 111.79, 123.81, 124.11, 124.86, 142.30, 145.87, 151.00, 154.65, 155.12, 156.07, 176.48. MS (EI, 70 eV): 372 (42), 340 (13), 303 (33), 256 (28), 213 (26), 167 (44), 129 (19), 84 (94), 77 (100).
3. Results and discussion
3.1. Synthesis of tricyclic guanidines
Recently, Alizadeh et al. reported a two-step reaction for the synthesis of highly functionalized bicyclic guanidines 4 using cyclic thiourea 1, ketene dithioacetal 2 and dialkyl acetylenedicarboxylates 3 (Scheme 3) [51].
Conversion from bicyclic to tricyclic guanidines.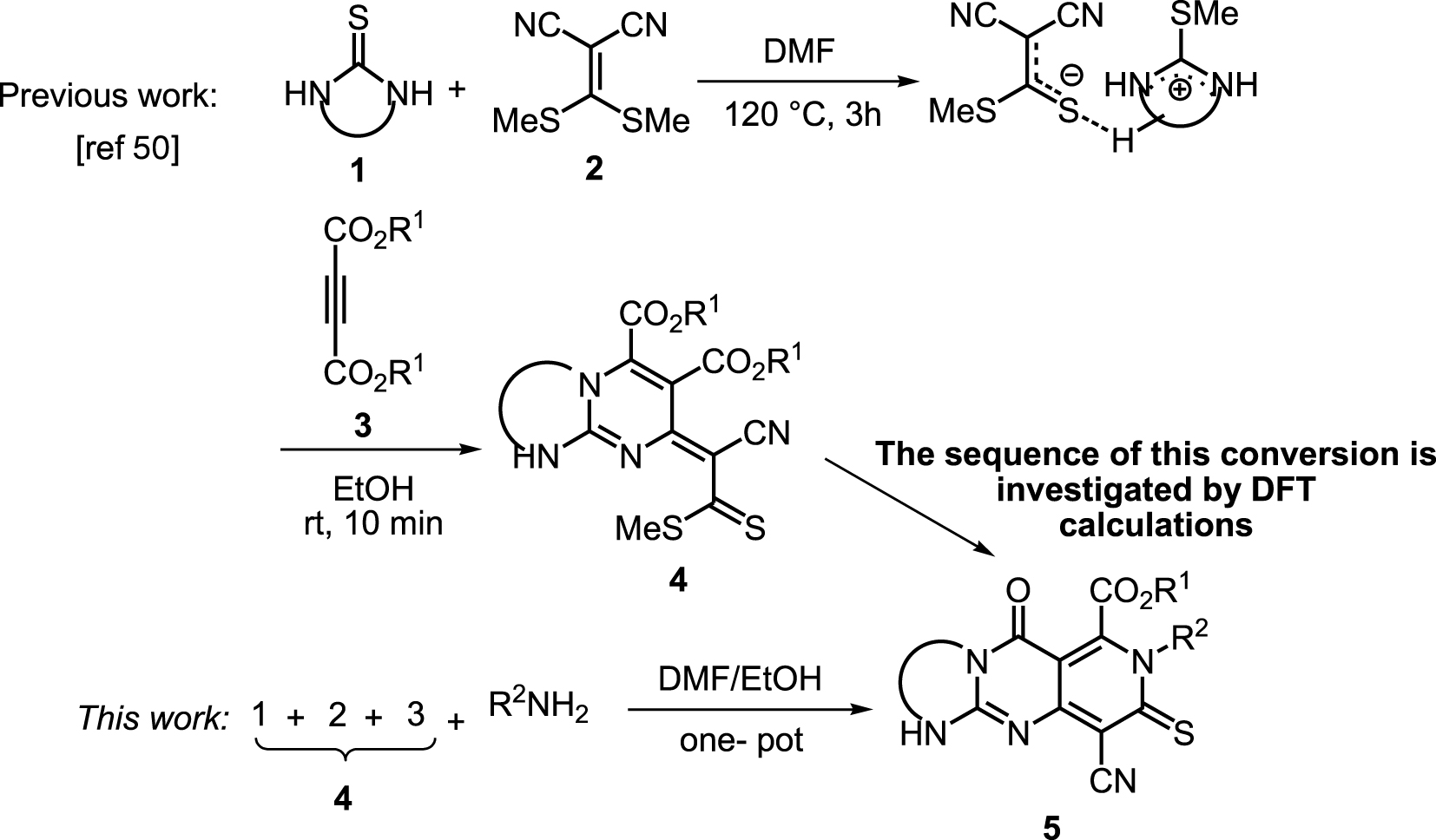
Due to the advantages of one-pot reactions, we began the current study by adding the bicyclic adducts 4 to a tandem one-pot process. Next, the addition of N-nucleophiles to the mixture of this reaction, containing the in situ generated bicyclic guanidines, led to the tricyclic guaidine adducts 5a-m in good yields (Table 1).
One-pot, four-component synthesis of tricyclic guanidines[a]
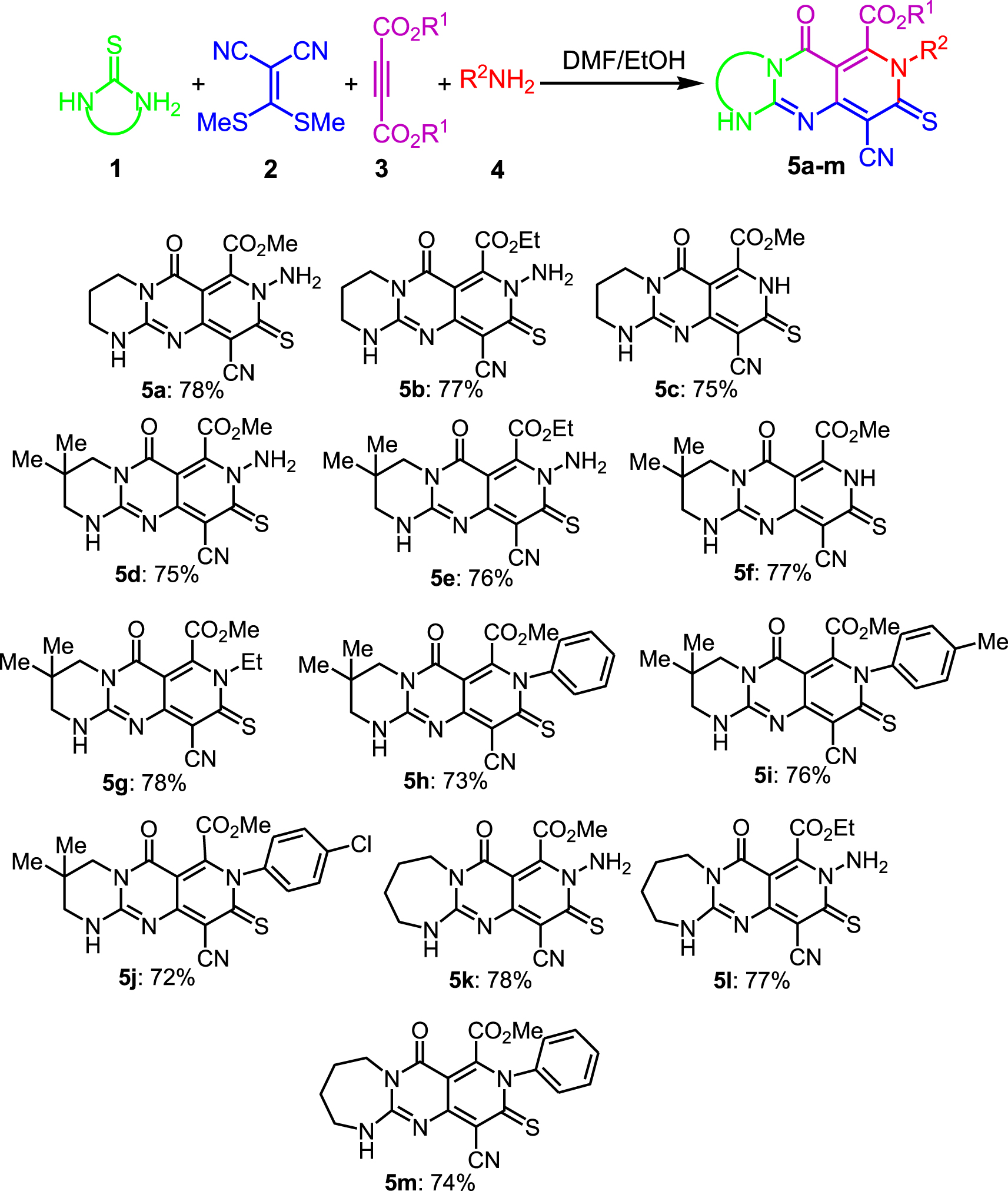 |
a Reaction conditions: one-pot, 1 (1 mmol), 2 (1 mmol), 2 mL of DMF, 120 °C, 3 h; next 3 (1 mmol), 2 mL of EtOH, rt, 1 h, next 4 (1.2 mmol), rt, 1h.
We did find that the efficiency of the procedure is not sensitive to the electronics of the precursors. In addition, further experiments demonstrated that the presence of an alcoholic solvent and the excessive amount of nucleophile (1.2 equivalent) are critical to the final transformation (conversion from bicyclic to tricyclic guanidine). Apart from CHN, IR, mass, 1H NMR and 13C NMR analyses for all compounds, the structure of 5k as a representative example is further confirmed by X-ray crystallography (Scheme 4).
ORTEP diagram of 5k.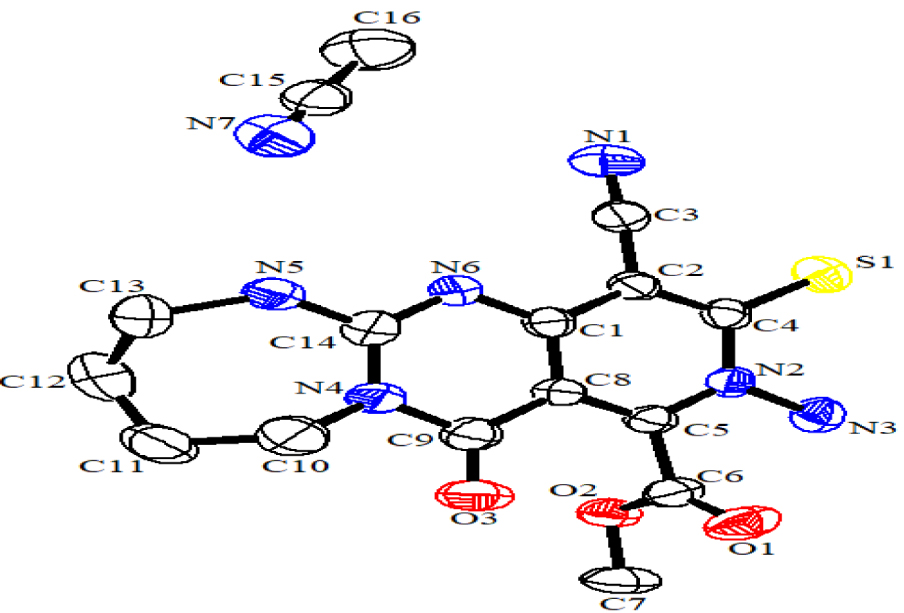
3.2. Computational methods
To investigate the origin of this transformation, the in situ generated dimethyl 2-[(Z)-1-cyano-2-(methylsulfanyl)-2-thioxoethylidene]-6,7,8,9-tetrahydro-2H-pyrimido[1,2-a]pyrimidine-3,4-dicarboxylate and hydrazine (lead to 5a in Table 1) were selected as the model reaction. The computational investigation of the reaction is classified into two main sections including thioamidation and N-Michael addition. M06−2X is demonstrated to be an efficient empirical function to investigate the non-covalent interactions, including the long-range zwitterionic interactions in the guanidinium-catalyzed reactions [16, 49, 52, 53]. Therefore, all geometries of reactant complexes (RCs), intermediates (INTs), transition states (TSs), product complexes (PrCs) and products (Prs) were optimized at the M06−2X level of theory in conjunction with the 6-31G* basis set. The intrinsic reaction coordinate (IRC) was performed to verify all of the TSs. The solvation model based on density (SMD) [54] was applied on the optimized geometries through the M06−2X/6-311+G** single-point calculation to examine the solvation effect of ethanol. The charge density on atoms was computed by natural bond orbital (NBO) [55] analysis based on the M06−2X/6-31G* method in the gas phase. All optimizations were carried out using the Orca 4 software [56].
3.2.1. Thioamidation step
Energy profile of the thioamidation step. The values are relative energies (𝛥E298 in ethanol solvent, kcal⋅mol−1) calculated at M06−2X/6-311+G**//M06 − 2X/6-31G* level.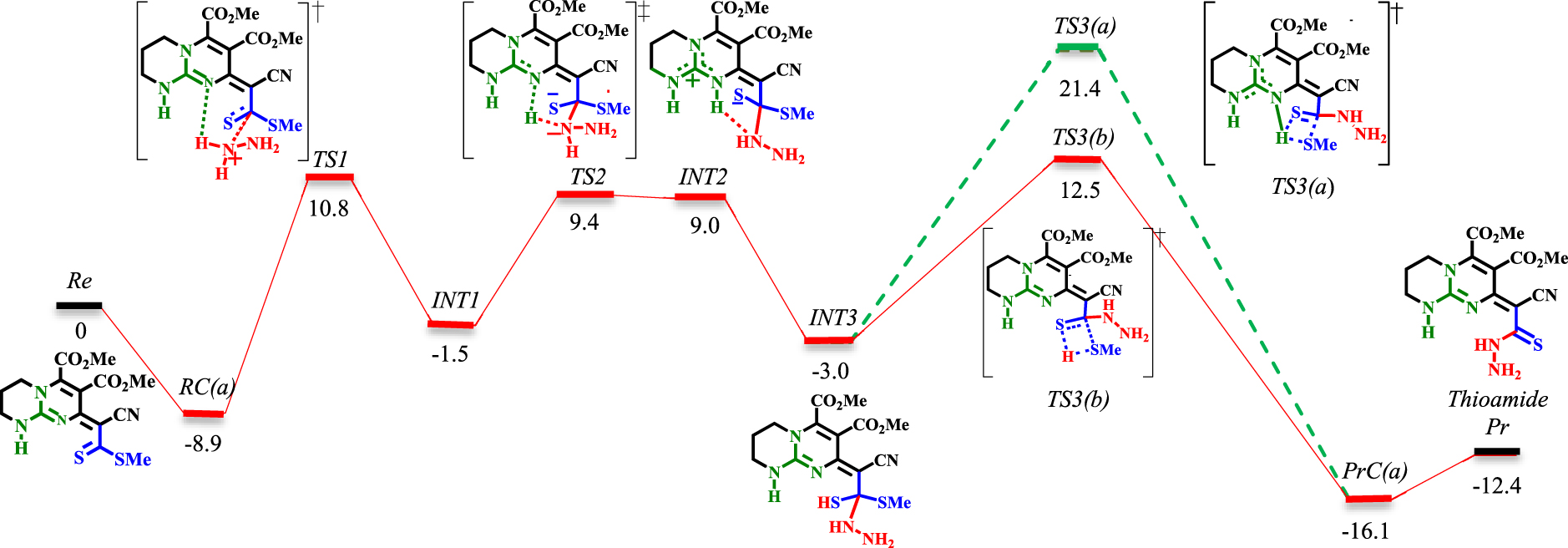
Energy profile of the thioamidation step under explicit solvation of MeOH (blue and purple lines). The values are relative energies (𝛥E298 in ethanol solvent, kcal⋅mol−1) calculated at M06−2X/6-311+G**//M06−2X/6-31G* level.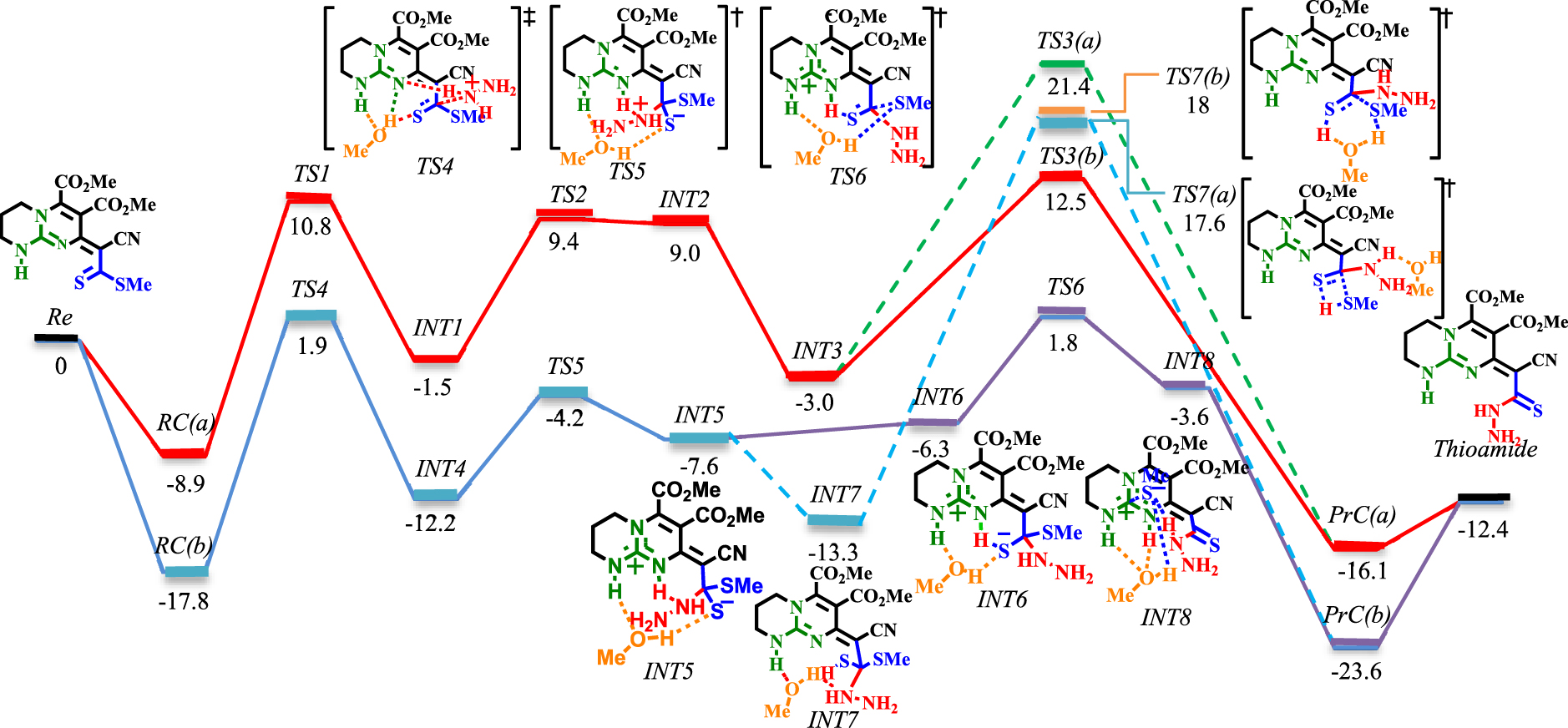
The reaction is studied via two main approaches (see Schemes 5 and 6). In the first, only bicyclic guanidine and hydrazine are considered (red and green lines in Schemes 5 and 6). In the second, methanol (to simplify the calculations, MeOH is replaced by EtOH) is explicitly included as the solvent (blue and purple lines in Scheme 6). For simplicity, only the structures of TSs and some of the important INTs are given (a full sequence of reaction involving the structures of all the INTs can be found in the Supplementary data).
The reaction starts with the bimolecular reactant’s complex RC(a), which is then proceeded by C−N bond formation ([TS1]‡). This step (RC(a)→[TS1]‡→INT1) requires crossing an energy barrier of 19.7 kcal⋅mol−1. Involving methanol as the solvent requires the same amount of activation energy (RC(b)→[TS4]‡→INT4). However, the presence of an H-bond donor solvent significantly reduces the energy surfaces of RC(b), [TS4]‡, INT4 relative to RC(a), [TS1]‡, INT1. The next step is proton transformation from an ammonium ion to an imine group through a Brønsted-base mechanism. This step has a 10.9 kcal⋅mol−1 barrier (INT1→[TS2]‡→INT2). However, when methanol is involved, the barrier is lowered to 8 kcal⋅mol−1 (INT4→[TS5]‡→INT5). The elimination of methanethiol (MeSH) is the final step in the thioamidation process. In both conditions, the proton is transferred from the Brønsted acid (NH of guanidinium) to the thiolate (INT2→INT3 and INT5→INT7). The elimination of MeSH could proceed via [TS3(a)]‡ or [TS3(b)]‡ with 24.4 and 15.5 kcal⋅mol−1 barriers, respectively (via the 1,3-H-shift mechanism). In contrast, when proceeding from [TS7(a)]‡ (via 1,3-H-shift mechanism) or [TS7(b)]‡ (via the proton shuttle mechanism), higher barriers (30.9 kcal⋅mol−1 for [TS7(a)]‡ and 31.3 kcal⋅mol−1 for [TS7(b)]‡) are encountered. Due to the involvement of barriers greater than 30 kcal⋅mol−1, the possibility of proceeding from [TS7(a)]‡ and [TS7(b)]‡ decreases. Fortunately, the investigation of an alternative pathway (from INT5 to the thioamide product, purple line in Scheme 6) via [TS6]‡ led to the most favorable reaction through a 9.4 kcal⋅mol−1 barrier. This step is exergonic with energy 4.8 kcal⋅mol−1 with reference to INT5. Here, INT5 will easily evolve to INT6 with a 1.3 kcal⋅mol−1 barrier. INT6 then undergoes C−SMe bond cleavage as a result of MeOH assistance upon isomerization from thiolate to thiocarbonyl (8.1 kcal⋅mol−1 for INT6→[TS6]‡).
As seen in Scheme 6, the lower barrier [TS6]‡ is characterized by an intramolecular H-bonded chain-like network between the polarized substrate and the MeOH molecule. Every part of this chain concerts to redistribute the electron changes on the involved heteroatoms, which leads to tautomerization and leaving of the MeS− group. This cooperation is similar to the ‘proton hopping’ mechanism, first suggested by Grotthuss [57]. The thion-thiolate species is stabilized by H-bonding with the NH of guanidinium (Brønsted acid activation). Concomitantly, as the NH⋯S bond is weakening, the C−SMe bond cleavage is encouraged by intramolecular H-bonding with MeOH. This will increase the electron density in the lone-pair region of MeOH, which persuades the H-bond acceptance from the second NH of guanidinium. Hence, if the two NH are considered as the two conductors of this chain, weakening of the H-donation on one side is compensated by the strengthening of the donation on the other side. Therefore, the bifunctional Brønsted acidity of guanidinium in cooperation with MeOH generates the driving force for C−SMe bond cleavage.
Observing INT8 (Scheme 6) gives instructive insight into the mechanism of MeSH elimination. As theoretically evidenced, the stepwise elimination of MeSH via [TS6]‡ is energetically more favored than the concerted elimination processes (to be compared with [TS3(a)]‡, [TS3(b)]‡, [TS7(a)]‡ and [TS7(b)]‡). This finding is contrary to the manner of ROH dissociation in the ring-opening polymerization of lactone [58] and transesterification/amidation of aromatic ester [59]. In these reactions, the computed results demonstrated that the cleavage of C−OR bond and proton transformation is concerted. To further validate the Lewis acid–base complex in INT8, diagrams of the optimized structure, molecular orbitals and NBO analysis are given in Schemes S1 and S2 (see Supplementary data).
The optimized geometry of INT8 shows that the guanidinium’s Lewis acidic center leans toward the MeS− with an interaction distance of 3.07 Å. Concomitantly, MeS− forms an H-bond with MeOH (2.11 Å). On the other hand, MeOH forms a bidentate H-bond (1.83 Å and 1.93 Å) with both NH protons. The former H-bonding reinforces the interaction between the Lewis acid (CN3) and the Lewis base (MeS−). Similarly to the case of [TS6]‡, the same discussion can be carried out for INT8. The H-bonding between MeOH and MeS− increases the electron density on the oxygen atom. This leads to an increase in the H-bond acceptance capability of MeOH from NH groups. Consequently, the central carbon of CN3 becomes more electron-deficient and a better anion receptor for MeS−. Therefore, an unconventional mode of activation, namely bifunctional Lewis–Brønsted acid activation, is established for MeSH elimination.
Overall, the thioamidation step is exergonic by −12.4 kcal⋅mol−1, suggesting that there is a significant driving force for the formation of a thioamide adduct. As theoretically evidenced, explicit inclusion of MeOH significantly affects the thioamidation step. It does not only lower the activation barriers but also affects the nature of the rate determining step (RDS) relative to the bimolecular mechanism. Accordingly, the elimination of MeSH ([TS3(b)]‡) is the RDS in the bimolecular mechanism. In comparison, the C−N bond formation ([TS4]‡) corresponds to the RDS when MeOH is involved as the third partner.
Energy profile of the N-Michael addition. The values are relative energies (𝛥E298 in ethanol solvent, kcal⋅mol−1) calculated at M06−2X/6-311+G**//M06−2X/6-31G* level.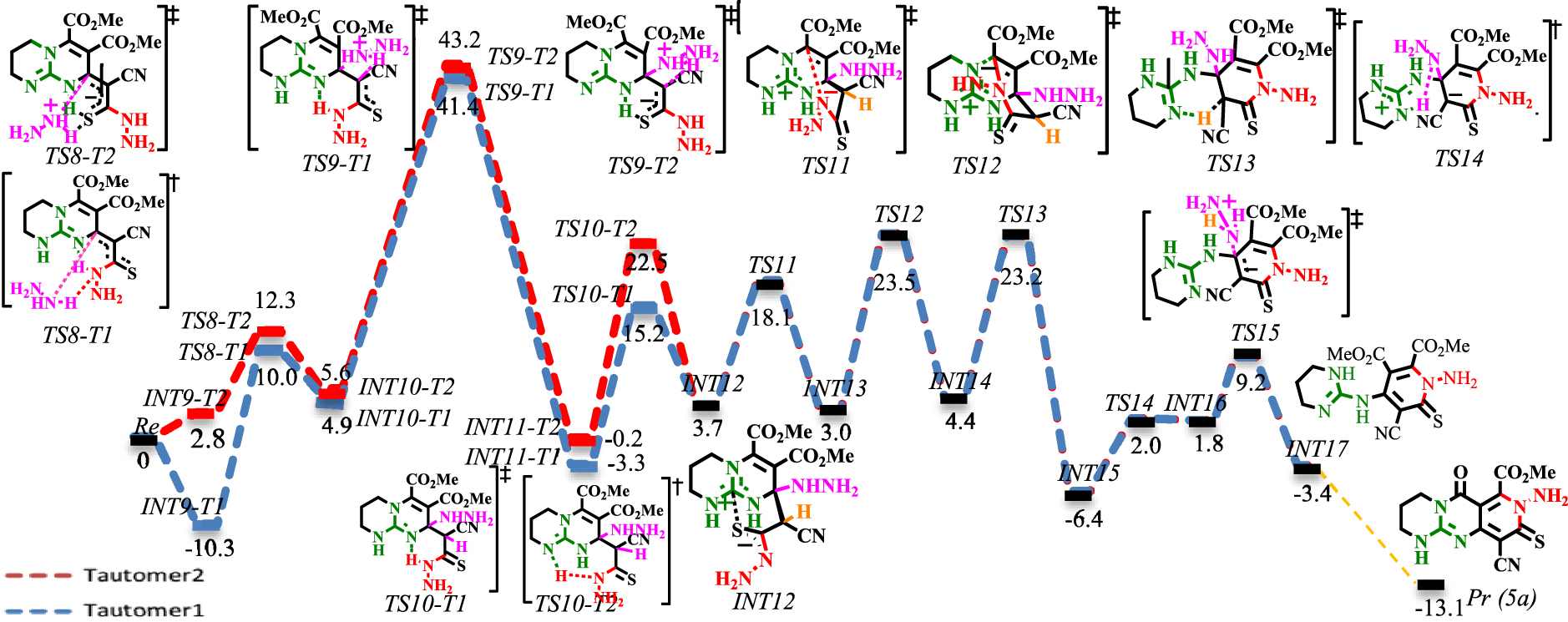
Energy profile of the N-Michael addition under explicit solvation of MeOH (green and purple lines). The values are relative energies (𝛥E298 in ethanol solvent, kcal⋅mol−1) calculated at M06−2X/6-311+G**//M06−2X/6-31G* level.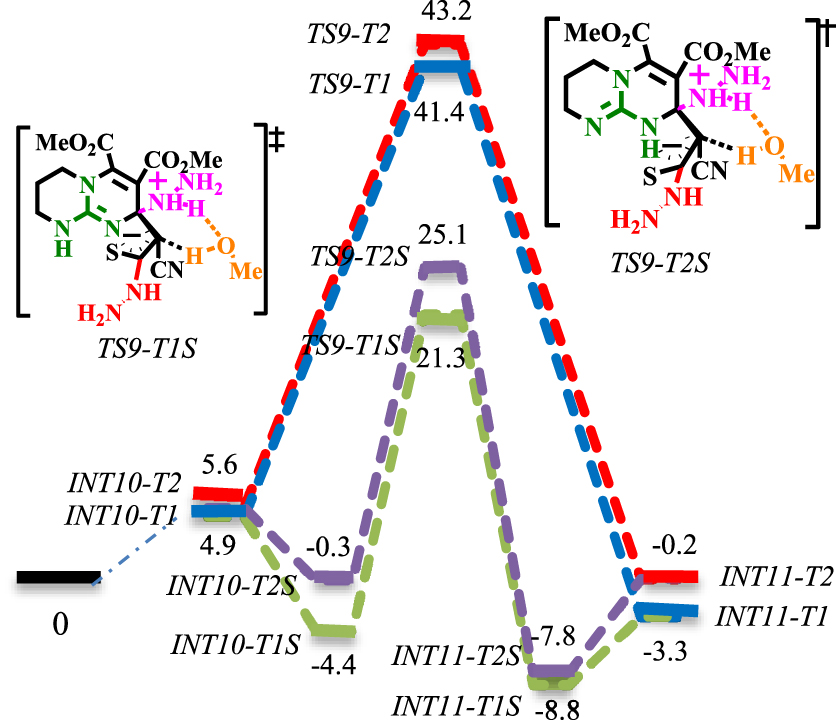
3.2.2. N-Michael addition step
The reaction profiles for N-Michael addition are shown in Schemes 7 and 8. The bimolecular mechanism (red and blue lines in Schemes 7 and 8) has been compared with the termolecular mechanism involving explicit solvation of MeOH (green and purple lines in Scheme 8).
Schematic presentation of CN3 functionalities.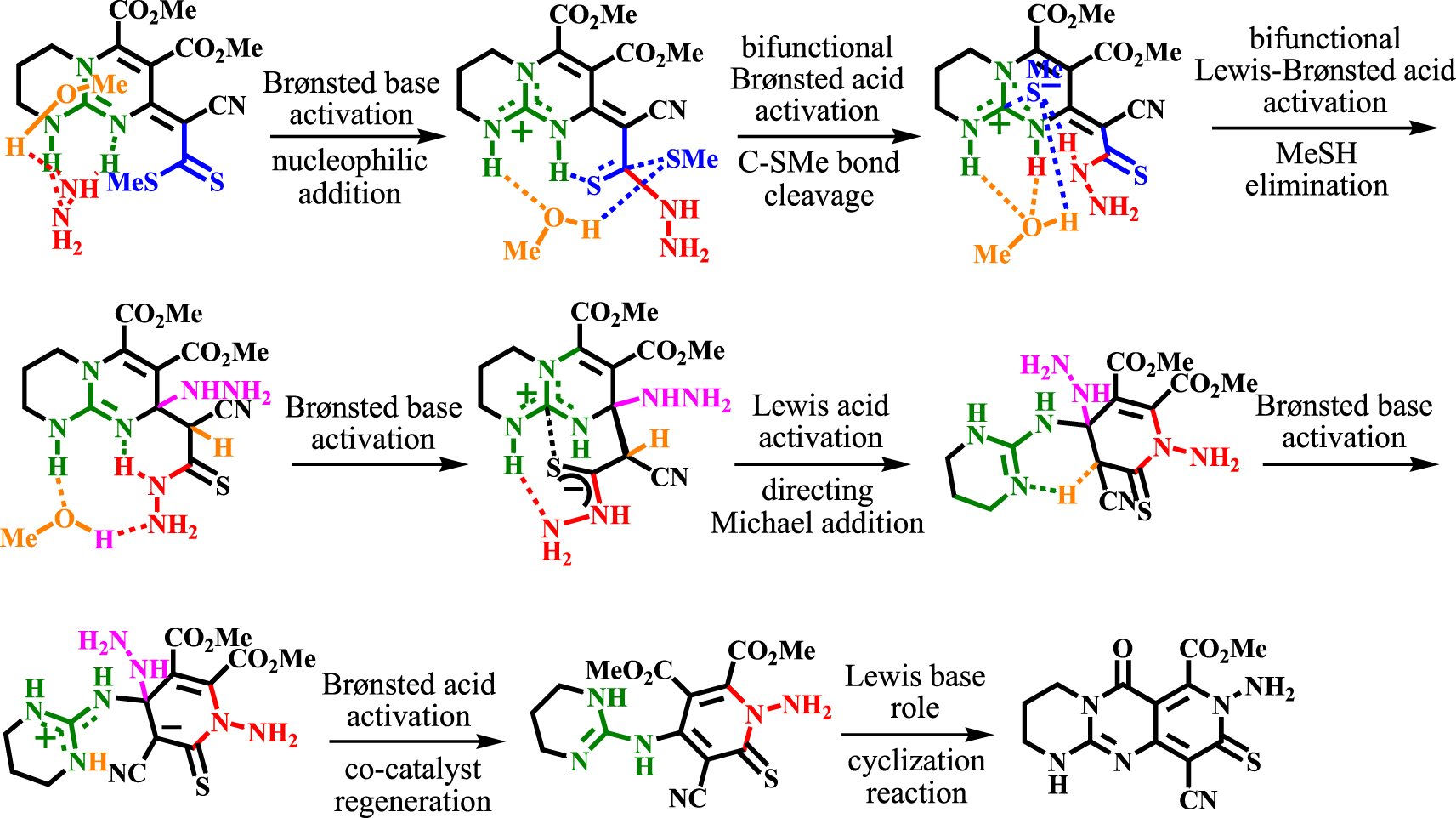
As a result of 1,3-H-shift on the nitrogen in the CN3 core, it is envisioned that the resulting thioamide can advance the reaction via tautomers 1 and 2 (for the tautomerization mechanism, see Scheme S3 in Supplementary data). The corresponding intermediates and transition states are addressed by adding T1 and T2 suffixes to their names. As seen in Scheme 7, the intermolecular Michael addition of an excessive amount of hydrazine (co-catalyst) to the β-unsaturated carbon of thiocarbonyl occurs via [TS8-T1]‡ with a 20.3 kcal⋅mol−1 barrier. The proton transformation from an ammonium cation to the 𝛼-carbon of thiocarbonyl from [TS9-T1]‡ and [TS9-T2]‡ encounters a high barrier (36.5 and 37.6 kcal⋅mol−1, respectively) and cannot take place. As the corresponding intermediate and transition states are ionic complexes, it was envisioned that explicit solvation by methanol may fix the high-barrier problem (the methanol-solvated complexes will be denoted by adding a suffix (S) to their names). As seen in Scheme 8, the MeOH-solvated energy surfaces at [TS9-T1S]‡ and [TS9-T2S]‡ are comparably different relative to their non-solvated counterparts ([TS9-T1]‡ and [TS9-T2]‡). On the other hand, the results suggest that the proton transformation via the proton shuttle mechanism, assisted by MeOH, is significantly more favored than 1,3-H-shift mechanisms. The solvation of [TS9-T1S]‡ is more favorable: the solvated transition state [TS9-T1S]‡ lies at 21.3 kcal⋅mol−1 and crosses a barrier of 25.43 kcal⋅mol−1 to yield INT11-T2S. Next, the Brønsted-base functionality activates the nucleophilic motif by proton abstraction from the NH of thioamide. The calculated activation energy for proton abstraction at [TS10-T1]‡ is 24 kcal⋅mol−1, while it is 30.4 kcal⋅mol−1 at [TS10-T2]‡. The easier deprotonation at [TS10-T1]‡ is attributed to the formation of a six-membered ring transition state, which is less strained than [TS10-T2]‡. Next, the regioselective N-Michael addition proceeds at [TS11]‡ with a 14.4 kcal⋅mol−1 barrier. Then, the delocalized negative charge at [TS12]‡ triggers the ring-opening reaction with a 20.5 kcal⋅mol−1 barrier. Following this, Brønsted basic deprotonation of the acidic proton of the methine group (via [TS13]‡), Brønsted acidic prototropy to NH2NH (via [TS14]‡) and regeneration of the co-catalyst (via [TS15]‡) delivers INT17. Finally, INT17 bears an intramolecular substitution reaction between the guanidine functionality (Lewis base) and the ester functional group to yield 5a (not investigated here: orange line in Scheme 7) [60, 61]. This reaction implies the reagent activity of the CN3 core.
The ionic complex INT12 involving Lewis acid–base interaction has a determinative role in the Michael addition sequence. The positively charged central carbon atom in INT12 works as an anion receptor site, which accommodates the delocalized negative charge perpendicular to the guanidinium plane. This activity will bring the reactionary sites in close contact, leading to N-Michael addition. Therefore, the requirement of an excessive amount of the nucleophile as a co-catalyst is rationalized. To further evidence the Lewis acid interaction in INT12, the optimized geometry, molecular orbitals and NBO analysis are depicted in Schemes S4 and S5 (see Supplementary data).
To provide a brief description of the catalytic–reagent activities of the triaza CN3 core, a pictorial presentation is given in Scheme 9.
4. Conclusion
In summary, DFT calculations were employed to gain knowledge of the catalytic–reagent activities of the guanidine functionality during the transformation from bicyclic to tricyclic guanidines. It is demonstrated that the strong Lewis acidity of the central carbon atom in the CN3 core has a major influence on advancing the reaction. In the thioamidation step, the stepwise elimination of MeSH involving Lewis acid–base complexation with MeS− and, following this, the proton abstraction from guanidinium’s acidic N−H are energetically more favored than the concerted processes in which the C−SMe bond cleavage is concomitant with the 1,3-hydrogen shift or the proton shuttle assisted by a solvent. However, in the N-Michael addition step, the Lewis acidity of guanidinium, by effectively assembling both the nitrogen and the Michael acceptor site close to each other, plays a more important role. Unlike the previous studies, in which the Lewis acid interaction was included in asymmetric transformations to provide different stereochemical outcomes, the interaction involved in the N-Michael addition step not only provides selectivity but also triggers reactivity. Otherwise, in the absence of such an activation mode, the process must be stopped in the thioamidation step. In addition, calculations in good agreement with experimental results revealed the pivotal role of the methanol solvent as well as an excessive amount of N-nucleophiles as the co-catalyst in performing the reaction.
Conflict of interest
The authors declare no conflict of interest.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.16 or from the author. The experimental details and randomization protocols are provided.