


 CC-BY 4.0
CC-BY 4.0
1. Introduction
The increasing concentration of carbon dioxide (CO2) in the atmosphere is partly responsible for climate change. CO2 is often considered as useless and detrimental waste. Nonetheless, CO2 can also be regarded as a cheap, abundant, and renewable carbon source. It is non-toxic (even though it is an asphyxiant) and is especially useful as a phosgene substitute [1]. Many interesting organic compounds, including dimethyl carbonate [2], cyclic carbonates (CCs) [3], and urethanes [4], can be synthesized using CO2 as a building block [5, 6].
Even though the synthesis of CCs was first reported in the 1930s, the synthesis of five-membered (or more) CCs is gaining increased attention. They are used to synthesize polycarbonates [7, 8, 9], as aprotic polar solvents for organic reactions [10], electrolytes [11] or solvents for lithium-ion batteries [12, 13], and as intermediates in the manufacture of fine chemicals [14, 15, 16]. CCs are popularly implemented in polymer science, for ring-opening polymerizations and reactions with amines, alcohols, thiols, and carboxylic acids [17]. For example, difunctional CCs can be advantageously reacted with diamines to produce polyhydroxyurethanes, promising substitutes to polyurethanes, without the recourse to highly toxic and carcinogenic isocyanates [14, 18, 19, 20, 21, 22].
Several approaches can be used to synthesize five-membered CCs [23, 24, 25]. The most efficient method is the insertion of CO2 into epoxide rings. This reaction requires use of a catalyst, such as alkali metal salts [26, 27, 28], metal oxides [29], Schiff base [30], ion-exchange resins [31], polymers such as modified polystyrene [32], ionic liquids [33], SiO2 modified by quaternary ammonium or phosphonium salts [34], gold nanoparticles supported on resins [35], or CO2 adducts of N-heterocyclic carbenes [36]. Endo et al. reported that alkali metal salts could be used as catalysts to synthesize CCs using crown ethers as co-catalysts [27]. Huang and Shi found that alkali metal salts can catalyze cycloaddition of CO2 with epoxides efficiently in the presence of triphenylphosphane (PPh3) and phenol [28]. Cellulose could also improve the coupling reaction of CO2 and epoxides catalyzed by KI [37]. Recently, a few studies reported the excellent catalytic performance of ligand-metal-organic frameworks in the cycloaddition reaction of epoxides with CO2 [38, 39].
Cyclic xanthates (XAs) constitute another family of interesting five-membered cyclic reactive species. They are highly reactive and can be easily prepared from epoxides and carbon disulfide (CS2) at low temperatures (20–25 °C) [40, 41]. This reaction is usually carried out in organic solvent or water in the presence of a catalyst, such as amines, alkali metal salts, quaternary ammonium salts, potassium alcoholates, and transition metal complexes [42, 43, 44, 45, 46]. Petrov et al. demonstrated that fluorinated epoxides react with CS2 selectively, leading to the corresponding cyclic XAs [47]. XAs are potential solvents for liquid electrolytes in Lithium-ion batteries [13] and efficient chain transfer agents in controlled radical (co)polymerization of fluorinated monomers [48]. They could also be transformed into bis-cyclocarbonates, producing novel fluorinated functional polycarbonates or polyhydroxyurethanes. Xanthates can be used as precursors for poly(thio)urethane materials via ring-opening reaction using primary or secondary amines.
Although research on the synthesis and use of CCs- and to a lesser degree on cyclic XAs-based compounds is extensive, few fluorinated derivatives bearing such functional groups have been reported [47]. Fluorinated derivatives usually exhibit outstanding properties [49, 50, 51], such as chemical resistance, thermal stability, low dielectric constants and dissipation factors, hydrophobic and oleophobic properties, excellent weathering, and interesting surface properties fulfilling the requirements for various high value-added applications. Therefore, the synthesis of compounds that carry both a fluorinated segment and cyclic carbonates or xanthates moiety would be highly interesting as reactive fluorinated intermediates.
This work originated from the work of Galiano et al. [13] and presents the results of optimization and reproducibility of the cycloaddition of CX2 (carbon dioxide (X=O) or carbon disulfide (X=S)) with two fluorinated epoxides: hexafluoropropene oxide (HFPO) and 2-(trifluoromethyl)oxirane (TFEO) (Figure 1). Moreover, it describes the synthesis of original bis-CCs and bis-XAs by reacting CO2 and CS2, respectively, with 1,4-bis(2′,3′-epoxypropyl)perfluorobutane (BEPFB) (Figure 1) using the optimum conditions in the presence of LiBr.
Structures of HFPO, TFEO, and BEPFB used in this work.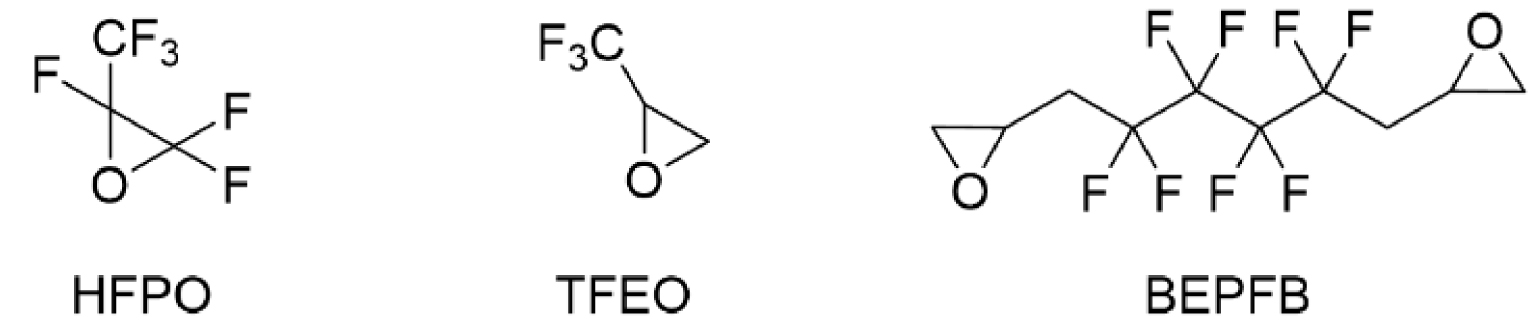
2. Results and discussion
CCs can be prepared through the coupling reaction of CO2 and strained heterocycles using a suitable catalyst [16, 24]. This approach benefits from eliminating phosgene as a reagent and is 100% atom economical, making it a highly desirable transformation.
The insertion of CO2 and CS2 into HFPO was examined (Scheme 1), and various experimental conditions (choice of solvent, reaction duration, and temperature) have been investigated to improve the carbonation reaction. Then, the optimized conditions were tried on another substrate, TFEO, for reproducibility (Scheme 1). Furthermore, synthesizing the original bis-CCs and bis-XAs was accomplished by adding CO2 and CS2 to BEPFB (Scheme 1). The results are summarized in Table 1.
Synthesis of original five-membered fluorinated CCs (1, 3, and 5) and XAs (2, 4, and 6).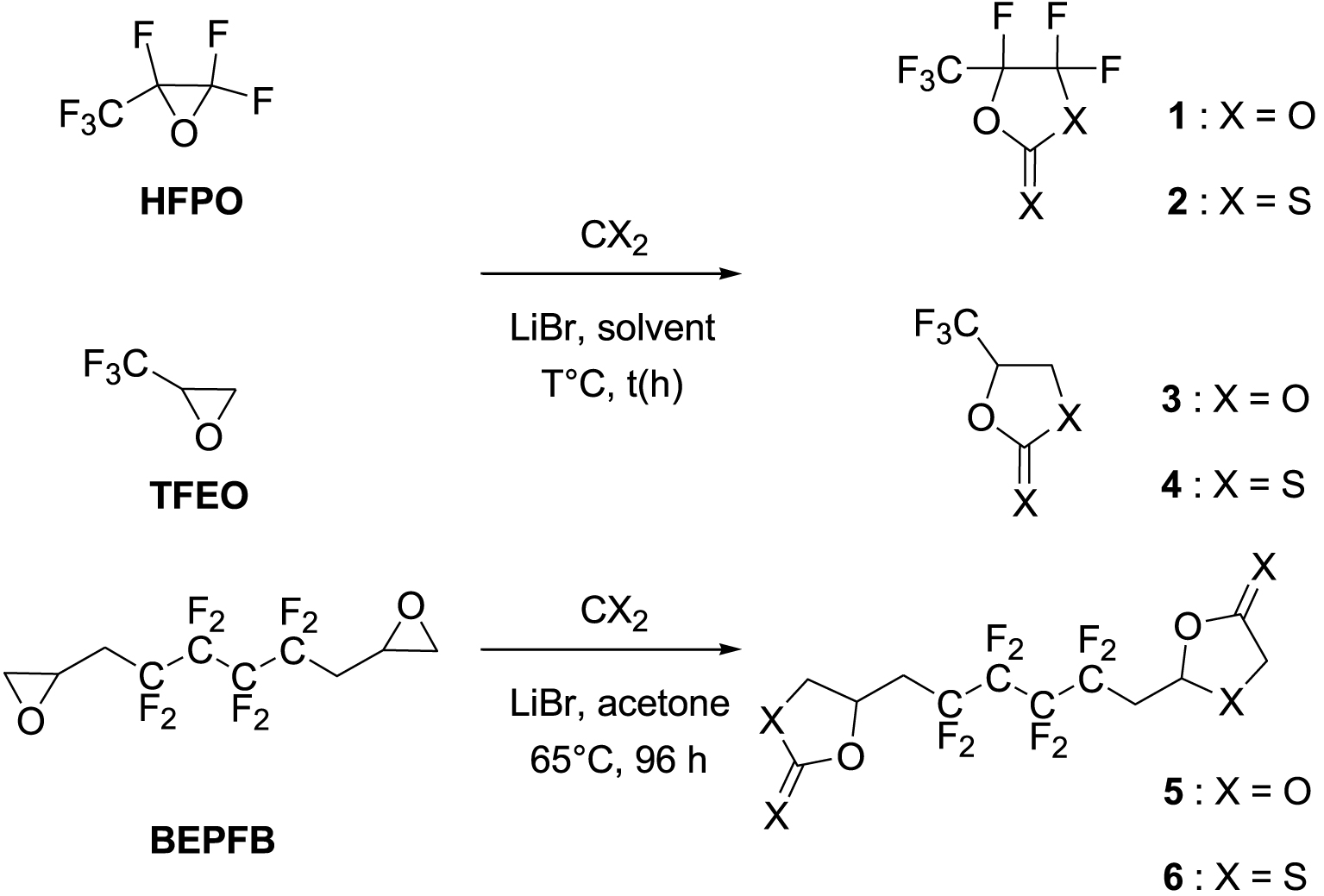
Reactions of HFPO, TFEO, and BEPFB with CO2 or CS2a
| Run # | Epoxide | X in CX2 | Solvent | T (°C) | t (h) | Yield (%) |
|---|---|---|---|---|---|---|
| 1 |
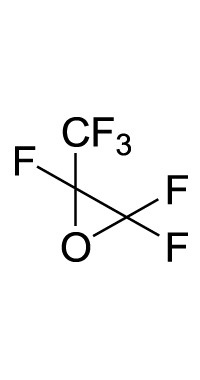 |
O | DMF | 25 | 24 | 25 |
| 2 | O | DMF | 50 | 24 | 45 | |
| 3 | O | DMF | 80 | 24 | 74 | |
| 4 | O | DMF | 80 | 14 | 66 | |
| 5 | O | 1,4-dioxane | 50 | 24 | 63 | |
| 6 | O | 1,4-dioxane | 70 | 24 | 72 | |
| 7 | O | THF | 50 | 24 | 56 | |
| 8b | O | Neat | 80 | 24 | 0 | |
| 9 | O | Acetone | 65 | 24 | 85 | |
| 10 | O | Acetone | 65 | 14 | 81 | |
| 11c | S | Acetone | 65 | 14 | 83 | |
| 12c | S | Acetone | 65 | 24 | 83 | |
| 13 |
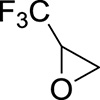 |
O | Acetone | 65 | 14 | 88 |
| 14c | S | Acetone | 65 | 14 | 91 | |
| 15d |
 |
O | Acetone | 65 | 14 | 24e |
| 16d | O | Acetone | 65 | 24 | 33e | |
| 17d | O | Acetone | 65 | 96 | 85e | |
| 18d | S | Acetone | 65 | 96 | 87e |
aExperimental conditions: The carbonation reactions were performed in pressure reactors under 4 bar of CO2 and in the presence of 5 mol% of LiBr, or with 3 eq. of CS2, T (°C), t (h). bReaction conducted without solvent in the presence of KBr/Lecithin as catalyst [52]. cThis reaction was performed with 16 mol% of LiBr. dThese reactions were conducted with 10 mol% of LiBr. eObtained after purification.
First, the synthesis of 1 was carried out with CO2 in DMF at 25 °C and 50 °C and proceeded with relatively modest yields (Table 1, runs 1–2). Subsequently, increasing the temperature to 80 °C did prove beneficial, but the yields reached 74% at best (Table 1, runs 3 and 4). In addition, using 1,4-dioxane or THF as solvent (Table 1, runs 5–7) did not improve the yield even after 24 h. The KBr/lecithin catalytic system, known to afford good results, was also tried in the carbonation of HFPO (Table 1, run 8). Surprisingly, this reaction did not yield any CCs under the implemented conditions without solvent. On the other hand, the carbonation in acetone at 65 °C gave the best yields (Table 1, runs 9–12), reaching 85% in 24 h and 81% in 14 h.
The analog HFPO-based cyclic XA 2 was successfully synthesized in high yields (83%) using the same conditions by refluxing acetone in excess of CS2 for 14 h (Table 1, run 11). However, this reaction led to the formation of a minor by-product, 2′ (Scheme 2). Earlier attempts to react oxiranes and carbon disulfide resulted in complex mixtures [7], and the formation of these regioisomers, 2 and 2′, depends on the catalyst and reaction conditions [53]. The selective formation of XAs (5-substituted 1,3-oxathiolane-2-thiones) was attained using LiBr in appropriate solvents [46]. Thus, the non-catalyzed reactions conducted in this work were not expected to lead to the regioselective formation of cyclic XA [54]. Previous work has shown that triethylamine as a catalyst led to mixtures of products under high pressure [52]. On the other hand, lithium bromide [46], hydrotalcite [55], sodium methoxide [56], tributylphosphine, tetradentate Schiff-base aluminum complexes [43], and lithium perchlorate [57] selectively produced 1,3-oxathiolane-2-thiones.
The reaction of HFPO with CS2 in the presence of LiBr catalyst.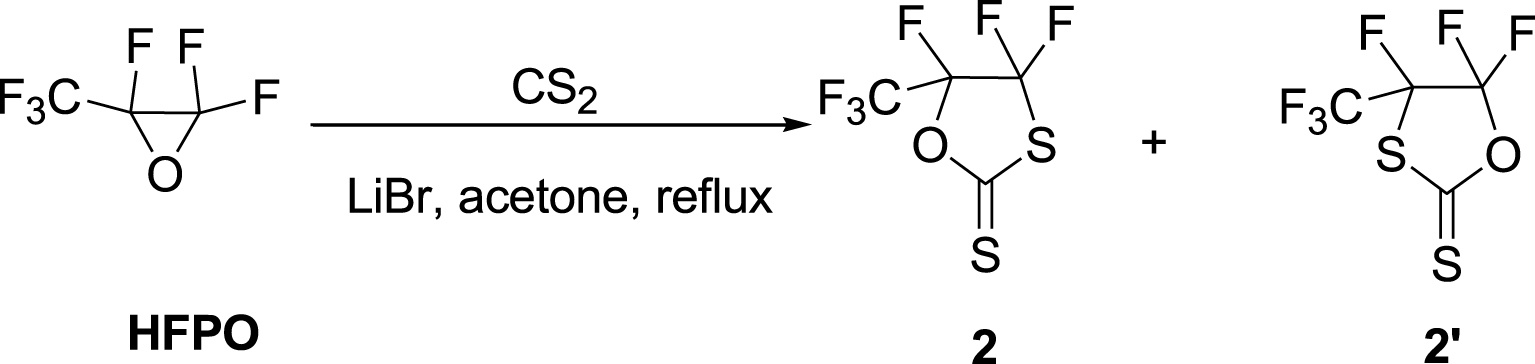
By implementing the optimal conditions obtained for products 1 and 2, trifluoromethyloxirane (TFEO) was predominantly (88% yield) transformed into the corresponding CCs, 3, by reacting with CO2 in acetone in the presence of LiBr at 65 °C (Table 1, run 10). Similar to the 2 case, the XAs counterpart (4) was easily obtained as a mixture of regioisomers in 91% yield using similar conditions as for HFPO (Table 1, run 14).
The original reactions of CX2 (X=O or S) with BEPFB proved slower than those performed with HFPO and TFEO (Table 1, runs 15–18). This could be ascribed to the strong electro-attractive effect of the fluorine atoms and the trifluoromethyl groups present on the epoxy rings of HFPO and TFEO; these strong electron-withdrawing groups induce a partial polarization of the carbon–carbon bond of the epoxide, which in turn enhances the electrophilic character of the carbon atom bearing the fluorine or trifluoromethyl group and its reactivity towards the bromide anion of the catalyst. The ring-opening of the epoxide by the bromide anion is believed to be the rate-determining step of the reaction of CO2 and CS2 on epoxides. HFPO with a fully fluorinated backbone reacts with CO2 or CS2 quantitatively in 14 h (81% and 83% yield, respectively). In comparison, the reaction with BEPFB needs 96 h to lead to carbonate 5 or xanthate 6 in high yields.
The FTIR spectrum of 5 (Figure S8 in SI) confirmed the formation of the CCs with the characteristic bands of the carbonate function: C=O bond stretching vibration at 1776 cm-1, and other vibrations corresponding to C–C and C–O bonds appear at 1047 cm-1 and 1106 cm-1, respectively. Similar observations are noted on the FTIR spectrum of 6 (Figure S11 in SI) with the characteristic bands of the XAs function: C=S bond stretching vibration at 1777 cm-1.
In the following section, we present NMR spectra of the original fluorinated bis-cyclic carbonates 5 and xanthates 6 (Figures 2–4). The other products synthesized in this study have been characterized by 1H, 19F, and 13C NMR spectroscopy, and the corresponding spectra are shown in the SI (Figures S1–S7 and S9–S10).
1H NMR spectra of 6 (upper spectrum), 5 (middle spectrum), and BEPFB (lower spectrum) (DMSO-d6, 20 °C, 400 MHz).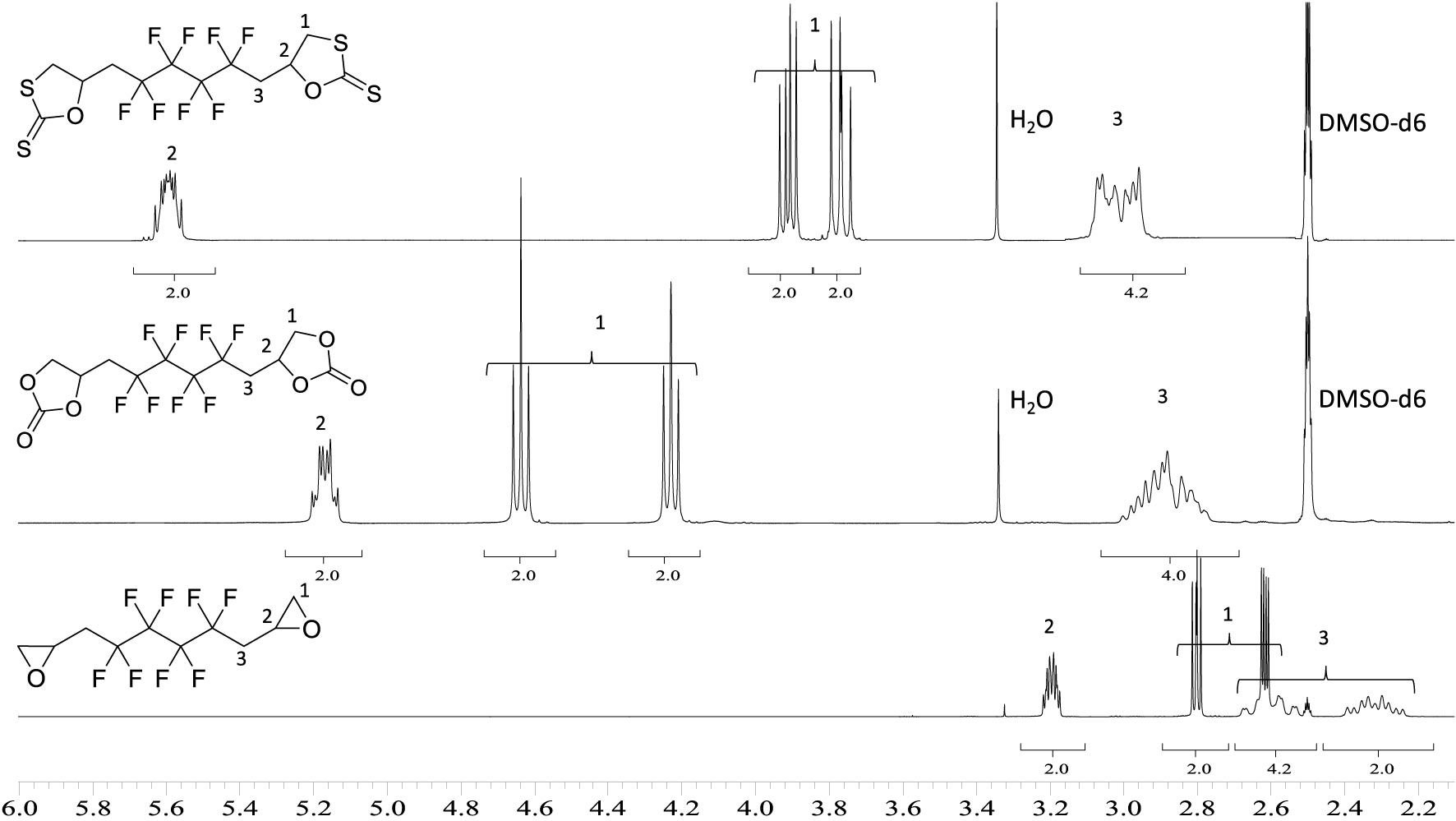
19F NMR spectrum of 5 (top) and BEPFB (bottom) (DMSO-d6, 20 °C, 235.2 MHz).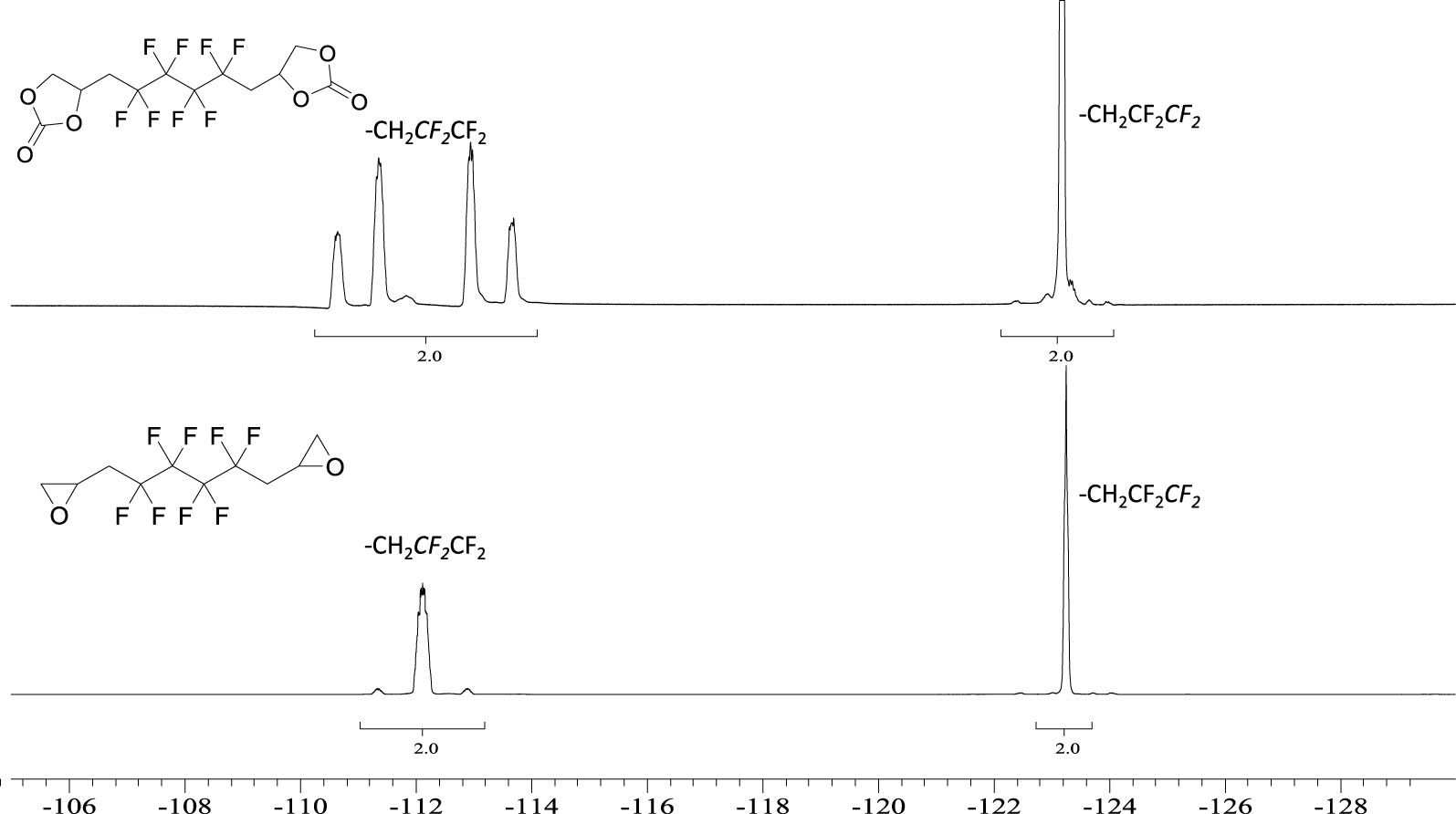
13C NMR spectrum of 5 (top) and BEPFB (bottom) (DMSO, 20 °C, 100.6 MHz).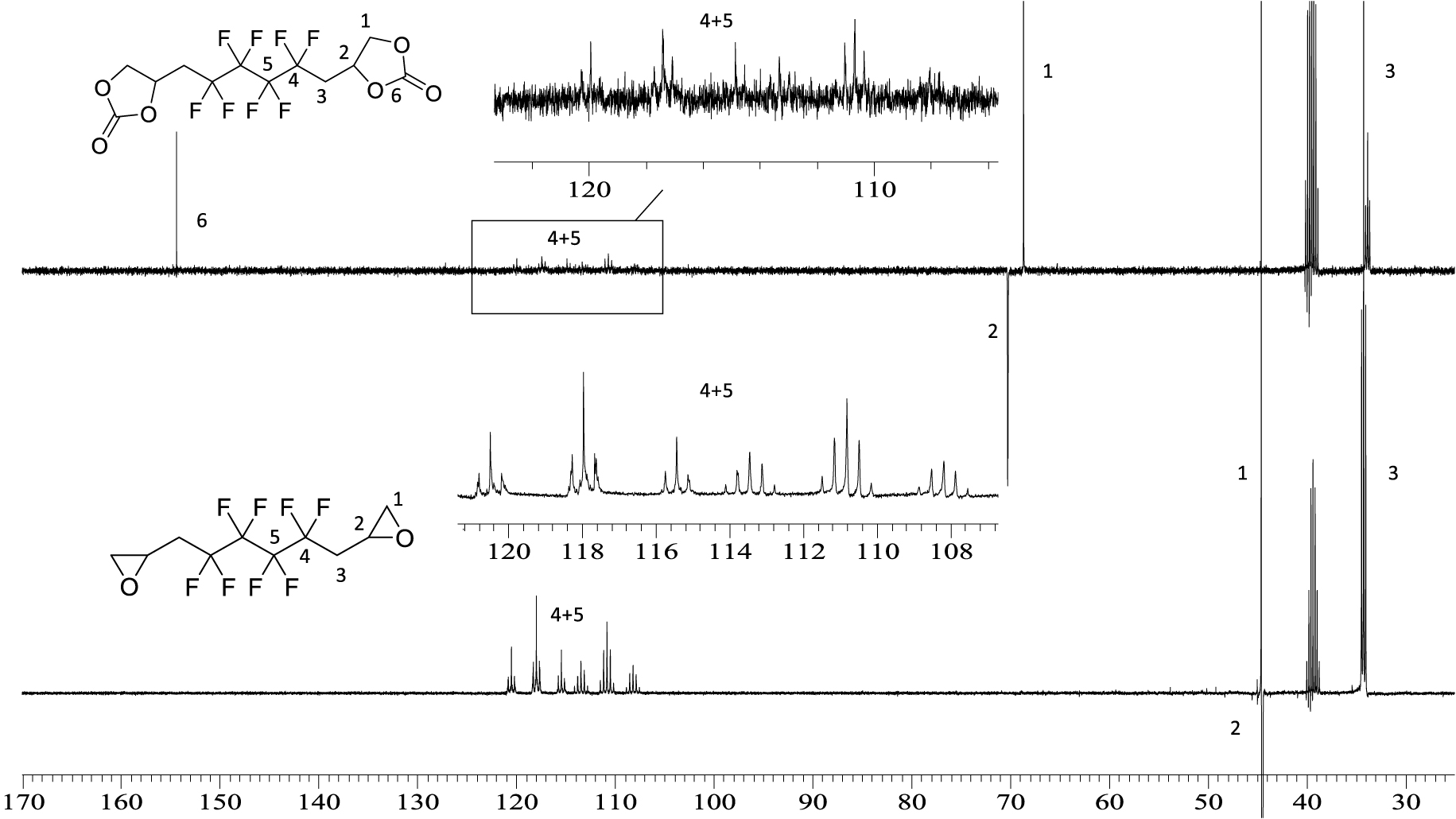
Figure 2 shows the 1H NMR spectra of cyclic-carbonates 5 and xanthates 6. These spectra clearly show the disappearance of the signals of the diepoxide at 2.6, 2.8, and 3.2 ppm replaced by AB systems (located at 4.6 and 4.2 ppm for 5; 3.9 and 3.7 ppm for 6), corresponding to the inequivalent hydrogen atoms CHaHb of the carbonate groups. The same observation can be made for the CH motif of the carbonate ring with the chemical shift at 5.2 ppm for 5 and 5.6 ppm for 5 and 6, respectively, low field-shifted by the presence of the carbonyl group compared to the epoxide (3.2 ppm). Both hydrogen atoms of CH2CF2 groups appear as one complex system at 2.9 ppm due to fluorine coupling in α and β positions.
The 19F NMR spectrum of 5 (Figure 3) displays an AB system at −112 ppm assigned to the CH2CFAFBCF2 groups of A and B diastereoisomers. Indeed, the chemical shifts are different because the product is a mixture of two diastereomers due to the presence of an asymmetric carbon on the cyclic carbonate. The signals at −123 ppm are assigned to the central difluoromethylene groups (CH2CF2CF2). Similar observations were seen on the 19F NMR spectrum of 6 (Figure S9 in the SI).
5 was also characterized by 13C NMR (Figure 4). The resulting spectrum shows the signals of the carbonyl groups of the CCs at 154 ppm. The CF2 peaks appear between 106 and 120 ppm. Due to the presence of the carbonyl group, the signal corresponding to the CH groups of the cyclocarbonate is deshielded to 70 ppm compared to the CH of the epoxide that appears at 44 ppm. Similar results were obtained in the 13C NMR spectrum of 6 (Figure S10 in the SI) except for the resonance of the XAs moiety, which appears at 212 ppm.
3. Experimental section
3.1. Materials
1,4-bis(2′,3′-epoxypropyl)perfluorobutane (BEPFB) was provided by TOSOH Finechemicals, INC (Shiba, Japan). 2-(trifluoromethyl)oxirane (TFEO) provided by SynQuest (Alachua, USA). Hexafluoropropene oxide (HFPO), carbon disulfide (CS2), lithium bromide (LiBr), sodium sulfate (Na2SO4), acetone (analytical grade), dichloromethane (DCM, analytical grade), methanol (analytical grade), N,N-dimethylformamide (DMF), 1,4-dioxane and tetrahydrofuran (THF, analytical grade) were purchased from Sigma-Aldrich. The deuterated solvents used in NMR spectroscopy were purchased from Euroiso-top (purity > 99.8%).
3.2. Characterization
3.2.1. Nuclear magnetic resonance (NMR)
The NMR spectra were recorded on a Bruker AC 400 instrument, using deuterated chloroform (CDCl3), deuterated dimethylsulfoxide (DMSO-d6), or deuterated acetone (acetone-d6) as the solvents. Tetramethylsilane (TMS) or trichloromonofluoromethane (CFCl3) were used as references for 1H (or 19F) nuclei. Coupling constants and chemical shifts are given in hertz (Hz) and part per million (ppm), respectively. The experimental conditions for recording 1H, 13C, (or 19F) NMR spectra were as follows: flip angle 90° (or 30°), acquisition time 4.5 s (or 0.7 s), pulse delay 2 s (or 2 s), number of scans 128 (or 512), and a pulse width of 5 μs for 19F NMR.
3.2.2. Fourier transform infrared spectroscopy (FTIR)
The FTIR spectra were recorded on a Nicolet 210 FTIR spectrometer. The characteristic absorptions mentioned in the text were strong bands reported in cm-1.
3.2.3. Autoclave operations
The carbonation reactions were performed in Hastelloy Parr autoclave systems (HC 276, 100 mL) equipped with a manometer, a mechanical Hastelloy anchor, a rupture disk (3000 PSI), inlet, and outlet valves. An electronic device regulated and controlled both the stirring and heating of the autoclave. Before reaction, the autoclave was pressurized with 30 bars of nitrogen to check for leaks. The autoclave was then conditioned for the reaction with a vacuum of 10-2 mbar for 40 min to remove any trace of oxygen. The liquid and dissolved solid phases were introduced via a funnel. Then, the gases were introduced by double weighing (i.e., the weight difference before and after filling the autoclave with the gas).
3.3. Synthetic procedures
3.3.1. Synthesis of 4,4,5-trifluoro-5-(trifluoromethyl)-1,3-dioxolan-2-one (1)
In a round-bottom flask (100 mL), LiBr (209 mg, 2.4 mmol) was dissolved in acetone (30 mL). The solution was transferred into a reactor via a funnel. The reactor was cooled to −30 °C, and HFPO (8 g, 4.8 mmol) was introduced by double weighing and then CO2 (4 bar). The reaction was carried out at 65 °C for 14 h. Then, a pressure drop (30 bar) was noticed, probably due to the consumption of the HFPO and CO2. At 35 °C, a rapid increase in temperature to 65 °C was observed, which shows that the reaction is exothermic. During the hour that followed this exothermic event, the pressure decreased from 30 to 12 bar for a temperature maintained at 65 °C. After the reaction, the autoclave was degassed (releasing unreacted HFPO and CO2), and acetone was evaporated under a vacuum. The remaining crude was dispersed in deionized water (50 mL). The aqueous mixture was extracted three times with dichloromethane (200 mL). The organic phase was dried with Na2SO4, filtered, and the dichloromethane evaporated. The final product is a yellow liquid obtained in 81% yield (19F NMR spectrum, Figure S1 in the SI).
13C NMR (100.6 MHz, CDCl3, 𝜹): 99.68–102.11 (C–F), 114.94–116.90 (CF2), 123.81–125.70 (CF3), 159.45 and 162.69 (C=O).
19F NMR (235.2 MHz, CDCl3, 𝜹): −135.1, −132.8–129.8, −122.5 (CF), −87.2, −83.6, −78.2 (CF2), −81.8 (CF3).
3.3.2. Synthesis of 4,4,5-trifluoro-5-(trifluoromethyl)-1,3-oxathiolane-2-thione (2)
In a round-bottom flask (100 mL), LiBr (209 mg, 2.4 mmol) and carbon disulfide (CS2, 11.00 g, 144 mmol) were dissolved in acetone (30 mL). The solution was transferred into a reactor via a funnel. The reactor was cooled to −30 °C, and HFPO (8 g, 481 mmol) was introduced by double weighing. The reaction was carried out at 65 °C for 14 h. The purification was similar to that used for 1. The product was obtained as a colorless oil with 83% yield (19F and 13C NMR spectra, Figures S2 and S3 in the SI).
13C NMR (100.6 MHz, CDCl3, 𝜹): 101.9–108.9 (C–F), 113.6–125.5 (CF2 and CF3), 159.1 and 162.5 (C=S).
19F NMR (235.2 MHz, CDCl3, 𝜹): −132.8, −130.1, −122.8 (CF), −89.0, −87.4, −83.9, −78.5 (CF), −82.1 (CF3).
3.3.3. Synthesis of 4-(trifluoromethyl)-1,3-dioxolan-2-one (3)
Trifluoroethyleneoxide (15.01 g, 153 mmol) and LiBr (644 mg, 7.7 mmol) were dissolved in acetone (40 mL). The solution was transferred into a reactor, and the atmosphere was replaced with CO2 (4 bar). The reaction was carried out at 65 °C for 14 h. The reactor was degassed after reaction and cooling to release the unreacted CO2. The crude reaction product was then distilled with a Kugelrohr distillation apparatus. After purification, the product was obtained as a colorless liquid (1H, and 19F NMR spectra, Figures S4 and S5 in the SI).
1H NMR (400.1 MHz, CDCl3, 𝜹): 4.53 (m, 1H, H3a), 4.68 (m, 1H, H3b), 5.03 (m, 1H, H2).
13C NMR (100.6 MHz, CDCl3, 𝜹): 31.1 (s, CF3), 36.2 (s, C3a), 36.3 (s, C3b), 162.4 (s, C1).
19F NMR (235.2 MHz, CDCl3, 𝜹): −80.2 (s, CF3).
3.3.4. Synthesis of 5-(trifluoromethyl)-1,3- oxathiolane-2-thione (4)
In a two-neck round-bottom flask (100 mL), trifluoroethyleneoxide (10.2 g, 104.1 mmol) and carbon disulfide (9.0 mL, 104.1 mmol, 1 eq) were introduced at 0 °C with a catalytic amount of LiBr (1.48 g, 17.2 mmol, 16.5 mol%) in acetone (50 mL). The reaction was then allowed to proceed under reflux at 65 °C for 14 h. The mixture was then cooled, poured into ethyl acetate, and washed twice with water. The organic phase was dried over magnesium sulfate before evaporating the solvent under a vacuum to obtain the pure product as a yellowish liquid (1H and 19F NMR spectra, Figures S6 and S7 in the SI).
1H NMR (400.1 MHz, CDCl3, 𝜹): 3.84 (m, 2H, H3), 5.34 (m,1H, H2).
13C NMR (100.6 MHz, CDCl3, 𝜹): 31.1 (s, CF3), 36.2 (s, C3a), 36.3 (s, C3b), 162.4 (s, C1).
19F NMR (235.2 MHz, CDCl3, 𝜹): −77.6 (s, CF3).
3.3.5. Synthesis of 4-[2,2,3,3,4,4,5,5-octafluoro-6-(2-oxo-1,3-dioxolan-4-yl)hexyl]-1,3-dioxolan-2-one (5)
The procedure was similar to that of the synthesis of 1. 1,4-bis(2′,3′-epoxypropyl)perfluorobutane (10.02 g, 31.8 mmol) and LiBr (276 mg, 3.18 mmol) were dissolved in acetone (30 mL). The solution was transferred into a reactor and the atmosphere was replaced with CO2 (4 bar). The reaction was carried out at 65 °C for 96 h. During the reaction, an increase in the pressure inside the reactor was observed (up to 12 bar), followed by a decreased pressure from 12 bar to 6 bar for a temperature maintained at 65 °C. After reaction and cooling, the reactor was degassed. The crude product was washed with acetone (100 mL), filtered and evaporated under a vacuum, and led to the desired product. The product was obtained as a white powder (1H, 19F, 13C NMR spectra, and FTIR spectrum, Figures 2–4 and S8 in the SI).
Melting Point: 129 ± 2 °C.
1H NMR (400.1 MHz, CDCl3, 𝜹): 2.90 (m, CH2CF2, 4H), 4.63 and 4.22 (m, OCH2, 4H), 5.20 (m, CH, 1H).
19F NMR (235.2 MHz, CDCl3, 𝜹): −112.3 (AB system, CH2CFAFBCF2), −123.2 (s, CH2CF2CF2).
13C NMR (100.6 MHz, CDCl3, 𝜹): 154.4 (C=O), 106.6–120.72 (CF2), 70.2 (CH), 60.2 (OCH2), 39.3 (CH2CF2).
3.3.6. Synthesis of 4-[2,2,3,3,4,4,5,5-octafluoro- 6-(2-thioxo-1,3-oxathiolan-4-yl)hexyl]-1,3-oxathiolane-2-thione (6)
The procedure was similar to that of the synthesis of 2. LiBr (276 mg, 1.59 mmol), carbon disulfide (7.00 g, 10 mmol) and 1,4-bis(2′,3′-epoxypropyl)perfluorobutane (5.03 g, 15 mmol) were dissolved in acetone (30 mL). The solution was introduced into a reactor via a tight funnel. The reaction was carried out at 65 °C for 96 h. After opening the reactor, the crude reaction product was washed with dichloromethane (100 mL), filtered, and dried under a vacuum to lead to the desired product. The pure product was obtained as a white powder (1H, 19F, 13C NMR spectra, and FTIR spectrum, Figures 2 and S9–S11 in the SI).
Melting Point: 136 ± 2 °C.
1H NMR (400.1 MHz, CDCl3, 𝜹): 3.02 (m, CH2CF2, 4H), 3.92 and 3.77 (m, OCH2, 4H), 5.60 (m, CH, 1H).
13C NMR (100.6 MHz, CDCl3, 𝜹): 212.8 (C=S), 107.5–120.2 (CF2), 84.6 (CH); 62.0 (OCH2), 39.1 (CH2CF2).
19F NMR (235.2 MHz, CDCl3, 𝜹): −111.9 (AB system, CH2CFAFBCF2), −124.0 (s, CH2CF2CF2).
4. Conclusions
In this study, three fluorinated epoxides were efficiently carbonated or xanthated using mild conditions. The reaction conditions were optimized to produce the desired cyclic carbonates (1 and 3) and cyclic xanthates (2 and 4) in high yields (81–91%) in acetone at 65 °C catalyzed by LiBr. Then, original five-membered bis-CC (5) and bis-XA (6) were synthesized in high yields (85 and 87%, respectively) by reacting CX2 with 1,4-bis(2′,3′-epoxypropyl)perfluorobutane using the optimum conditions in the presence of LiBr. All the carbonates and xanthates (1–6) produced in this work were confirmed and characterized by 1H, 19F, 13C NMR, and FTIR analysis.
Conflicts of interest
The authors have no conflict of interest to declare.
Acknowledgments
The authors are grateful to the Institut de Chimie of the CNRS and CEA Le Ripault, France for financial support by supplying us with free materials, TOSOH Finechemicals and INC (Shiba, Japan), and SynQuest (Alachua, USA) that made this research possible. The authors thank Ecole Nationale Supérieure de chimie de Montpellier (ENSCM) and the Université Libanaise (UL). The authors also thank Prof. Bernard Boutevin (ENSCM), Dr. Vincent Besse, Dr. Sandy Gosset (ENSCM) and Dr. Daoud Naoufal (UL).