

 CC-BY 4.0
CC-BY 4.0
1. Introduction
In many essential chemical or biological reactions, various amines containing phenyl or methyl groups are utilized as precursors for preparing drugs and biological compounds as well as ligands in organic synthesis [1, 2]. Among the many findings in the formation of a new carbon–carbon bond, the addition of an aryl or an alkyl group to a C=N bond using organometallic reagents, producing amine products, is known to be a significant pathway. These additions are widely catalyzed in the presence of different transition metal complexes of Rh(I)/(II) [3], Pd(II) [4], Ni(0)/(II) [5], Ru(II) [6], Co(II), Fe(III), and Cu(I)/(II).
Model reaction of copper–phosphine-catalyzed phenylation of N-Ts-aldimines.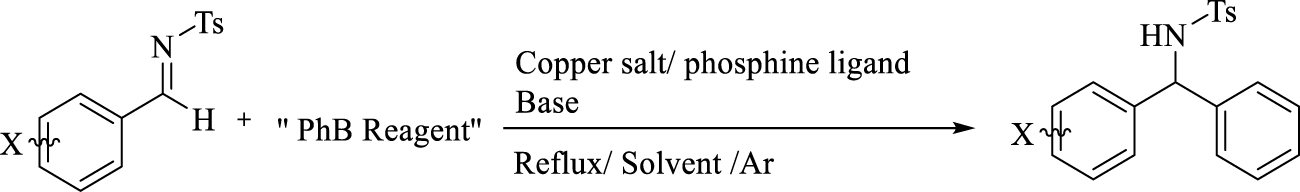
Formation of 4-chloro-N-tosylbenzamide and 4-chloro-N-tosylbenzamide by-products during the phenylation of aldimines using CuCl2 (Equation (1)) and Cu(OAc)2 (Equation (2)).
The use of air-stable organoboron reagents is preferred compared to zinc, tin [7, 8], silane [9], and titanium [10], which are toxic and difficult to manage. Consequently, as a result of the work by Hayashi [11], Miyaura [12], Lin [13], Xu [14], Zhang [15], Manolikakes [16], and others, the addition of organoboron reagents using noble metal catalysts has increased considerably. However, most of these additions were carried out by means of rare and costly rhodium and palladium catalysts [17, 18, 19]. Currently, there is a growing trend toward the use of copper. Copper is an earth-abundant transition metal with a magnificently diverse chemistry and coordination ability with respect to heteroatoms or pi bonds when used as a catalyst in various organic reactions [20]. Despite many successful copper-catalyzed additions on N-protected aldimines using stannane and silane reagents, there have been few reports on the use of aryl or alkyl boron reagents in the literature [21]. Schaus and co-workers reported the efficient addition of aryl boron reagents to active α-iminoesters using chiral 1,1′-biphenols and thiourea [22]. Hu and co-workers reported the addition of CuCl/bipyridine-catalyzed arylboroxines to N-Ts-aldimines using microwave energy, achieving a conversion rate up to 84% [23]. Zhou and co-workers reported the asymmetric arylation of N-azaaryl aldimines with arylboroxines in the presence of copper–monodentate phosphoramidite complexes [24]. Highlighting the importance of cost-effective first-row transition metals in direct 1,2-addition, in this work, phenylation of aldimines along with methylation is developed by utilizing cheap copper salts and air-stable organoboron reagents (Scheme 1).
Initially, the addition of phenylboronic acid (2a) to aldimine was investigated in the presence of a copper complex of common monodentate and bidentate phosphine ligands (Table 1). The reaction did not proceed with dppm, dppp, dppf, and dppb. Dppe, PMe3, and P(OMe)3 produced a low yield of the desired product, while PPh3 generated approximately 55%. When different copper salts were used, the addition of 2a to N-(4-chlorobenzylidene)-4-methylbenzenesulfonamide did not proceed well. The use of copper(II) chloride resulted in the formation of chloride 4-chloro-N-tosylbenzamide as a by-product (4; 85% for 10 h) most probably through the phenylation of sulfonamide from aldimine hydrolysis (Scheme 2, Equation (1)) [25, 26, 27, 28]. Replacing the copper salt by copper(II) acetate produced 4-chloro-N-tosylbenzamide (5; 95% for 30 min) (Scheme 2, Equation (2)) [29, 30, 31, 32].
Effect of various phosphine ligands
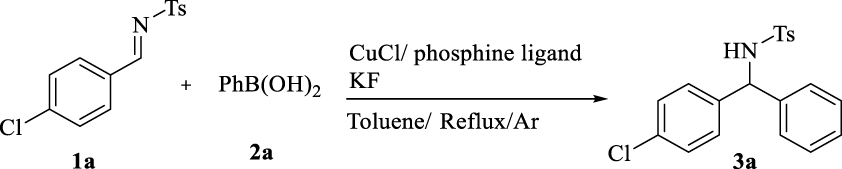
| P(OPh)3 (40) | dppf (trace) | dppm (trace)a |
| PMe3 (12) | PPh3 (55) | dppp (trace) |
| P(OMe)3 ( <10) | dppe (25) | dppb (trace) |
Reaction conditions: p-chlorobenzaldimine (0.1 mmol), 2a (0.2 mmol), copper salt (10 mol%), ligand (20 mol%), KF (0.3 mmol), and toluene (0.5 mL) for 24 h under reflux in Ar atmosphere. aIsolated yield (%).
Optimization of condition

| Entry | Copper salt | Base | Yielda |
|---|---|---|---|
| 1 | Cu(OTf)2 | KF | 20 |
| 2 | KF | CuSO4 | Trace |
| 3 | KF | Cu(NO3)2⋅6H2O | Trace |
| 4 | KF | CuCl2 | Trace |
| 5 | KF | Cu(OAc)2 | Trace |
| 6 | KF | CuCl | 55 |
| 7 | KF | Cu(OTf) | 28 |
| 8 | KF | CuF | 25 |
| 9 | KF | CuBr | 15 |
| 10 | KF | CuI | 20 |
| 11b | KF | CuCl | - |
| 12c | KF | CuCl | 65 |
| 13d | KF | CuCl | 83 |
| 14 | Et3N | CuCl | - |
| 15 | K2CO3 | CuCl | 20 |
| 16e | K2CO3 | CuCl | 50 |
| 17e | KF | CuCl | 45 |
| 18f | KF | CuCl | 40 |
Reaction conditions: p-chlorobenzaldimine (0.1 mmol), 2a (0.2 mmol), KF (0.3 mmol), and toluene (0.5 mL) for 24 h under reflux in Ar atmosphere. aIsolated yield (%). bNo ligand was used. cCopper salt (5 mol%) and PPh3 (10 mol%). dCopper salt (2.5 mol%) and PPh3 (5 mol%). eEt3N used as additive. f2b (0.1 mmol) was used after 48 h.
In such side reactions, the presence of copper(II) salts prevents the formation of active copper species and the occurrence of a catalytic cycle (Table 2, entries 1–5). Although the reaction is run with some copper(I) salts, catalyzation by copper(I) chloride is the most efficient for a time of 24 h with a much higher yield (entries 6–10, Table 2). Using 5 mol% of copper(I) chloride with 10 mol% PPh3 results in the desired product 3a with a 65% yield (entry 12, Table 2). Surprisingly, when the reaction is conducted using 2.5 mol% CuCl with 5 mol% PPh3 at 110 °C, the yield of the favored product 3a reaches 83% with no observation of by-products 4 and 5 (entry 13, Table 2; see Supplementary information). When the model reaction is performed by using triphenylboroxine (2b) with many different catalytic systems and conditions, the same result is produced (up to 40% yield; entry 18, Table 2). However, the former is more active and produces an adduct in less time. Noncoordinate bases such as potassium carbonate do not efficiently promote the reaction, while triethylamine together with potassium carbonate as an additive increases both the yield and the rate significantly (entries 15 and 16, Table 2). Much stronger bases such as potassium fluoride and potassium hydroxide (2:1 ratio to boron reagent) convert aldimine to the required product with a 90% yield. Nonetheless, potassium hydroxide is not suitable for converting copper salts to copper(I) hydroxide, thereby accelerating the aldimine hydrolysis. Although the best base is potassium fluoride in excess, obtaining a trace amount of the product using PhBF3K prevents the formation of this reagent during the reaction. Therefore, as reported in other studies, the PhBF3K salt cannot be an active compound in the catalytic cycle (see Supplementary information). Owing to the effects of different solvents, the progress of the reaction followed a complex route. Protic solvents lead to complete hydrolysis of aldimines. Among other compounds, toluene provided promising outcomes with an 83% yield. Although the CuCl salt is insoluble in toluene, the copper–phosphine complex is soluble in organic solvents (see Supplementary information). These reactions are sensitive to the order of addition. Premixing the copper salt and ligand followed by the addition of a boron reagent and substrates generates the addition product in acceptable yields. Despite using dried reagents, imine was hydrolyzed under all conditions by the water released in the boronic acid–boroxine equilibrium.
Under optimum reaction conditions, the scope of phenylation of aromatic N-Ts-aldimines was studied, ranging from moderate to good (entries 1–11, Table 3). Benzaldehyde derivatives with electron-donating substituents as well as electron-withdrawing substituents generated the desired products in acceptable yields (entries 8–10, Table 3).
Scope of phenylation
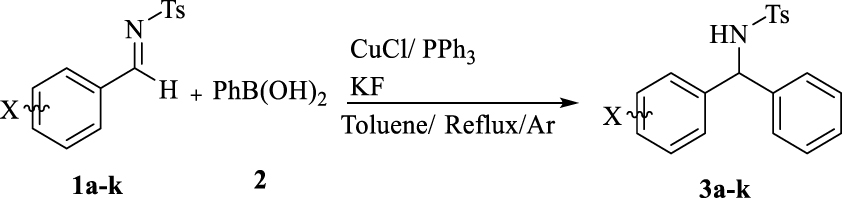
| Entry | X | Yielda | mp (°C) | mp (°C)b |
|---|---|---|---|---|
| 1 | p-Cl | 83 | 114.5–115 | 114–115 |
| 2 | o-Cl | 72 | 170–173 | 172–173.2 [33 ] |
| 3 | m-Cl | 75 | 132–133 | 135–136 [16 ] |
| 4 | p-F | 80 | 122–124 | 120–121 |
| 5 | p-NO2 | 75 | 124–126 | 121–123 [34 ] |
| 6 | p-CF3 | 80 | 145–146 | 142–144 |
| 7 | p-Br | 78 | 120–121 | 121–123 |
| 8 | p-OMe | 65 | 139–140 | 139–141 [35 ] |
| 9 | o–OMe | 45 | 124–125 | 126.6–127.8 [33 ] |
| 10 | p-CH3 | 51 | 133–136 | 132–134 [36 ] |
| 11 | –H | 60 | 155–156 | 157–159 |
Reaction conditions: p-chlorobenzaldimine (0.1 mmol), 2a (0.2 mmol), CuCl (2.5 mol%), PPh3 (5 mol%), KF (0.3 mmol), and toluene (0.5 mL) for 24 h under reflux in Ar atmosphere. aIsolated yield (%). bReported melting point.
To continue further development of this study, copper-catalyzed methylation of aldimines was studied under similar conditions. Different aromatic aldimines treated with methylboronic acid (2b) led to the desired products in lower yields and at lower rates, showing the broad scope of methylation (Table 4).
Scope of methylation
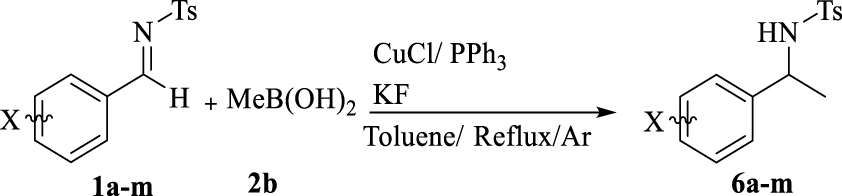
| Entry | X | Yielda | mp (°C) | mp (°C)b | |
|---|---|---|---|---|---|
| 1 | p-Cl | 65 | 127–130 | 128–130 | |
| 2 | o-Cl | 30 | Oily product | - | |
| 3 | m-Cl | 43 | 64–65 | 66.7–67.8 | |
| 4 | p-F | 72 | 112–114 | 115–116 | |
| 5 | p-NO2 | 75 | 156–150 | 156–159 [37 ] | |
| 6 | p-CH3 | 50 | 113–115 | 114–116 | |
| 7 | p-Br | 59 | 136–140 | 139–141 | |
| 8 | o-OMe | 21 | 101–104 | 105–108 | |
| 9 | p-OMe | 25 | 85–88 | 88–89 | |
| 10 | p-CF3 | 78 | 119–121 | 122–123 | |
| 11 | -H | 55 | 80–83 | 82–84 [38] | |
| 12 | β-naphthyl | 65 | 144–146 | 146–148 | |
| 13 | α-naphthyl | 51 | 121–123 | 122.5–123.5 | |
Reaction conditions: p-chlorobenzaldimine (0.1 mmol), 2b (0.2 mmol), CuCl (2.5 mol%), PPh3 (5 mol%), KF (0.3 mmol), and toluene (0.5 mL) for 48 h under reflux in Ar atmosphere. aIsolated yield (%). bReported melting point.
Based on observations, a catalytic cycle is proposed for the reaction (Scheme 3). In toluene (S), the starting point of the catalytic cycle is the solvated complex [Cu(PPh3)2S2]Cl, abbreviated to [Cu(PPh3)2]+ (A), having a copper atom in tetrahedral coordination. The oxidative addition of the phenyl group to copper(I) complex A gives copper(III) phenyl complex B. Insertion of imine into the resulting boron–copper bond of B yields an α-boroxyalkylcopper phenyl intermediate D, which then is converted to the boron ether of a sec-alcohol by reductive elimination [39, 40].
Proposed mechanism: solvated complex [Cu(PPh3)2]+ (A), copper(III) phenyl complex (B), and α-boroxyalkylcopper phenyl intermediate (D).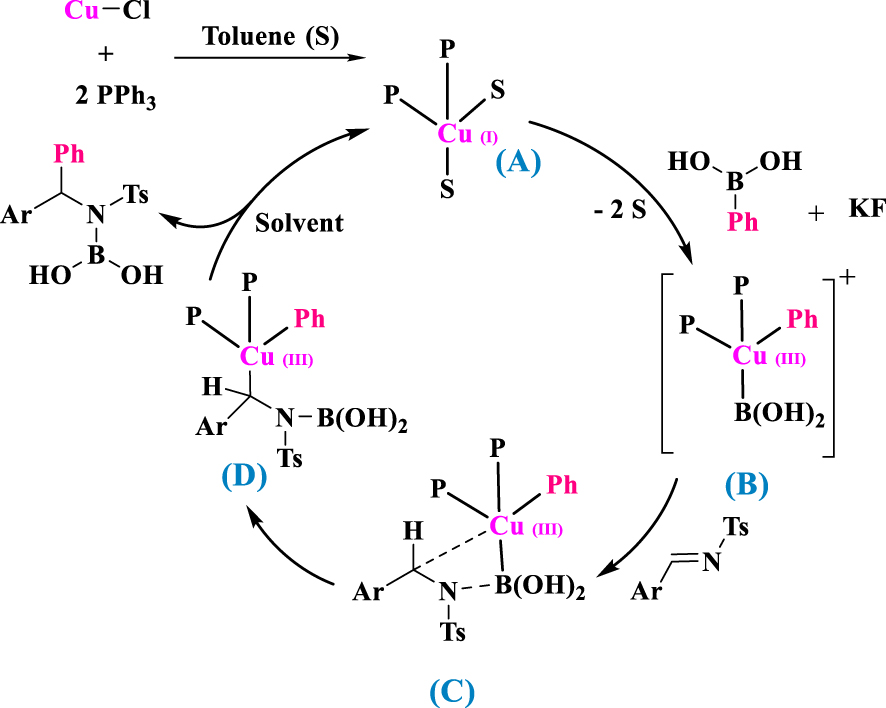
In summary, the addition of aryl and alkyl boron reagents to N-Ts-aldimines was performed in the presence of copper/phosphine catalysts. The coordination of copper(I) chloride with PPh3 was determined as the most efficient combination for this addition, delivering the desired products in moderate-to-good yield. The potential of direct synthesis of the N–C amino bond along with the broad scope of the reaction has implications for the fields of organic chemistry and medicine. Despite the lower yield of this catalytic system in comparison to that of rhodium and palladium, the use of economical and available copper salt is the main highlight of this protocol. Further studies, including an investigation into the enantioselective procedure, are being actively pursued by our research group.
All the chemicals were procured from Sigma-Aldrich and Merck Chemicals. Et2O and 1,4-dioxane were distilled from benzophenone/sodium under nitrogen atmosphere prior to use. Aldimines, phenylboronic acids, and methylboronic acids, triphenylboroxine and trimethylboroxine were prepared according to reported procedures [41, 42]. The 1H NMR spectra were recorded on Bruker Avance III HD 500 MHz using TMS as the internal standard.
2. General procedure
Under argon atmosphere, triphenylphosphine (0.01 mmol, 2.6 mg) was placed in a flame-dried Schlenk tube. CuCl (0.005 mmol, 0.5 mg) and toluene (0.5 mL) were added. The contents of the Schlenk tube were stirred at room temperature for 30 min, and aldimine 1 (0.1 mmol) and 2a (0.2 mmol) and potassium fluoride (0.3 mmol, 17.4 mg) were added sequentially. The Schlenk tube was placed in an oil bath at 110 °C. Then, the mixture was stirred for 24 h, cooled to room temperature, and filtered through a short silica gel column with Et2O (5 mL) as the eluent. The mixture was concentrated via evaporation, and the residue was purified by chromatography on silica gel (eluent: petroleum ether/ethyl acetate 10:1) to obtain the corresponding product (3).
N-((4-chlorophenyl)(phenyl)methyl)-4-methylbenzenesulfonamide (3a)
Yield: 30.80 mg (83%); white solid; mp 114.5– 115 °C.
1H NMR (500 MHz, CDCl3): 𝛿 2.42 (s, 3H), 5.01 (d, J = 6.9 Hz, 1H), 5.55 (d, J = 6.9 Hz, 1H), 7.04–7.10 (m, 4H), 7.16–7.22 (m, 4H), 7.23–7.26 (m, 3H), 7.57 (d, J = 8.1 Hz, 2H) [43].
N-((4-fluorophenyl)(phenyl)methyl)-4-methylbenzenesulfonamide (3d)
Yield: 28.43 mg (80%); white solid; mp 122–124 °C.
1H NMR (500 MHz, CDCl3): 𝛿 2.39 (s, 3H), 5.16 (d, J = 7.0 Hz, 1H), 5.54 (d, J = 7.1 Hz, 1H), 6.92–6.96 (m, 4H), 7.02–7.07 (m, 4H), 7.17–7.21 (m, 3H), 7.55 (d, J = 8.3 Hz, 2H) [34].
N-((4-trifluorophenyl)(phenyl)methyl)-4-methylbenzenesulfonamide (3f)
Yield: 32.43 mg (80%); white solid; mp 145–146 °C.
1H NMR (500 MHz, CDCl3): 𝛿 2.35 (s, 3H), 5.34 (d, J = 7.2 Hz, 1H), 5.59 (d, J = 7.2 Hz, 1H), 7.01–7.05 (m, 2H), 7.10 (d, J = 8.0 Hz, 2H), 7.23 (s, 1H), 7.19–7.23 (m, 3H), 7.25 (s, 1H), 7.42 (d, J = 8.2 Hz, 2H), 7.51 (d, J = 8.3 Hz, 2H) [34].
N-((4-bromophenyl)(phenyl)methyl)-4-methylbenzenesulfonamide (3g)
Yield: 32.47 mg (78%); white solid; mp 120–121 °C.
1H NMR (500 MHz, CDCl3): 𝛿 2.41 (s, 3H), 5.24 (d, J = 7.1 Hz, 1H), 5.56 (d, J = 7.1 Hz, 1H), 7.04 (d, J = 8.4 Hz, 2H), 7.09–7.15 (m, 4H), 7.16–7.20 (m, 2H), 7.24–7.27 (m, 3H), 7.58 (d, J = 8.2 Hz, 2H) [35].
N-benzhydryl-4-methylbenzenesulfonamide (3k)
Yield: 20.224 mg (60%); yellowish white solid; mp 155–156 °C.
1H NMR (500 MHz, CDCl3): 𝛿 2.38 (s, 3H), 5.32 (d, J = 7.1 Hz, 1H), 5.58 (d, J = 7.2 Hz, 1H), 7.09–7.16 (m, 6H), 7.19–7.24 (m, 6H), 7.57 (d, J = 8.0 Hz, 2H) [38].
4-chloro-N-tosylbenzamide (4)
Yield: 22.25 mg (90%); white solid; mp 170–171 °C.
1H NMR (500 MHz, CDCl3): 𝛿 2.40 (s, 3H), 6.55 (br, 1H), 7.07 (d, J = 7.69, 2H), 7.13 (t, J = 7.5, 1H), 7.2–7.27 (m, 4H), 7.66 (d, J = 8.3, 2H) [44].
4-chloro-N-tosylbenzamide (5)
Yield: 24.78 mg (80%); white solid; mp 99–101 °C.
1H NMR (500 MHz, CDCl3): 𝛿 2.44 (s, 3H), 7.32 (d, J = 8.2 Hz, 2H), 7.43 (d, J = 8.6, 2H), 7.75 (d, J = 8.6 Hz, 2H), 7.91 (d, J = 8.4, 2H), 8.38 (br, 1H) [45].
N-(1-(4-chlorophenyl)ethyl)-4- methylbenzenesulfonamide (6a)
Yield: 20.13 mg (65%); white solid; mp 127–130 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.37 (d, J = 6.9 Hz, 3H), 2.39 (s, 3H), 4.43 (qui, J = 6.9 Hz, 1H), 5.12 (d, J = 6.9 Hz, 1H), 7.02 (d, J = 8.5 Hz, 2H), 7.13 (d, J = 8.3 Hz, 2H), 7.17 (d, J = 8.1 Hz, 2H), 7.57 (d, J = 8.1 Hz, 2H) [46].
N-(1-(2-chlorophenyl)ethyl)-4- methylbenzenesulfonamide (6b)
Yield: 9.30 mg (30%). Oily product.
1H NMR (600 MHz, CDCl3): 𝛿 1.42 (d, J = 6.9 Hz, 3H), 2.34 (s, 3H), 4.89 (m, 1H), 5.13 (d, J = 7.0 Hz, 1H), 7.06–7.10 (m, 2H), 7.12 (d, J = 8.2 Hz, 2H), 7.16–7.20 (m, 2H), 7.59 (d, J = 8.3 Hz, 2H).
N-(1-(3-chlorophenyl)ethyl)-4- methylbenzenesulfonamide (6c)
Yield: 13.32 mg (43%); white solid; mp 64–65 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.37 (d, J = 6.9 Hz, 3H), 2.37 (s, 3H), 4.42 (qui, J = 6.9 Hz, 1H), 5.24 (d, J = 7 Hz, 1H), 6.96 (br, 1H), 6.98–7.02 (m, 1H), 7.09–7.11 (m, 2H), 7.15 (d, J = 8.1 Hz, 2H), 7.56 (d, J = 8.1 Hz, 2H) [47].
N-(1-(4-fluorophenyl)ethyl)-4- methylbenzenesulfonamide (6d)
Yield: 21.12 mg (72%); white solid; mp 112–114 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.37 (d, J = 6.9 Hz, 3H), 2.39 (s, 3H), 4.41 (qui, J = 6.9 Hz, 1H), 5.21 (d, J = 6.9 Hz, 1H), 7.02 (d, J = 8.5 Hz, 2H), 7.12 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.2 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H) [48].
N-(1-(4-methylphenyl)ethyl)benzenesulfonamide (6f)
Yield: 14.47 mg (50%); white solid; mp 113–115 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.50 (d, J = 6.8 Hz, 3H), 2.23 (s, 3H), 4.63 (q, J = 6.9 Hz, 1H), 5.14 (dd, J = 7.0, 1H), 7.01 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 8.2 Hz, 1H), 7.40–7.48 (m, 3H), 7.57 (d, J = 8.1 Hz, 2H), 7.65 (d, J = 8.2 Hz, 2H), 7.72–7.76 (m, 1H) [51].
N-(1-(4-bromophenyl)ethyl)benzenesulfonamide (6g)
Yield: 20.90 mg (59%); white solid; mp 136–140 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.37 (d, J = 6.9, 3H), 2.39 (s, 3H), 4.43 (q, J = 6.9 Hz, 1H), 5.21 (d, J = 6.9 Hz, 1H), 7.02 (d, J = 8.4 Hz, 2H), 7.12 (d, J = 8.3 Hz, 2H), 7.16 (d, J = 8.1 Hz, 2H), 7.57 (d, J = 8.2 Hz, 2H) [49].
N-(1-(2-methoxyphenyl)ethyl)-4- methylbenzenesulfonamide (6h)
Yield: 6.40 mg (21%); white solid; mp 101–104 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.44 (d, 6.9 Hz, 3H), 2.30 (s, 3H), 3.69 (s, 3H), 4.51–4.57 (m, 1H), 5.52 (d, J = 9.3 Hz, 1H), 6.60 (d, J = 8.2 Hz, 1H), 6.73 (t, J = 7.5 Hz, 1H), 6.91 (d, J = 7.5 Hz, 1H), 7.02 (d, J = 8.1 Hz, 2H), 7.06–7.11 (m, 1H), 7.49 (d, J = 8.2 Hz, 2H) [47].
N-(1-(4-methoxyphenyl)ethyl)-4- methylbenzenesulfonamide (6i)
Yield: 9.16 mg (30%); white solid; mp 85–88 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.39 (d, J = 6.8 Hz, 3H), 2.38 (s, 3H), 3.74 (s, 3H), 4.40 (qui, J = 6.8 Hz, 1H), 4.82–4.97 (br, 1H), 6.71 (d, J = 8.7 Hz, 2H), 7.01 (d, J = 8.7 Hz, 2H), 7.18 (d, J = 7.6 Hz, 2H), 7.61 (d, J = 8.3 Hz, 2H) [37].
N-(1-(β-naphthyl)ethyl)-4- methylbenzenesulfonamide (6l)
Yield: 21.15 mg (65%); white solid; mp 144–146 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.58 (d, J = 6.8 Hz, 3H), 2.32 (s, 3H), 5.18 (dq, 6.8 Hz, 1H), 5.28 (qui, J = 6.8 Hz, 1H), 7.05 (d, J = 8.0 Hz, 2H), 7.28–3.18 (m, 1H), 7.37 (d, J = 7.0 Hz, 1H), 7.40–7.47 (m, 1H), 7.56 (d, J = 8.1 Hz, 2H), 7.61 (d, J = 8.2 Hz, 2H), 7.68 (d, J = 8.1 Hz, 1H), 7.79 (d, J = 7.3 Hz, 1H), 7.89 (d, J = 8.0 Hz, 1H) [46].
4-methyl-N-(α-(naphthalen-2-yl)ethyl) benzenesulfonamide (6m)
Yield: 16.60 mg (51%); white solid; mp 121–123 °C.
1H NMR (600 MHz, CDCl3): 𝛿 1.50 (d, J = 6.8 Hz, 3H), 2.23 (s, 3H), 4.63 (qui, J = 6.9 Hz, 1H), 5.14 (dd, J = 7.0, 1H), 7.01 (d, J = 8.1 Hz, 2H), 7.21 (d, J = 8.2 Hz, 1H), 7.40–7.48 (m, 3H), 7.57 (d, J = 8.3 Hz, 2H), 7.65 (d, J = 8.2 Hz, 2H), 7.72–7.76 (m, 1H) [50].
Acknowledgment
The authors are grateful to the University of Kurdistan Research Councils for the support of this work.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.35 or from the author.