



 CC-BY 4.0
CC-BY 4.0
1. Introduction
Solvent environment and, particularly, the presence of hydrogen bonds-donating or -accepting interactions as well as the occurrence of stacking and dipole–dipole interactions are expected to have a significant impact on the photophysics of fluorophores like coumarin 153 (C153) [1, 2, 3, 4]. For this reason, the effect of the solvent on the photophysics of C153 has been explored in a range of solvents, including alcohols [5, 6, 7], acetonitrile [2, 8, 9, 10], propylene carbonate [6], dimethyl sulfoxide, formamide, nitromethane, acetone [2], methanol, ethanol and fluorinated ethanol solvents [11], in mixtures such as dioxane–water [12], acetonitrile–benzene [13], hexane–propionitrile [12], tert-butyl alcohol–water and trimethylamine N-oxide–water [14], acetonitrile–propylene carbonate [15] dimethyl sulfoxide–glycerol [16, 17], toluene–acetonitrile [18], and cyclohexane–phenol solvent [19]. These studies were extended to aqueous and non-aqueous reverse micelles [20], Triton X-100-cyclohexane microemulsions [21], and also to various ionic liquids, such as 1-butyl-3-methylimidazolium hexafluorophosphate [22], 1-hexyl-3-methylimidazolium hexafluorophosphate [8], 1-butyl-3-methylimidazolium tetrafluoroborate [10], hydroxylfunctionalized ionic liquids [23], ionic liquids containing fluoroalkylphosphate [24] and tetraalkylammonium bromide [25], 1-dodecyl-3-methylimidazolium bis(trifluoromethylsulfonyl)amide with benzene, chloroform, propylene carbonate solvents [26], and deep eutectic solvents such as choline chloride [27] and acetamide–urea [28]. One should notice that the properties of the mixtures are modulated through the change of the mixture composition. The aforementioned works showed that the strength and nature of the hydrogen bonds can influence the electronic transitions of C153, resulting in either fluorescence quenching or enhancement. Indeed, the efficiency of fluorescence, expressed in terms of quantum yield, can be affected by hydrogen bond interactions as they may promote non-radiative decay processes and reduce the quantum yield. It is therefore essential to take into account the specific molecular structure of C153 (namely, the donor or acceptor nature of its atoms) and the characteristics of the solvent in experimental studies to understand how hydrogen bond interactions influence C153 photophysics in a given environment. This can be illustrated through the following examples. First, Maronceli et al. showed that the presence of hydrogen bonds between C153 and alcohols results (beyond the effects of solvent polarity) in a small additional shift in both the absorption and emission spectra of C153. However, when examining the solvation dynamics of C153 in N-methylpropionamide, a solvent with hydrogen bond donor ability, distinct differences in dynamics were observed compared to the case when alcohol solvents were considered [6]. On the other hand, in propylene carbonate, a non-associated solvent lacking hydrogen bond donor capability, the behavior of the solvation function closely resembles that observed in alcohols. Other studies, instead, showed that coumarin 102 and C153 have different photophysical behaviors in the same solvent and that these different behaviors are due to their methyl CH3 and trifluoryl CF3 substituents, respectively [29, 30]. Computer molecular modeling could corroborate these experimental results, since simulation approaches can provide deep atomistic insights into the local structure of the solvent around the donor and acceptor regions of C153, insights that cannot be obtained by any kind of experimental strategy. As a matter of fact, the microscopic environment surrounding the C153 molecule was also investigated through quantum-chemical calculations and molecular dynamics (MD) simulations [11, 31], which permitted to analyze interatomic and intermolecular radial distribution functions (RDF)—considering either the center-of-masses or specific atoms like O, F, N, and C—without explicitly accounting for the hydrogen atoms of C153 and the solvent molecules. Notably, these investigations were conducted for both the ground state (GS) and excited state (ES) of C153. Based on molecular dynamics simulations of C153 in 1,4-dioxane, Cinacchi et al. [32] suggested that changes in the solvation shell around the GS and ES of C153 are primarily due to alterations in the orientation of the solvent molecules. In other MD studies, the local structure around the hydrogen atoms of C153 has been explored [26, 31]. It has been observed that C153 is solvated by alcohols or water through hydrogen bonds, specifically between the hydroxyl OH group of the solvents and the carbonyl oxygen atom of C153. Furthermore, MD simulations of C153 in dimethyl sulfoxide (DMSO)–water mixtures have shown that the hydration structures in GS and ES differ from those in pure water due to the significant influence of DMSO molecules [27]. This was attributed to the formation of a hydrogen bond network between DMSO and water molecules upon mixing. In the case of imidazolium ionic liquids (ILs), MD studies of C153 in 1-ethyl-3-methylimidazolium tetrafluoroborate (EmimBF4) [22] demonstrated a preferential solvation of C153 by the Emim+ cation. In a study by Barman [33], MD simulations, quantum-chemical calculations and infrared (IR) spectroscopy were combined to investigate the formation of hydrogen bonds between aniline and C153. It was concluded that the carbonyl group of C153 serves as a primary hydrogen bond-accepting site and shows a higher bond strength in the ES. Furthermore, the formation of a C=O⋯ H–N hydrogen bond was confirmed by the observation of the IR absorption band at 1736 cm−1 in the IR spectrum of C153 in the aniline–cyclohexane mixture, shifted compared to the stretching band of the C=O group (1748 cm−1) which is commonly measured in pure cyclohexane. Similar experiments were conducted for the coumarin C102–aniline system [34], and the formation of the stronger hydrogen bond for C153 in the ES was also observed in methanol solutions [35]. Furthermore, the solvation of C153 in ionic 1-butyl-3-methylimidazolium tetrafluoroborate–propylene carbonate mixtures was investigated by coupling MD simulations, time-correlated photon counting and fluorescence upconversion techniques. It was established that the solvation of C153 is determined by its interaction with the ions at high IL content, and with the solvent molecules at a IL molar fraction lower than 0.2 [36]. Xu et al. [37] conducted B3LYP/TZVP calculations to thoroughly explore the formation of hydrogen bonds between C153 and ethanol in both GS and ES. They observed that the hydrogen bond C=O⋯ H–O is strengthened in the electronic ES, i.e., the bond length decreases from 1.867 Å in GS to 1.813 Å in ES, as also shown by Han and coworkers [38], This observation suggests that the hydrogen bond O⋯ H–O in the C153–EtOH complex in the ES must be strengthened. This suggestion was also substantiated by IR experiments conducted in this study [38].
In order to retrieve essential information about hydrogen bond interactions, geometric data [39, 40], spectroscopic data [41, 42, 43, 44, 45, 46, 47], physical chemical data [48], and MD simulation data [49, 50, 51, 52, 53] have been usually analyzed by means of principal components analysis (PCA). PCA can provide a comprehensive overview of hydrogen bond interactions offering insights into dominant spectral features, their variations, global trends, and correlations, and aiding in the identification and understanding of hydrogen bond-related patterns in the collected data.
This article provides a thorough study of the hydrogen bond interactions between C153 and various solvents, including methanol, ethanol, 1-propanol, 1-butanol, acetonitrile, γ-butyrolactone, and propylene carbonate. These solvents exhibit distinct properties such as dipole moment, viscosity, dielectric constant, density, and the capacity to form hydrogen bonds, as well as stacking interactions and dipole–dipole interactions. In this study, we combine MD simulations with PCA. Atomistic simulations provide the coordinates of atoms that are involved in the hydrogen bond interactions. The generic hydrogen bond between a donor D–H and acceptor atom A is described by the configuration given in Figure 1.
Definition of the distances describing the hydrogen bond interactions between a generic donor D–H and a generic electronegative acceptor A. The distances dH…A and dD…A are calculated using molecular dynamic simulations relying on the nearest neighbor approach.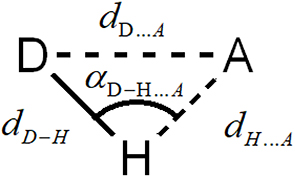
The D–H and A moieties may belong to any of the mixture’s constituents. The intramolecular distance dD–H is assumed to remain constant in our simulations, while dH…A and dD…A are determined as the average distances of the nearest neighbor radial distributions of the first neighbor electronegative atom A with respect to the H and D, respectively. It is worth noting that once dD–H, dH…A and dD…A are determined, the angle 𝛼 can be calculated using the law of cosines (see Figure 1).
The average values of distances dH…A and dD…A were calculated for both GS and ES of C153 and serve as indicators of the strength of the hydrogen bond interactions between C153 and the solvent molecules. Importantly, the GS and ES of C153 were simulated by considering their corresponding charge distributions, which were determined through quantum calculations (see Figure 3). These distances were calculated for cases where C153 acted as either a hydrogen bond donor or acceptor. In the former scenario, the 14 C–H bonds of C153 were considered, and these distances were calculated from electronegative atoms such as the N1 atom of acetonitrile, the O1 atoms of the alcohol solvents, and the O1, O2, and O3 of the other solvents. In the latter case, these distances were calculated between each of the 39 C–H bonds of the solvent molecules and the 6 electronegative atoms of C153, i.e., the N1, O1–2 and F1–3 atoms (see Figure 2 for more details on the numbering of the atoms).
Atomic labels for Coumarin-153 (a) and the investigated solvents ((b) methanol, (c) ethanol, (d) 1-propanol, (e) 1-butanol, (f) γ-butyrolactone, (g) propylene carbonate, (h) acetonitrile).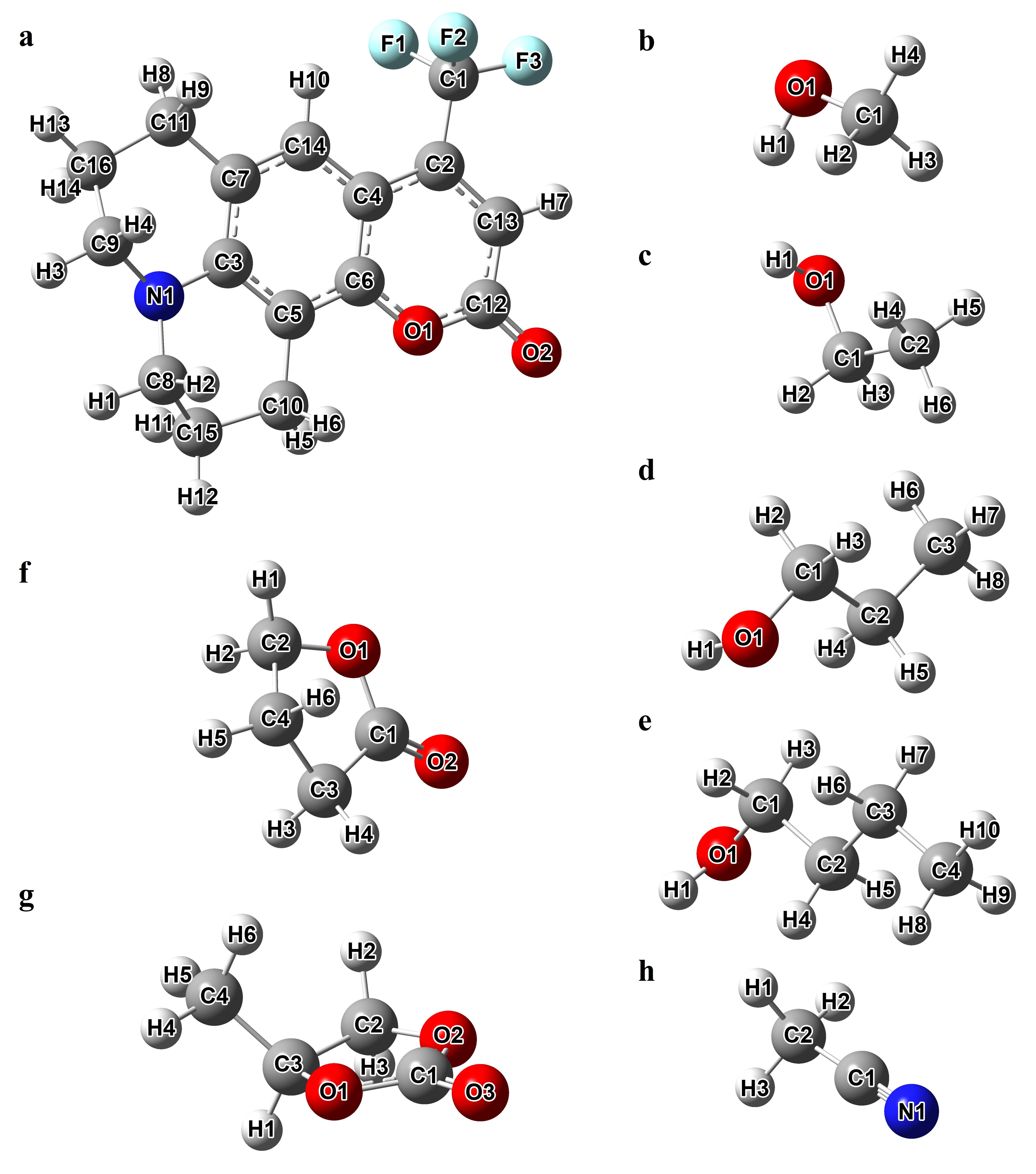
Charge values associated to the atoms of C153 in the ground state and excited state as determined from quantum-chemical calculations performed with B3LYP/6-311+G(d).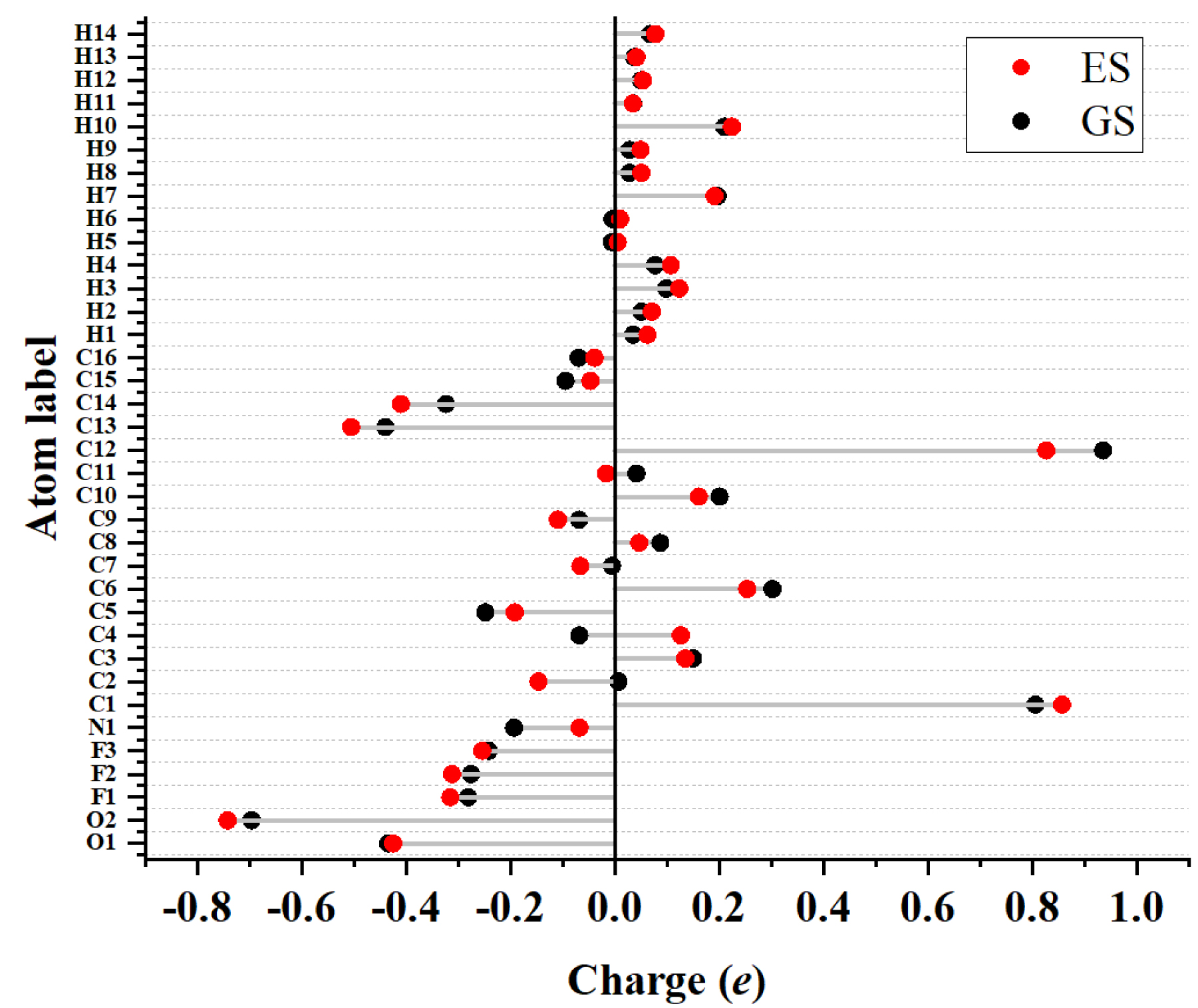
PCA was then applied to analyze the two distance matrices obtained in the two aforementioned cases. The first matrix contained 10 rows corresponding to the 10 electronegative atoms of the different solvents under study and 28 columns corresponding to the 14 C–H bonds of C153 in both GS and ES. The second matrix contained 43 rows corresponding to the 39 C–H and the 4 O–H bonds of the different solvents under study and 12 columns corresponding to the 6 electronegative atoms of C153 in both GS and ES. PCA helps in understanding the structural patterns encoded by the investigated data and extracting essential information regarding the nature of the hydrogen bond interactions as captured by the calculated distance values. The questions we would like to answer in this paper are: When C153 is acting as a hydrogen bond donor, are all its H atoms equivalent or do some of them exhibit a particular behavior? Is there any difference among the acceptor atoms of the solvents? What happens to the hydrogen atoms that are close to the electronegative atoms of C153? Is there a difference in the behavior of the H atoms of the solvents depending on their respective location (e.g., close to or far from the solvent electronegative atoms)?
The paper is organized as follows: the second section details the MD simulation procedure and the calculation of the distance matrices which were used to investigate the hydrogen bond interactions of C153 in the selected solvents. In the third section, the results of this study are discussed. The fourth section provides conclusions and future perspectives.
2. Methodology
A single C153 molecule was immersed into pure solvents-acetonitrile (AN), γ-butyrolactone (GBL), propylene carbonate (PC), methanol (MeOH), ethanol (EtOH), 1-propanol (PrOH), and 1-butanol (BuOH). Both solvent and solute molecules were modeled as non-polarizable, rigid bodies containing a collection of interaction sites. All-atom models were used for the solvents and the solute. Initial configuration and force field files were prepared using PACKMOL [86] and DL_FIELD (version 3.3), respectively [54], and the simulations were carried out in DL_POLY (version 4.07) [55]. These simulations were performed at constant values of the number of molecules, N, the pressure P, and the temperature T on systems containing 1 molecule of solute and 863 molecules of solvents, placed in a cubic box with periodic boundary conditions at an average temperature of 298 K and pressure 1 atm. The NPT ensemble was maintained using a Berendsen thermobarostat, with a relaxation time of 0.1 and 0.2 ps for the thermostat and the barostat, respectively. The Lennard-Jones forces were cut off at 15 Å and long-range Coulomb interactions were treated using the Ewald sum method. The equations of motion were solved using combined SHAKE and velocity algorithms. The equilibration of the systems as well as the production of the HISTORY file for further analysis were performed with a time step of 0.0005 ps and a total number of steps equal to 1,000,000. The configurations of the system were recorded in the HISTORY file every 10 steps, which produced 100,000 configurations that were treated by TRAVIS (version 17/08/12) [56]. This permitted better statistics and a noise level decrease for the radial distribution functions calculated between a single C153 molecule and the solvents.
All the calculations were performed in both GS and ES of C153. The charges of the equilibrated system in GS were switched to that of the ES (without changes in the C153 geometry) and the system was left to evolve under the new solute-solvent interactions for 1,000,000 steps with a time step of 0.0005 ps. The geometry of the molecule did not undergo any change.
We used the force field developed by Cinacchi for C153. The atoms’ numbering is given in Figure 2 [32].
Quantum-chemical calculations were performed with the B3LYP/6-311+G(d) level of theory using Gaussian09W [91] to obtain the atom positions as well as the Mulliken atomic charges for GS and ES of C153 [41]. The values of these charges are displayed in Figure 1. The OPLS2005 force field was used for alcohols [57], while the force field proposed by Koverga was used for propylene carbonate (PC), γ-butyrolactone (GBL), and acetonitrile (AN) [58].
PCA [96] was then used to get insights into the solvation structure of C153 in GS and ES. Indeed, for each possible hydrogen bond D–H⋯ A, the nearest neighbor radial distribution of the electronegative atom of a given compound around either the H or D atoms of the donor compound was calculated. Based on this distribution, the average interatomic distances dH…A and dD…A were computed and merged for the sake of simplicity in a unique distance metric d as follows:
The resulting distance values were finally gathered into two different data matrices, one containing the d values estimated when C153 was assumed to act as a donor of hydrogen bonds and the other when C153 was assumed to act as a hydrogen bond acceptor. The first matrix contains 10 rows corresponding to the 10 electronegative atoms of the different solvents under study and 28 columns corresponding to the 14 C–H bonds of C153 in both GS and ES. Each one of its elements represents the distance between a given electronegative atom and one of the 28 C–H bonds of C153. The second matrix contained 43 rows corresponding to the 39 C–H and the 4 O–H bonds of the different solvents under study and 12 columns corresponding to the 6 electronegative atoms of C153 in both GS and ES. Each one of its elements represents the distance between one of the 43 solvent bonds and a given electronegative atom of C153. Both distance matrices were double-centered and finally subjected to PCA by means of in-house-developed Matlab routines. Raw data are displayed in Figure 4.
Raw d values obtained through molecular dynamics simulations when considering C153 as a hydrogen bond donor (left) or acceptor (right). The numbering of atoms is given in Figure 2.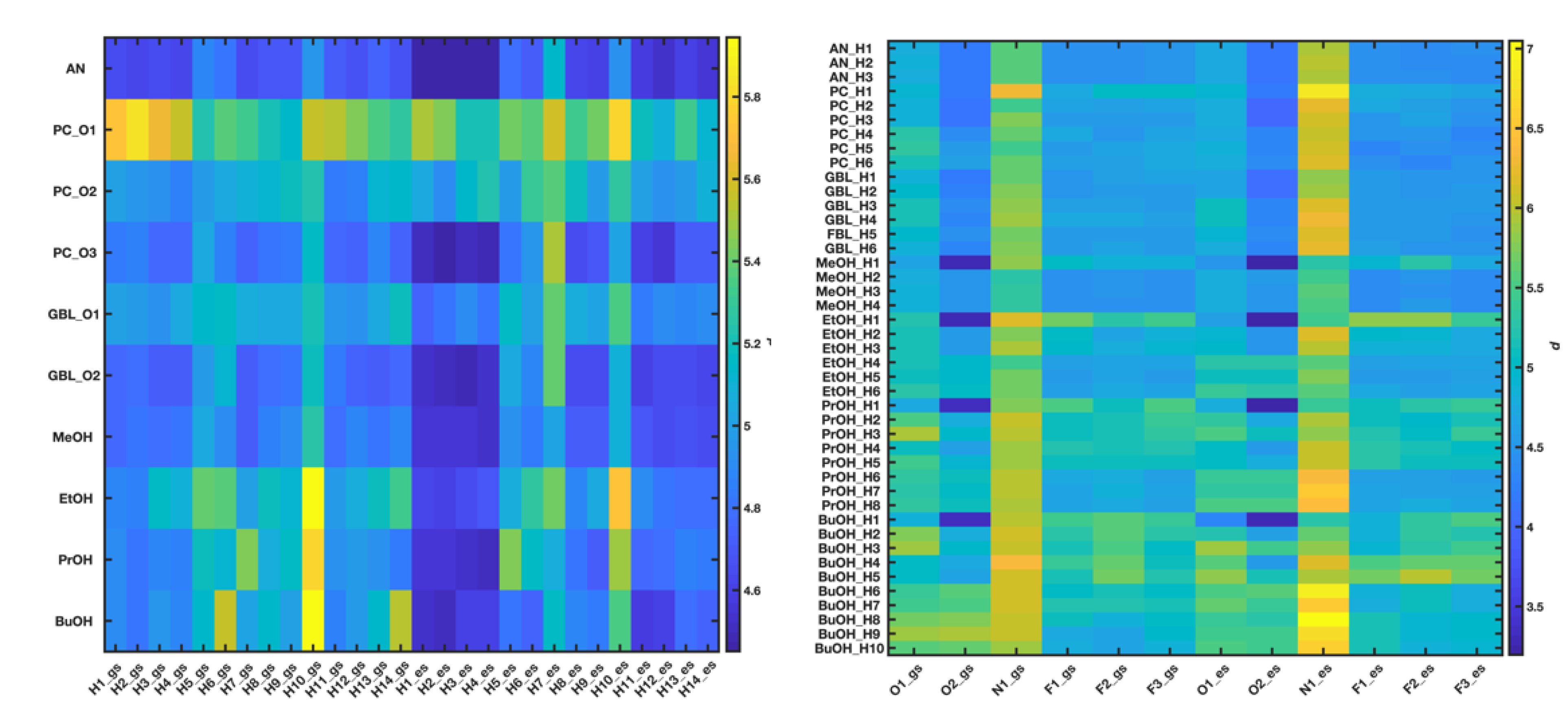
3. Results
3.1. Coumarin-153 as hydrogen bond donor
Figure 5 displays the first/second and the third/fourth principal component biplots resulting from the PCA decomposition of the distance matrix obtained in the case C153 acts as hydrogen bond donor1 . PC1 highlights a difference in the interacting trend of the H1 and H2 atoms located near the nitrogen atom of the quinolizidine heterobicyclic group of C153 with respect to the H10 atom that is close to the trifluoromethyl group of C153. The former seems to interact preferentially with the hydroxyl O1 atom of BuOH, EtOH, and PrOH and be located relatively far from the O atoms (O1 and O2) of the PC. The latter exhibits the opposite behavior. This is corroborated by the nearest neighbor distributions represented in Figure 6 and calculated using the following configurations: C14–H10⋯ A and C8–H2⋯ A (A = O1 or O2 of PC or O1 of BuOH), respectively.
First/second (left) and third/fourth (right) principal component biplots resulting from the PCA decomposition of the distance matrix obtained when considering C153 as a hydrogen bond donor in both its ground and excited states. Blue squares refer to the solvent’s electronegative atoms (acceptors), while red diamonds refer to C153’s hydrogen atoms (donors).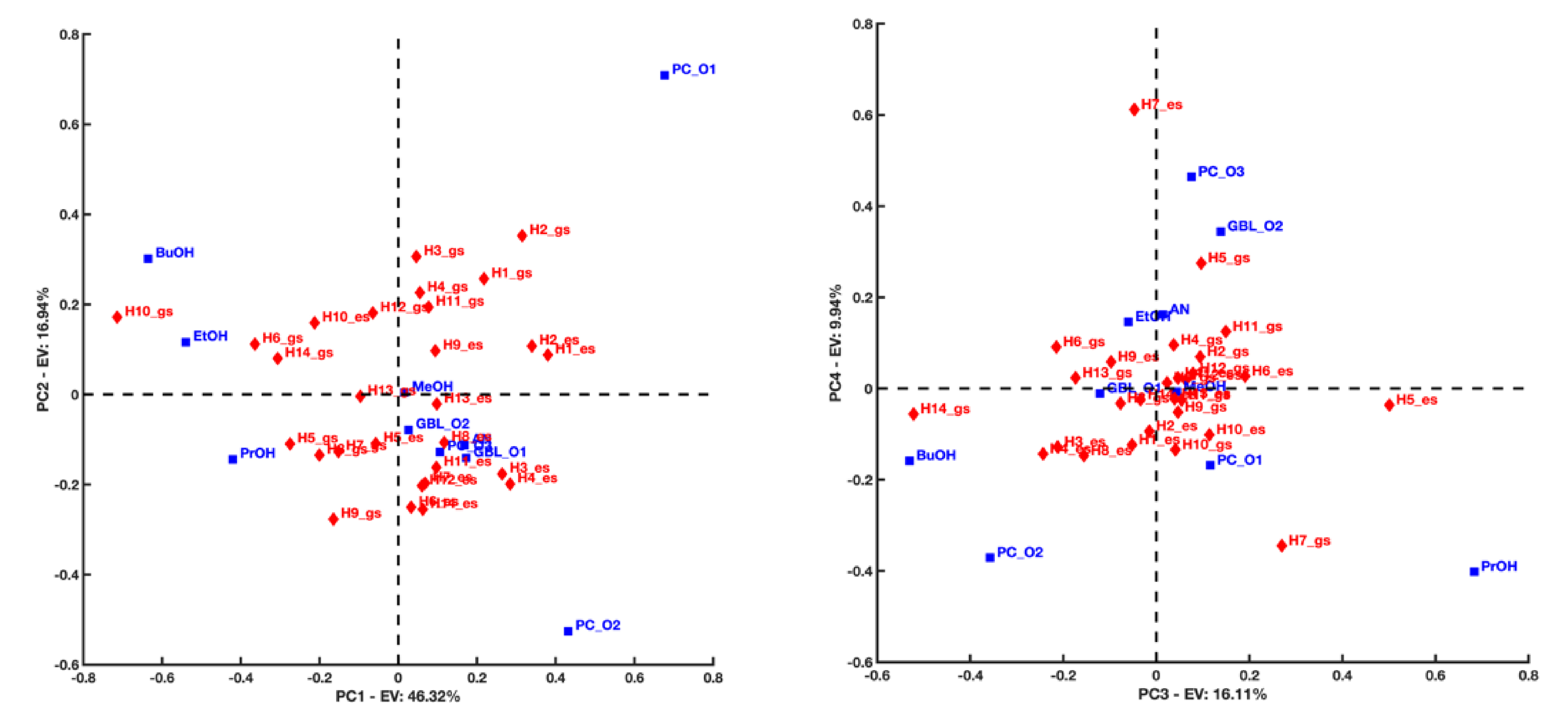
Radial distribution of the nearest neighbor for the atomic couples (i) O1 of BuOH/H10 of C153 (left black), (ii) O1 of PC/H10 of C153 (left red), (iii) O1 of BuOH/H2 of C153 (right black) and (iv) O1 of PC/H2 of C153.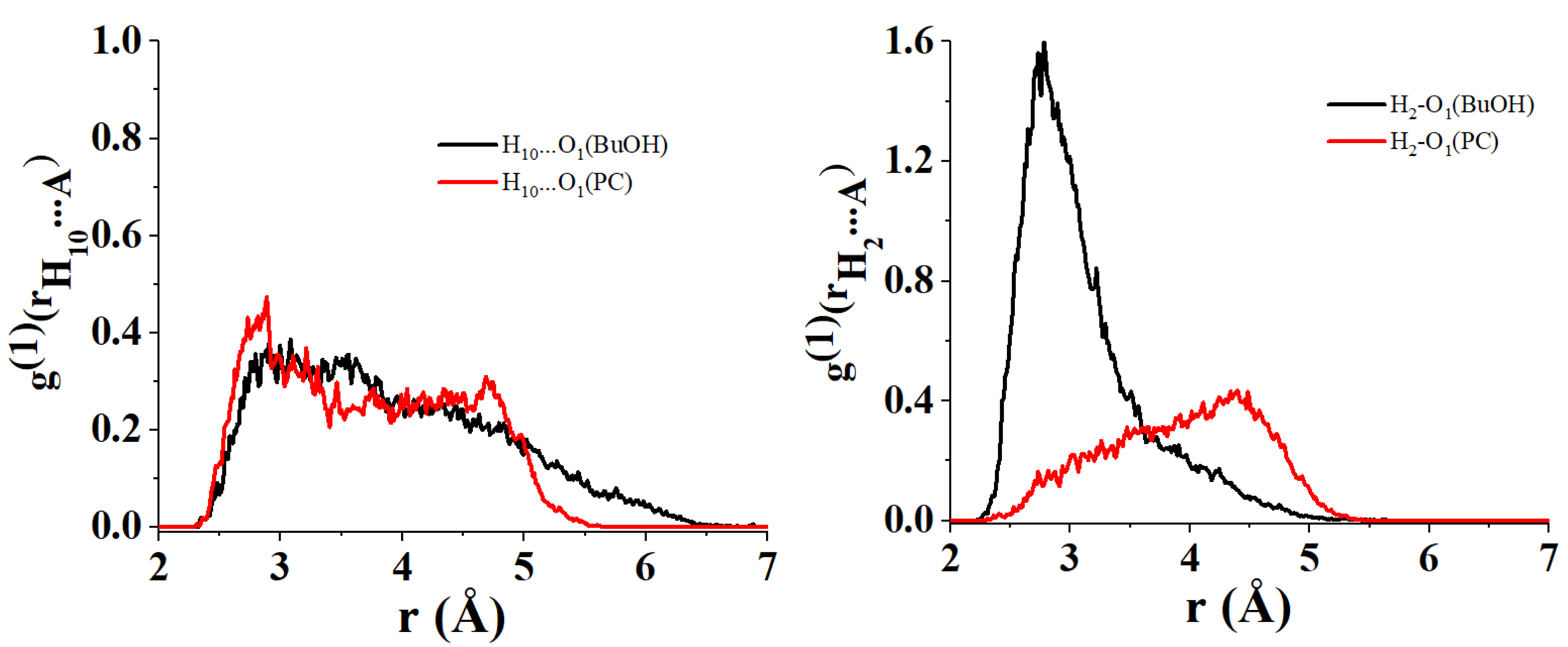
Interestingly, PC2 suggests that the distances between the H3 and H4 atoms of C153 to the O1 atom of PC are shortened when C153 goes from GS to ES. The opposite occurs when considering the distances of these H atoms to the O2 atom of PC. This is in good agreement with the changes observed for the nearest neighbor radial distributions concerning these H atoms and the O1 and O2 of PC (see Figure 7). Figure 7 left highlights, in fact, the emergence of a short-distance contribution that reduces the average distance between H3 and H4 to the O1 of PC. Conversely, from Figure 7 right, it is evident that a long-distance contribution makes the average distance between H3 and H4 to the O2 of PC increase. It is interesting to notice that the same conclusion cannot be drawn for the H1 and H2 atoms of C153 (their projection coordinates remain positive along PC2 when C153 goes from GS to ES). This suggests that the distance H1⋯O1, H1⋯O2, H2⋯O1 and H2⋯O2 do not vary as much as for H3 and H4 when C153 goes from GS to ES (see also Figure 8).
Radial distribution of the nearest neighbor for the atomic couples (i) O1 of PC/H3 of C153 in its ground state (GS) (left black), (ii) O1 of PC/H4 of C153 in GS (left red), (iii) O1 of PC/H3 of C153 in its excited state (ES) (left green), (iv) O1 of PC/H4 of C153 in ES (left blue), (v) O2 of PC/H3 of C153 in GS (right black), (vi) O2 of PC/H4 of C153 in GS (right red), (vii) O2 of PC/H3 of C153 in ES (right green) and (viii) O2 of PC/H4 of C153 in ES (right blue).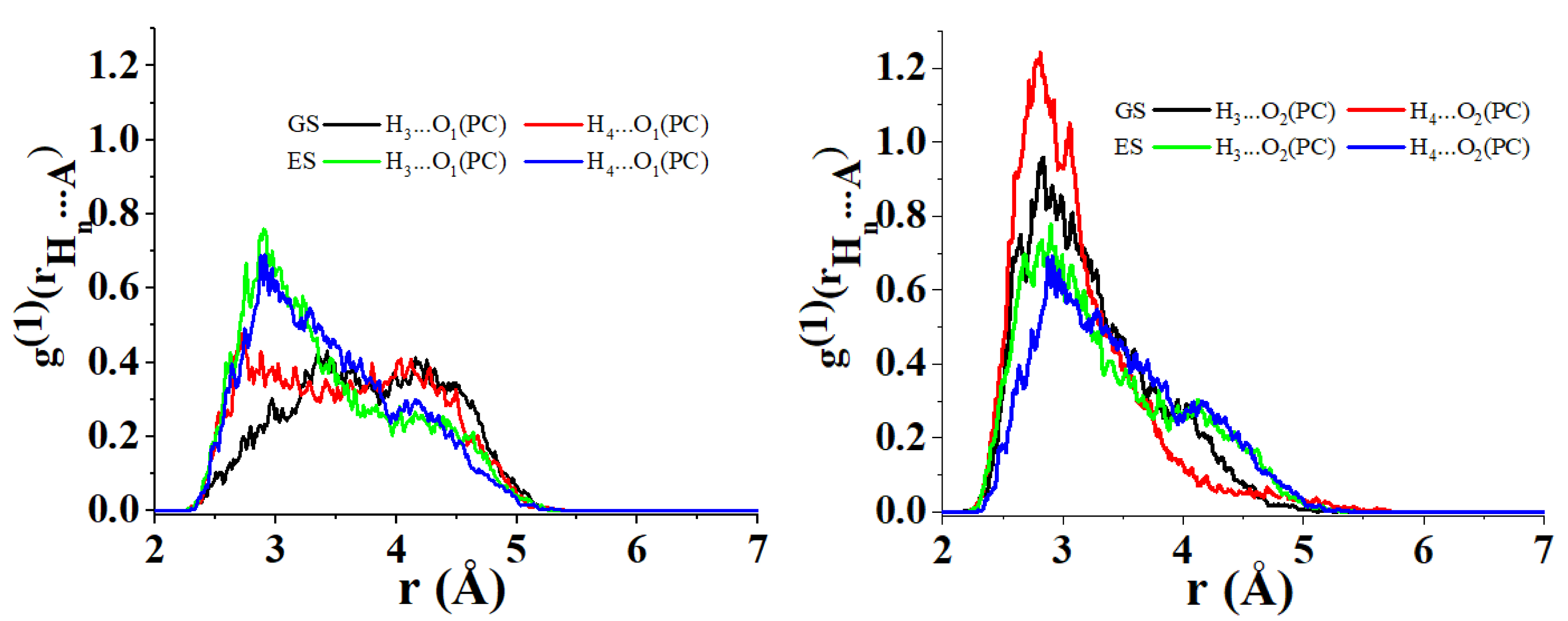
Radial distribution of the nearest neighbor for the atomic couples (i) O1 of PC/H1 of C153 in its ground state (GS) (left black), (ii) O1 of PC/H2 of C153 in GS (left red), (iii) O1 of PC/H1 of C153 in its excited state (ES) (left green), (iv) O1 of PC/H2 of C153 in ES (left blue), (v) O2 of PC/H1 of C153 in GS (right black), (vi) O2 of PC/H2 of C153 in GS (right red), (vii) O2 of PC/H1 of C153 in ES (right green) and (viii) O2 of PC/H2 of C153 in ES (right blue).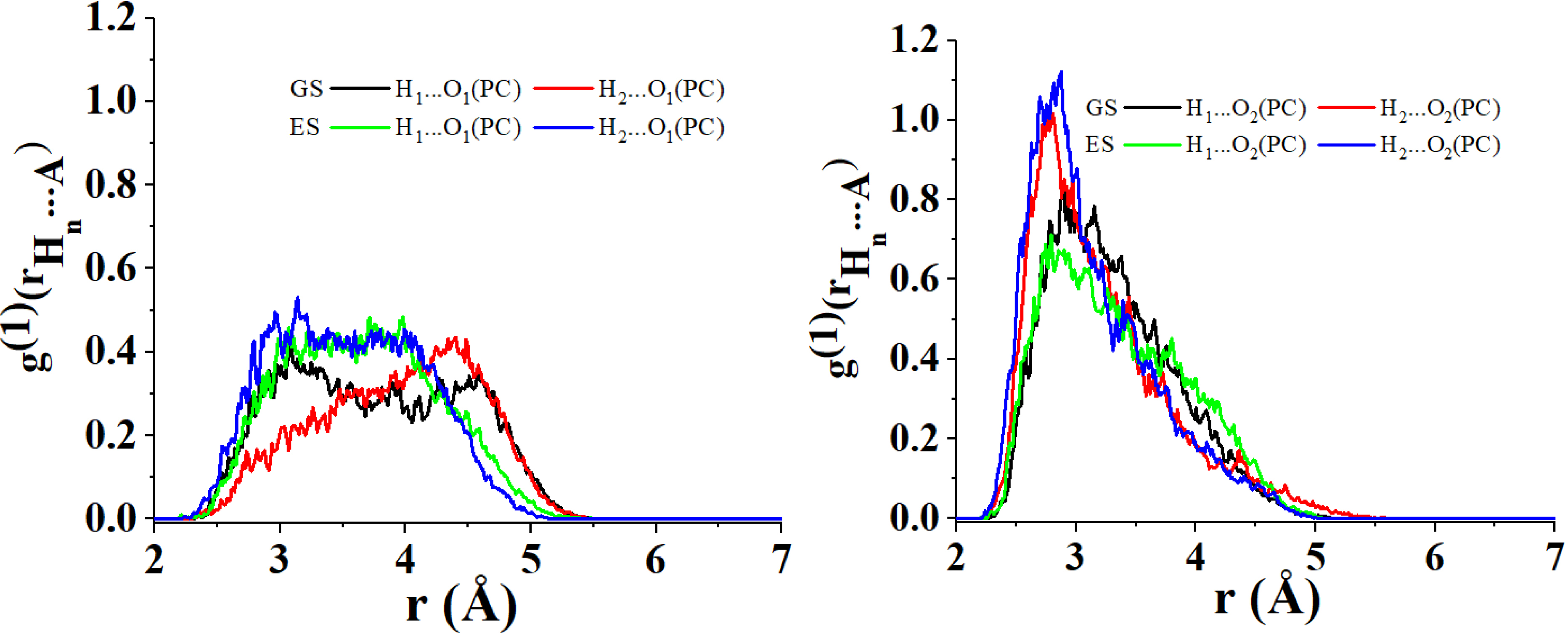
Finally, PC3 highlights a difference in the interacting behavior of the H5 atom of C153 in ES and the H14 atom of C153 in GS. It seems that H5 interacts preferentially with the O atom of BuOH and the O2 atom of PC and is located relatively far from the O atom of PrOH and the O1 atom of PC. An opposite interaction trend is instead observed for H14.
It is also interesting to notice that PC4 points out a difference in the interacting behavior of the H7 atom when C153 goes from GS to ES. When C153 is in GS, H7 seems to be located quite far from the O of PrOH. In contrast, this distance seems to significantly decrease when C153 is in ES. Additionally, the PC3–PC4 biplot suggests that the interacting behavior of the atoms O3 in PC and O2 in GBL might exhibit similarities.
3.2. Coumarin-153 as hydrogen bond acceptor
Figure 9 displays the first/second principal component biplot resulting from the PCA decomposition of the distance matrix obtained in the case C153 acts as a hydrogen bond acceptor2 . PC1 highlights a difference in the interacting trend of the carboxyl O2 atom of C153 with respect to the F atoms of the trifluoromethyl group of C153 in both GS and ES. The O2 atom, in fact, seems to interact preferentially with the H1 atoms of the hydroxyl groups of the alcohol solvents from which the F1−3 atoms appear relatively far. The latter, on the other hand, were found to generate preferential interactions with the terminal methyl or ethyl groups of some of the investigated solvents. Furthermore, PC1 also suggests that, overall, the distance between the N1 atom of C153 and the terminal H atoms of some of the investigated solvents (e.g., PrOH and BuOH) increases when C153 goes from GS to ES. This is clearly corroborated by the nearest neighbor distributions displayed in Figure 10.
First/second component biplot resulting from the PCA decomposition of the distance matrix obtained when considering C153 as a hydrogen bond acceptor in both its ground and excited states. Blue squares refer to the solvent’s hydrogen atoms (donors), while red diamonds refer to C153’s electronegative atoms (acceptors).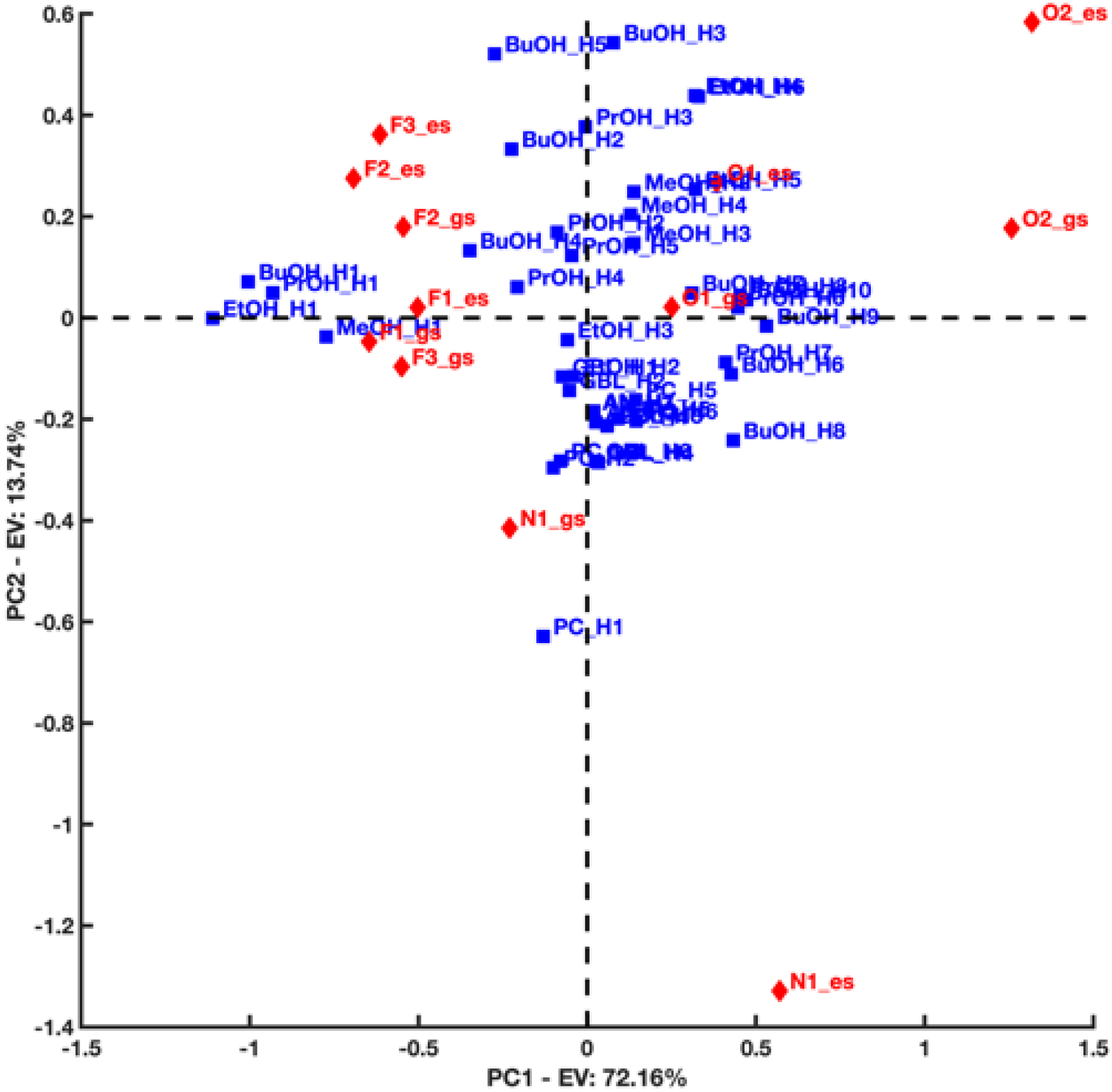
Radial distribution of the nearest neighbor for the atomic couples (i) O2 of C153 in its ground state (GS) and excited state (ES)/H1 of EtOH (left black), (ii) F1 of C153 in GS/ES/H1 of EtOH (left red), (iii) O2 of C153 in GS/ES/H5 of EtOH (left green), (iv) F1 of C153 in GS/ES/H5 of EtOH (left blue), (v) N1 of C153 in GS/H6 of BuOH (right black), (vi) N1 of C153 in GS/H8 of BuOH (right red), (vii) N1 of C153 in ES/H6 of BuOH (right green) and (viii) N1 of C153 in ES/H8 of BuOH (right blue).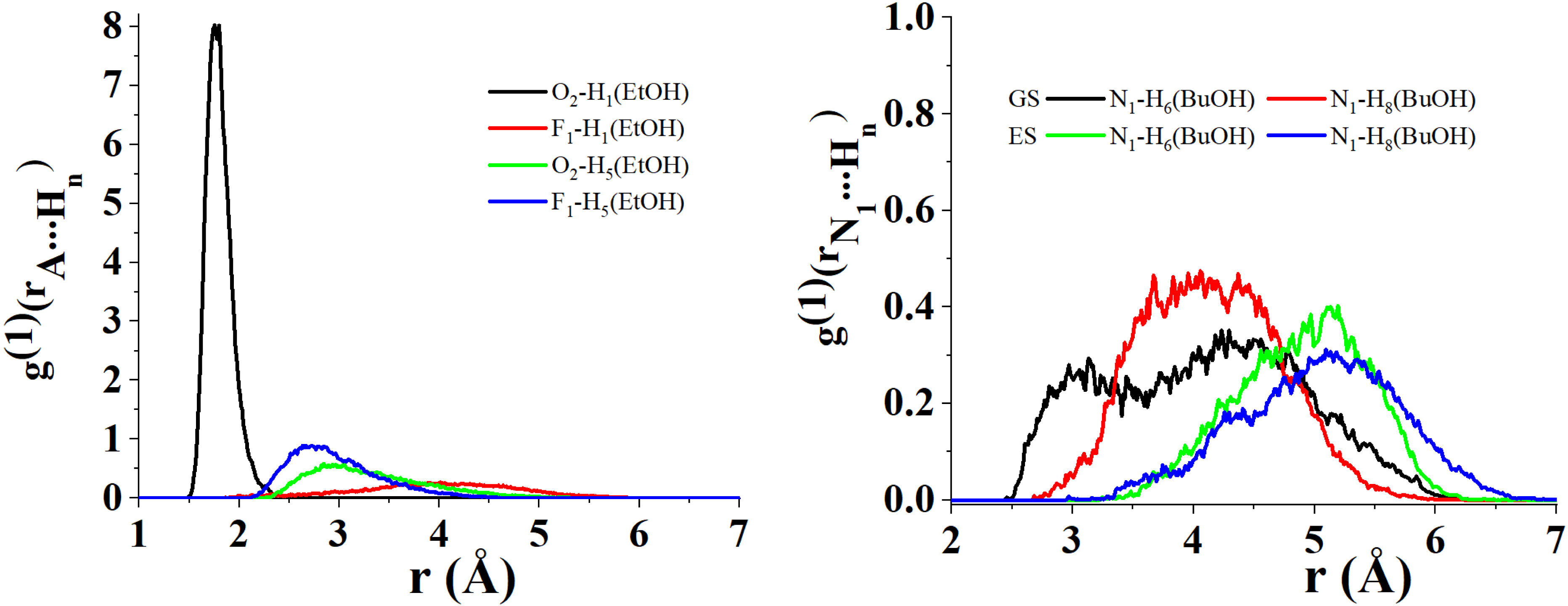
The following paragraphs will focus on the hydrogen-bonding interactions involving O2 of C153 and H1 of the hydroxyl group of the alcohol solvent on the one hand and H1 of the PC and GBL solvents on the other hand. We then calculated the values of the distances dH…A and dD…A describing their interactions. More precisely, we calculated these distances for the fifth neighbors. These distances were compared to those representative of two extreme configurations of D–H corresponding to linear and bent geometries. In the former, angle 𝛼 is equal to 180°, which implies dD…A = dD−H + dH…A. This corresponds to a strong interaction. In the latter, 𝛼 is equal to 90° implying . This corresponds to a weak interaction. Figure 11 shows that for all the alcohol solvents, the values of the first two neighbor distances are similar to those typical of a linear geometry of the O1–H1⋯ O2. In the ES, this similarity is even more pronounced, which indicates a general reinforcement of the interactions between the solvents and C153. This is in accordance with the findings of Cerezo et al. [33].
Graphical representation of the two distance values characteristics of the hydrogen-bonding interaction between the carbonyl O2 atom of C153 and the hydroxyl H1 atom of alcohols for the five nearest neighbors in the ground (a) and excited state (b) of C153. The line corresponding to 𝛼 = 180° reflects a linear O1–H1⋯ O2 configuration (i.e., dO 2…O1 = dO 2…H1 + dO 1–H1), while the line corresponding to 𝛼 = 90° reflects a bent O1–H1⋯ O2 configuration (i.e., ).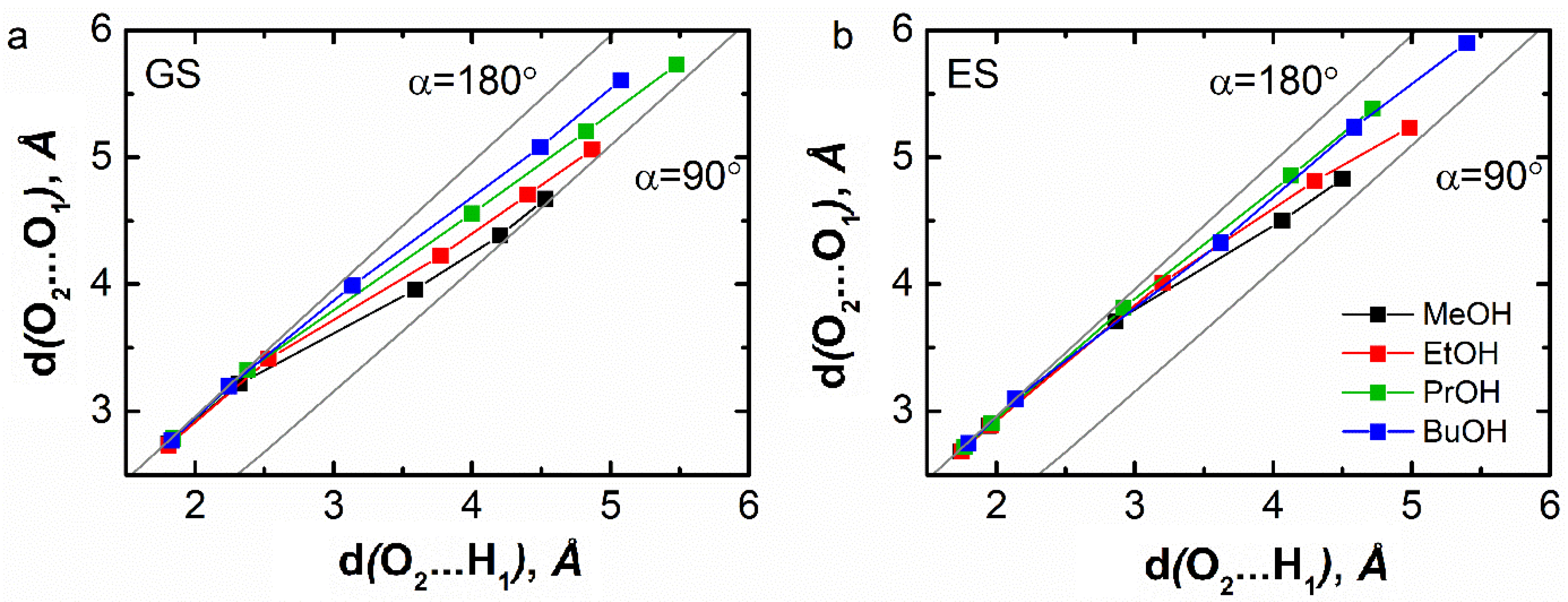
Conversely, regarding AN, GBL, and PC (see Figure 12), the calculated distances are very close to those characteristic of a bent geometry which emphasizes the occurrence of weak interactions between C153 and these solvents. Notice that when C153 is in ES, these distances do not vary and remain similar to those typical of a bent geometry.
Graphical representation of the two distance values characteristics of the hydrogen-bonding interaction between the carbonyl O2 atom of C153 and the H1 atom of propylene carbonate, γ-butyrolactone, and acetonitrile for the five nearest neighbors in the ground (a) and excited state (b) of C153. The line corresponding to 𝛼 = 180° reflects a linear C–H1⋯ O2 configuration (i.e., dO 2…O1 = dO 2…H1 + dO 1−H1), while the line corresponding to 𝛼 = 90° reflects a bent C–H1⋯ O2 configuration (i.e., ).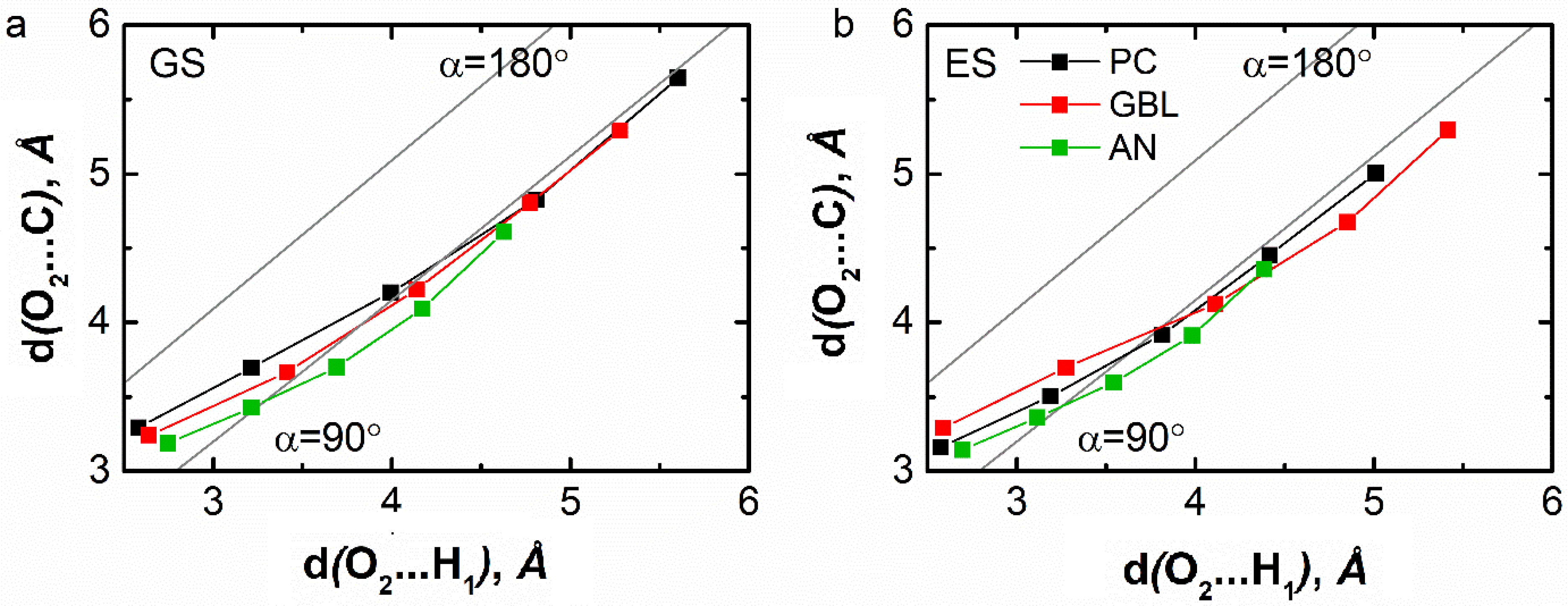
4. Conclusions
The aim of this work was to study the hydrogen bond interactions of C153 in various solvents, including alcohols, acetonitrile, γ-butyrolactone, and propylene carbonate. To do this and highlight the existence of similarity patterns in the interacting behavior of donor and acceptor moieties, we combined MD simulations and PCA. Hydrogen bonds were here described by the distances between donor and acceptor atoms. These distance values were calculated using the nearest neighbor radial distribution approach in both GS and ES of C153. These two states were modeled with different charge distributions but retained the geometrical structure of C153. PCA highlighted that, when C153 acts as a hydrogen bond donor:
- the H1 and H2 atoms of C153 interact preferentially with the hydroxyl O1 atom of BuOH, EtOH, and PrOH;
- the H10 atom of C153 is instead located at a short distance to the O1 and O2 of the PC;
- the distances between the H3 and H4 atoms of C153 to the O1 atom of PC are shortened when C153 goes from GS to ES. The opposite occurs when considering the distances of these H atoms to the O2 atom of PC;
- the H5 atom of C153 interacts preferentially with the O1 atom of BuOH and the O2 atom of PC and is located relatively far from the O1 atom of PrOH and the O1 atom of PC. An opposite interaction trend is instead observed for H14;
- when C153 is in GS, its H7 atom seems to be located quite far from the O1 of PrOH. In contrast, this distance seems to significantly decrease when C153 is in ES.
In addition, it was observed that when C153 acts as a hydrogen bond acceptor:
- the carboxyl O2 atom of C153 interacts preferentially with the H1 atoms of the hydroxyl groups of the alcohol solvents;
- the F1−3 atoms of C153 were found to generate preferential interactions with the terminal methyl or ethyl groups of some of the investigated solvents;
- the distance between the N1 atom of C153 and the terminal H atoms of several of the solvents under study (e.g., PrOH and BuOH) increases when C153 goes from GS to ES.
The hydrogen bond interactions of C153 were also assessed via the behavior of the fifth first neighbors. Our results show that the distance values involving the hydroxyl H atoms of the alcohols are close to those associated with strong hydrogen bond interactions (linear configuration of O2⋯H1–O2). These distances are even shortened in the ES of C153 which indicates a further reinforcement of the hydrogen bonds. On the other hand, the distance values involving the H atoms of PC and GBL are closer to those associated with weak hydrogen bond interactions (bent configuration of O2⋯H1–C). Moreover, they do not vary when C153 goes from GS to ES.
Declaration of interests
The authors do not work for advise, own shares in, or receive funds from any organization that could benefit from this article, and have declared no affiliations other than their research organizations.
Funding
The authors acknowledge financial support from the French National Agency for Research (ANR-19-CE05-0009-01 Ultrafast photoinduced processes of organic dyes in ionic liquid/molecular solvent mixtures designed for dye solar cells).
Acknowledgments
This work was performed using computational facilities of the Centre de Resources Informatiques (CRI) de l’Université de Lille and Centre Régional Informatique et d’Applications Numériques de Normandie (CRIANN) which are thankfully acknowledged for the CPU time allocation.
1 In this case, the first four principal components account for approximately 90% of the total data variation.
2 In this case, the first two principal components account for approximately 86% of the total data variation.