


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Lead(II) and bismuth(III) porphyrin complexes are known to form generally under harsh conditions [1,2,3] and to be mononuclear, that is one metal cation for one porphyrin unit although dimeric structures have also been reported [4,5]. However, these observations are valid for unfunctionalized macrocycles and the behavior of the porphyrin can be changed if not domesticated by additional functional groups around its binding site. With this target in mind, coordination of either lead(II) or bismuth(III) cations [6,7] have been significantly investigated in decorated porphyrin ligands, particularly those bearing one or two straps in diametrically opposed meso positions [8,9,10]. The formation of such a structure is due to the grafting of the strap(s) on the 5,15 or 10,20 meso positions of the macrocycle. Our group has previously studied the influence of different types of functionalities such as the overhanging carboxylic acid porphyrins that open the way to a new supramolecular coordination chemistry. These ligands exhibit a carboxylic acid group in the apical position above the N-core binding site. As a result, different coordination modes of lead(II) complex have been observed as either coordinated to the N-core, or bound at the level of the strap stabilized by an exogenous acetate group [9], further leading to unusual pentanuclear supramolecular assemblies (Scheme 1, top) [11].
These types of complexes are of importance in supramolecular chemistry of porphyrins which usually rely on the coordination/decoordination of ligand(s) on the N4-bound metal [12]. Indeed, with such dynamic bimetallic complexes, compartimentalized and non-compartimentalized translocations were observed and opened the way to photoredox processes [13,14,15] dynamic constitutional evolution [16] or stereoselective metalations [17] for instance. However, as suspected with bis-strapped “pearl oysterlike” porphyrins [18], the very same strap connected in adjacent meso positions (5,10 and/or 15,20) can also deliver an overhanging carboxylic acid but with an increased flexibility of the strap [19]. Indeed, comparison of “5,10-strapped” Zn(II) and Bi(III) complexes evidenced conformational adaptation of the strap leading to different orientations of the overhanging carboxylic acid (towards/outwards the N-core, Scheme 1, bottom). This is the reason why we decided to investigate the coordination properties of single strapped porphyrins 2 and 3 (Schemes 1 and 2) for which two stereoisomers are present depending on the position of the carboxylic acid group relative to the N-core. The coordination chemistry of overhanging carboxylic acid porphyrin 3 bearing an additional coordinating function (cyano) above the center of the macrocycle may be of interest to form adaptable supramolecular coordination assemblies [12,20,21].
2. Results and discussion
As described earlier for porphyrins 2 [19] the preparation of porphyrin 3 (Scheme 2) was performed via the same procedure by condensation of ethyl-2-cyanoacetate in basic medium on porphyrin 5 which led to the formation of porphyrin 4 as two different isomers (4o and 4i) separated by flash chromatography on silica gel column, and obtained with a yield of 10% and 75%, respectively. Porphyrins 4o and 4i were then saponified by treatment with KOH in ethanol for 2 h, to give porphyrins 3o and 3i in quantitative yields. The corresponding conformation of these isomers was determined at the stage of ester compounds, where the ethoxy carbonyl group is located in different positions (“in” or “out”).1 The formation of the two isomers in different yields (4o: 10% yield and 4i: 75% yield) can be explained by the steric hindrance generated by the cyano group, smaller than that of the carbethoxy group in the W-shaped conformation as reported for previous cyano compounds [22].
Top: dinuclear lead complex of 1 (left, Ar1 = 3,5-dimethoxyphenyl) which assemblies as a pentanuclear complex in the solid state (ball and stick view, X-ray structure, right). Bottom: zinc and bismuth complexes of porphyrin 2o (Ar2 = 4-methoxyphenyl) exhibiting two different conformations of the strap, either bent-over or straight-up, respectively.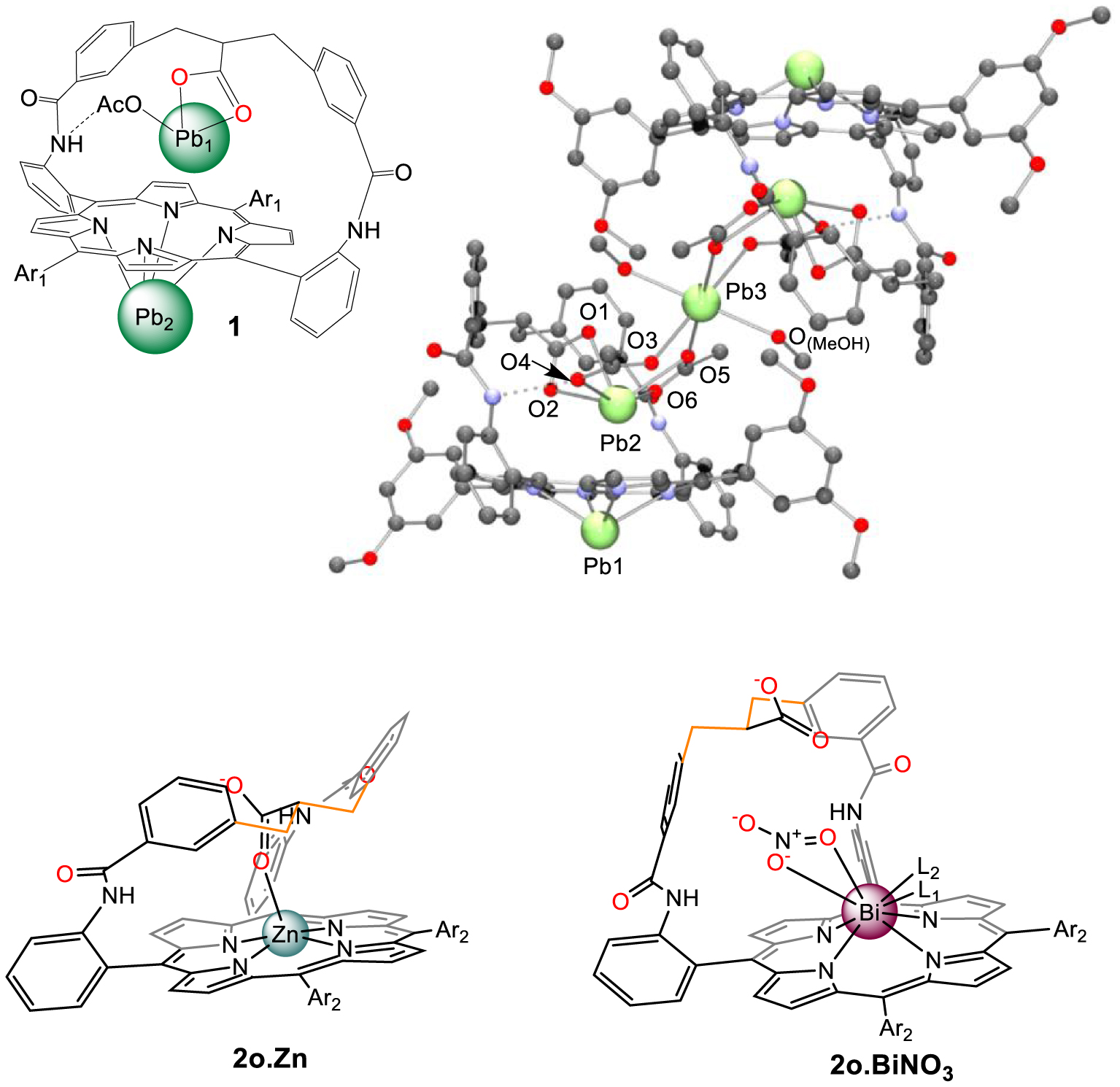
Conventionally, the overhanging ester group (i) (position “in”) represents the location of the group with respect to the macrocycle, which is closer to the macrocycle than ester (o) (position “out”) when the strap adopts a broken-shape conformation (footnote 1) [23,24]. 1H NMR shows that the ethoxy carbonyl group resonates at 2.97 ppm and 0.11 ppm in 4i but at 1.64 ppm and 0.5 ppm in 4o. This indicates that the “out” position is actually closer to the porphyrin plane than the “in” position. Moreover, proton Ha resonates at 3.50 ppm and 3.29 ppm for 4i and 4o respectively. This conformational analysis deduced from proton NMR spectroscopy is consistent with a bent conformation of the strap exhibiting a W-shaped structure as depicted in Scheme 2. Later on, this conformation was confirmed at the stage of the acid with compound 3i. Indeed, an X-ray single-crystal analysis was performed and confirmed a W-shaped structure of the CH2 benzylic carbon atoms C1 and C3 (Figure 1).
Synthesis of single strapped ligands (labeling detailed for 3i). (i) ethyl-2-cyanoacetate (50 eq), THF, EtONa, RT, 2 h (4o: 10% yield and 4i: 75% yield). (ii) EtOH, KOH (30 eq), THF, 2 h, RT, quantitative yields.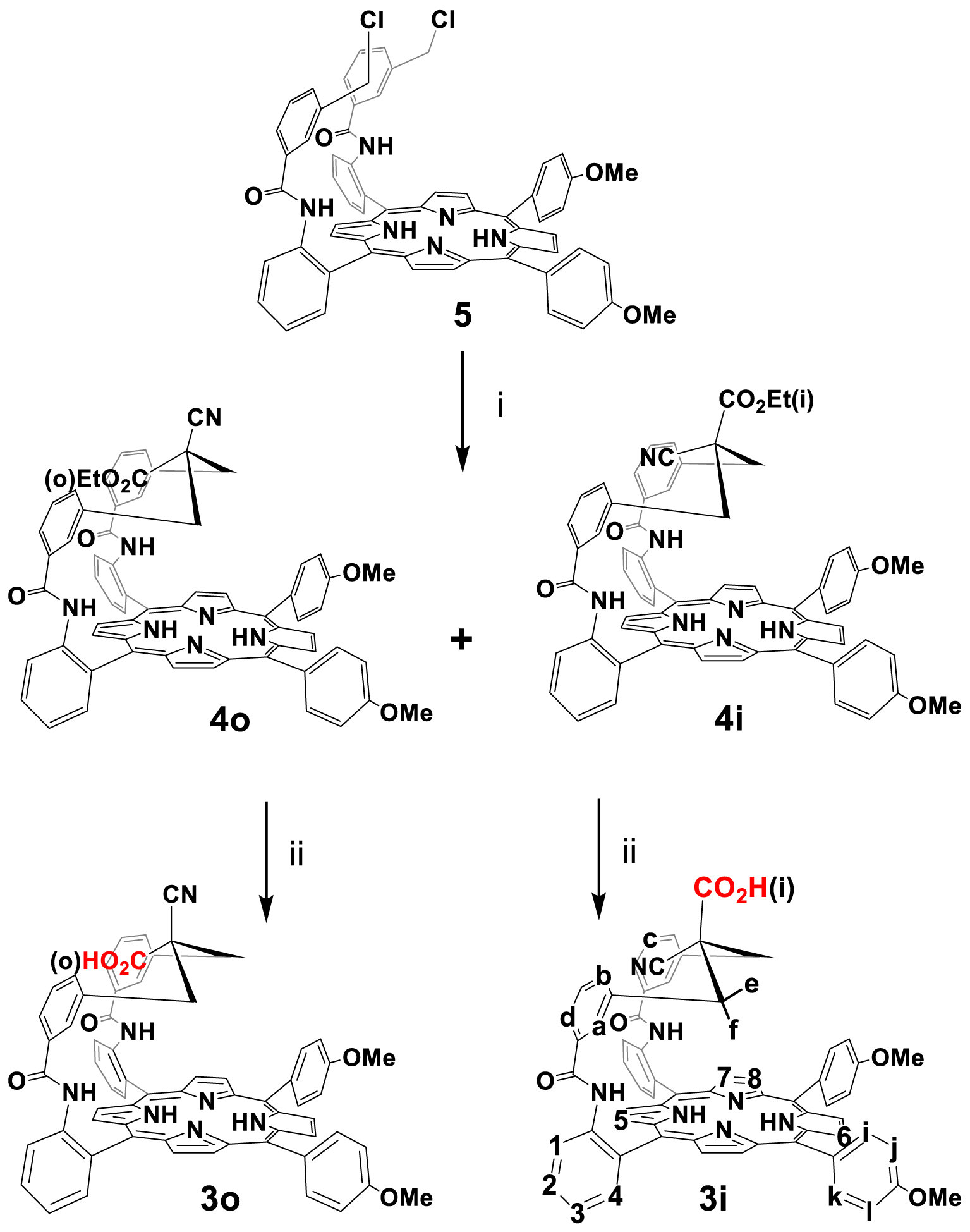
X-ray structure of 3i (CCDC 1971401): (a) side view and (b) apical view. Note: solvents of crystallization and hydrogen atoms were omitted for clarity.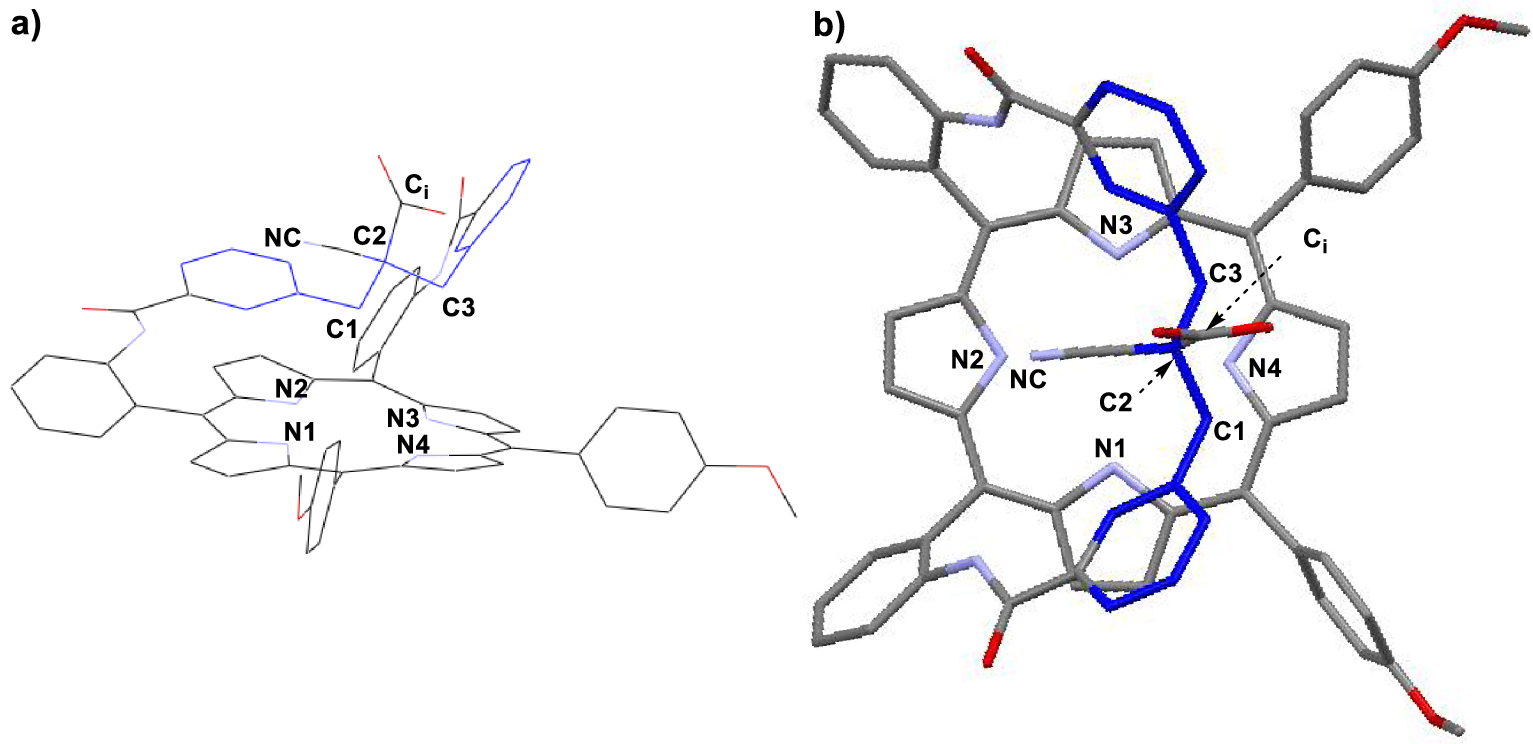
The X-ray structure of 3i also indicates that the porphyrin is saddle-shaped with an average displacement of four diametrically opposed β-pyrrolic carbon atoms above the 24-atom mean macrocyclic plane (24MP) (0.369 Å) while the four other ones are displaced of −0.368 Å below the 24MP of the porphyrin. This distortion is observed together with only a light ruffling (4°) [25]. The W-shaped strap is bent over the macrocycle with a mean angle of 40° relative to the 24MP. In this conformation of the free-base ligand, the carboxylic group is rejected away from the coordination site.
In order to compare the two series of ligands, with and without a cyano group, we first investigated the coordination of 2o and 2i toward Pb(II) (Scheme 3). Complexation properties of porphyrins 2o and 2i towards Pb(II) were studied at 298 K by 1H NMR titration experiments in DMSO-d6 with an excess of diisopropylethylamine (DIPEA). In contrast to harsh conditions usually required for non-functionalized porphyrins, the insertion of Pb(II) was instantaneous with 2o whereas it took 4 h with 2i (Scheme 3a, b). The addition of 1.5 equiv of Pb(NO3)2 to 2o in the NMR tube led to the observation of a broad 1H NMR spectrum (Figure 2b). No significant change in the spectrum was observed with an excess of this salt (3.5 eq) (Figure 2c), indicating the formation of a mononuclear species. A well-resolved spectrum was obtained by recording the NMR tube at 333 K (Figure 2d), and indicated the formation of two compounds in a 9:1 ratio. Two different conformations of the strap were observed based on the chemical shift changes of protons Ha, He and Hf. For instance, Ha which resonates at 4.42 ppm in 2o is split into two protons upon coordination, as Ha′ deshielded at 5.59 ppm (major, 𝛥𝛿Ha′ = 1.17 ppm) and Ha shielded at 4.37 ppm (minor, 𝛥𝛿Ha = −0.05 ppm) (Figure 2d). Similarly, He resonating at 1.07 ppm in 2o, is split at 1.45 ppm (major, 𝛥𝛿He′ = 0.38 ppm) and 0.14 ppm (minor, 𝛥𝛿He = −0.93 ppm). Finally, Hf resonating at −0.61 ppm in 2o, becomes downfield shifted to 1.32 ppm (major, 𝛥𝛿Hf′ = 0.71 ppm) and −0.24 ppm (minor, 𝛥𝛿Hf = 0.37 ppm) during the metal insertion. These observations are consistent with the presence of two major and minor compounds (2o.Pbos and 2o.Pbss). On the one hand, 2o.Pbss incorporates Pb(II) on the same side of the strap, promoting a conformational change of the strap from a shielded “bent-over” position to a deshielded “straight-up” one. On the other hand, 2o.Pbos has Pb(II) attached to the N-core of the macrocycle on the opposite side of the strap (Scheme 3a).2 It should be noted that the coordination of Pb(II) from the naked side of the macrocycle allows the strap to stay bent over the porphyrin plane.
1H NMR spectra (selected regions) in DMSO-d6 at 298 K of: (a) 2o, (b) 2o with 1.5 eq Pb(NO3)2, (c) 2o with 3.5 eq Pb(NO3)2, (d) 2o.Pb at 333 K, (e) 2i.Pb.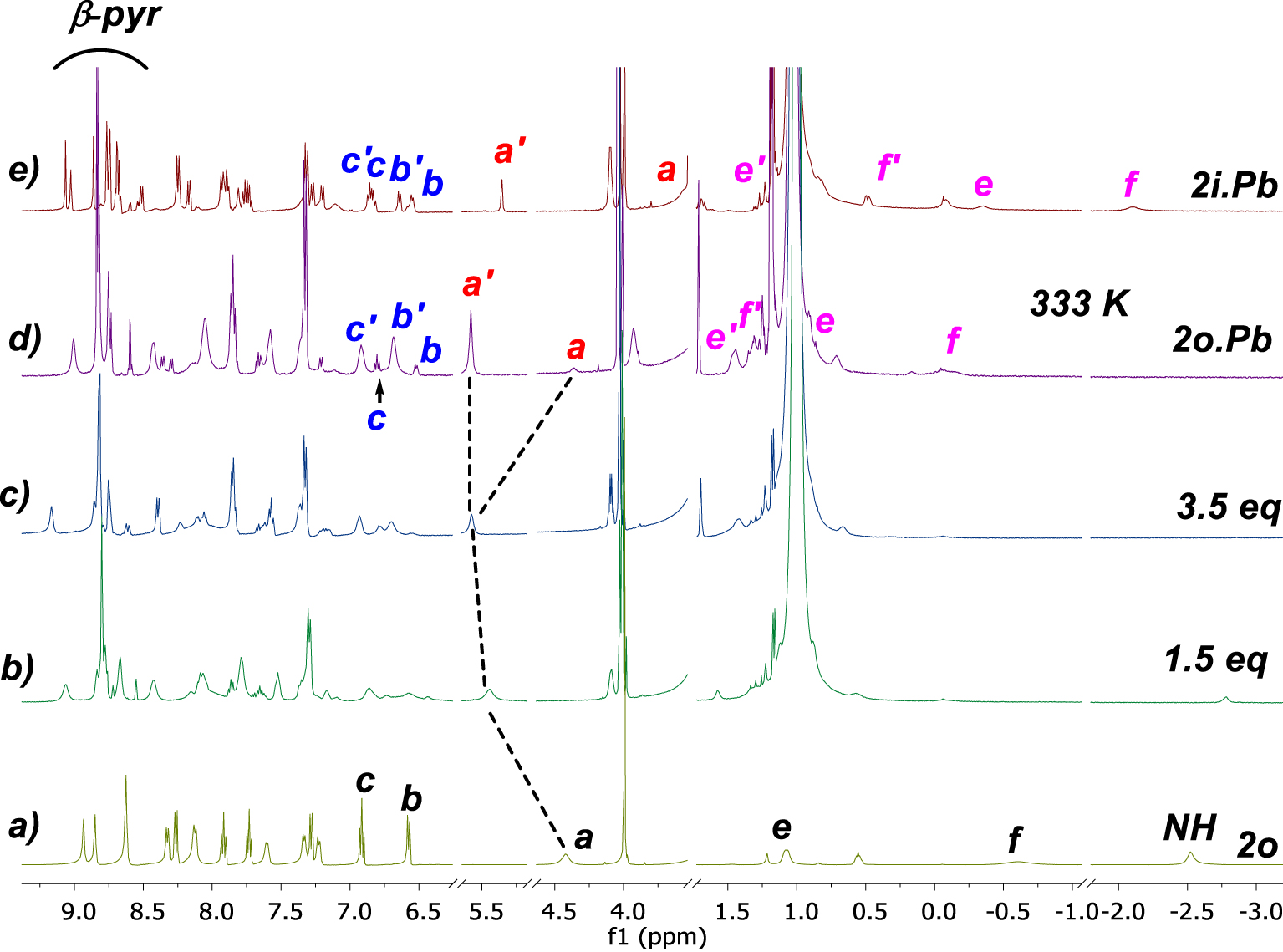
The complexation of Pb(II) with 2i is quite similar to 2o, where two compounds (2i.Pbss and 2i.Pbos) were observed but in equal proportions (Scheme 3b). One complex (2i.Pbss) has Pb(II) coordinated from the same side of the strap, while the second (2i.Pbos) has Pb(II) coordinated from naked side of the macrocycle. Although there is no evidence for a coordination bond between the overhanging carboxylic group and the lead cation, an assistance mechanism in the insertion of lead cation by the carboxylic acid group can easily explain the 9:1 ratio in the case of 2o in which the overhanging group could perform a deconvolution [26,27] of the metal salt with lead cation bound at the level of the strap before coordinating the N-core.
Having understood the coordination behaviors of the two isomers of 2 with lead, the coordination of Pb(NO3)2 was investigated with compound 3i bearing an additional cyano group as the more abundant isomer of porphyrins 3. Similarly to the case of 2i, titration with up to 3 equiv of Pb(NO3)2 showed the formation of a broad 1H NMR spectrum at 298 K after 5 h. No evolution occurred in the spectrum even after 24 h at room temperature. The coordination of Pb(II) occurred from the opposite side of the strap (Scheme 3c) as deduced from the shielding of Ha, He and Hf during coordination. For instance, Ha shifted from 3.35 to 2.66 ppm (𝛥𝛿Ha = −0.69 ppm), He shifted from 1.05 ppm to 0.59 ppm (𝛥𝛿He = −0.46 ppm) and Hf from −2.47 ppm to −3.30 ppm (𝛥𝛿Hf = −0.83 ppm).
Coordination of 2o (a), 2i (b), and 3i (c) with Pb(NO3)2 (3.5 eq) in DMSO-d6 at 298 K, followed by instantaneous formation of bismuth complex by transmetalation of Pb(II) by Bi(III) (3 eq of Bi(NO3)3 added). Note: ss = Pb(II) coordinates from the same side of the strap. os = Pb(II) coordinates from opposite side of the strap. Ar2 = 4-methoxyphenyl.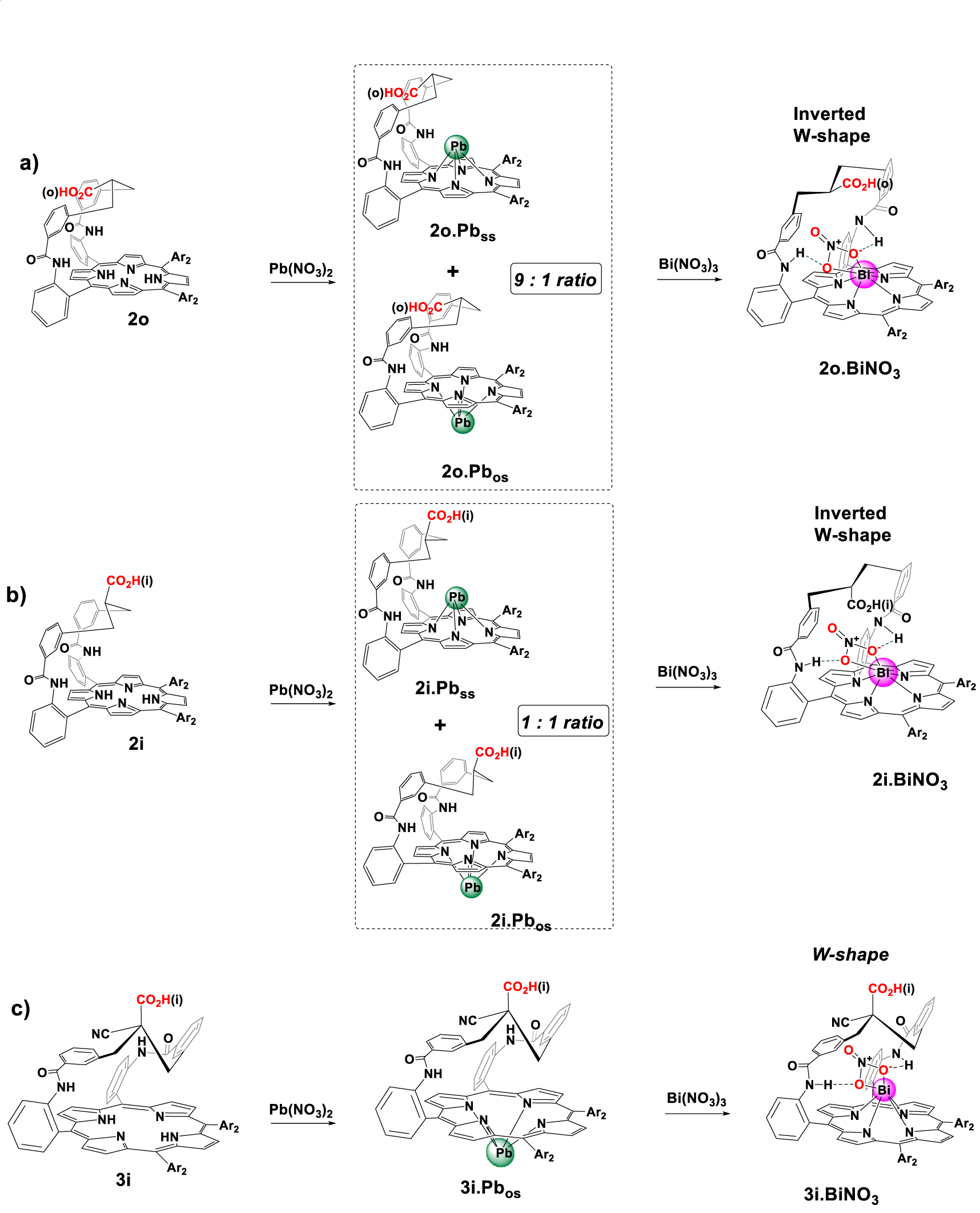
An X-ray structure was obtained and established the coordination of Pb(II) from the opposite side of the strap (Figure 3). The coordination sphere of lead in 3i.Pbos contains only the four nitrogen atoms of the macrocycle with an average bond length N–Pb of 2.382 Å with the metal cation standing 1.333 Å away from the 24MP. The strap exhibits a W-shaped structure and is bent over the coordination site with a mean angle of 40° relative to the 24MP, as in the free-base. It should be noted that the cyano group is located between two aromatic groups of the strap with its nitrogen atom at 3.354 Å from the 24MP whereas the carboxy residue is oriented outside of the cavity with its carbon atom 5.544 Å away from the 24MP. Therefore, the coordination of Pb(II) in the presence of an additional cyano group in α position of the carboxylic acid group (3i.Pbos) leads to the exclusive formation of a complex in which the metal coordinates from the opposite side of the strap. This can be explained by the steric hindrance created by the cyano group which forces the metal to approach the N-core of the macrocycle from the naked side.
X-ray structure of 3i.Pbos (CCDC 1971367): (a) side view and (b) apical view. Note: solvents of crystallization and hydrogen atoms were omitted for clarity. Selected distances: N1–Pb 2.387 Å, N2–Pb 2.387 Å, N3–Pb 2.392 Å, N4–Pb 2.365 Å, Pb–24MP 1.333 Å.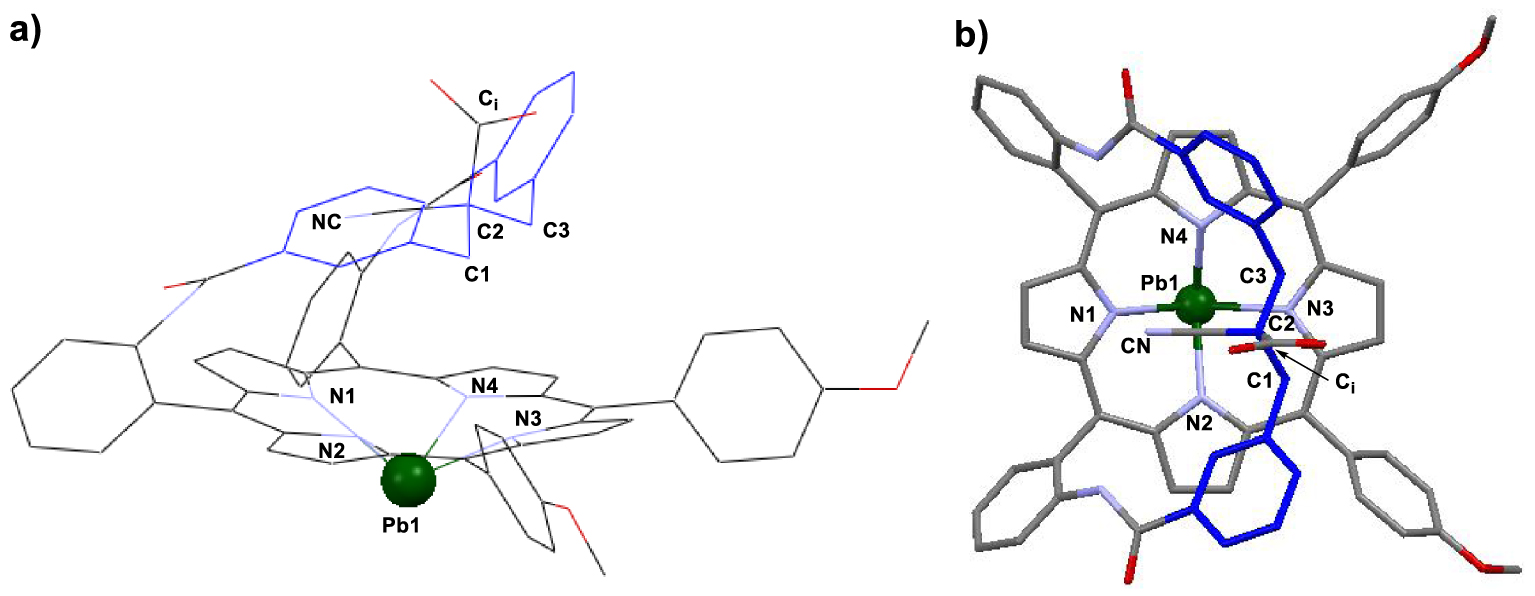
1H NMR (selected regions) in DMSO-d6 at 298 K of (a) 2i.BiNO3, (b) 2i.Pb (1:1 mixture), (c) 2o.Pb at 333 K (9:1 mixture), (d) 2o.BiNO3.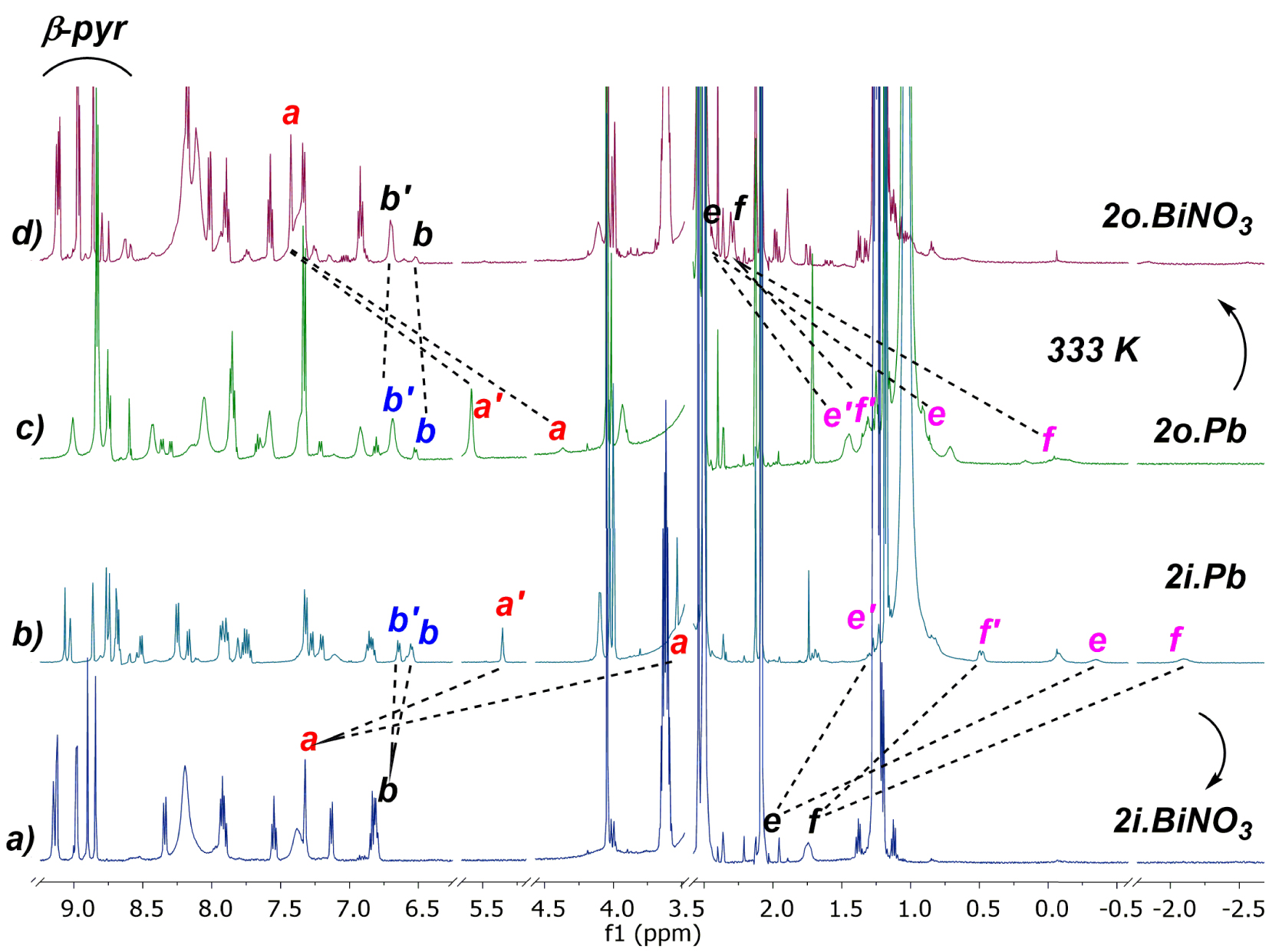
1H NMR spectra (selected regions) of 3i.Pbos treated with 3 eq Bi(NO3)3 in DMSO-d6 at 298 K, leading to the formation of 3i.BiNO3.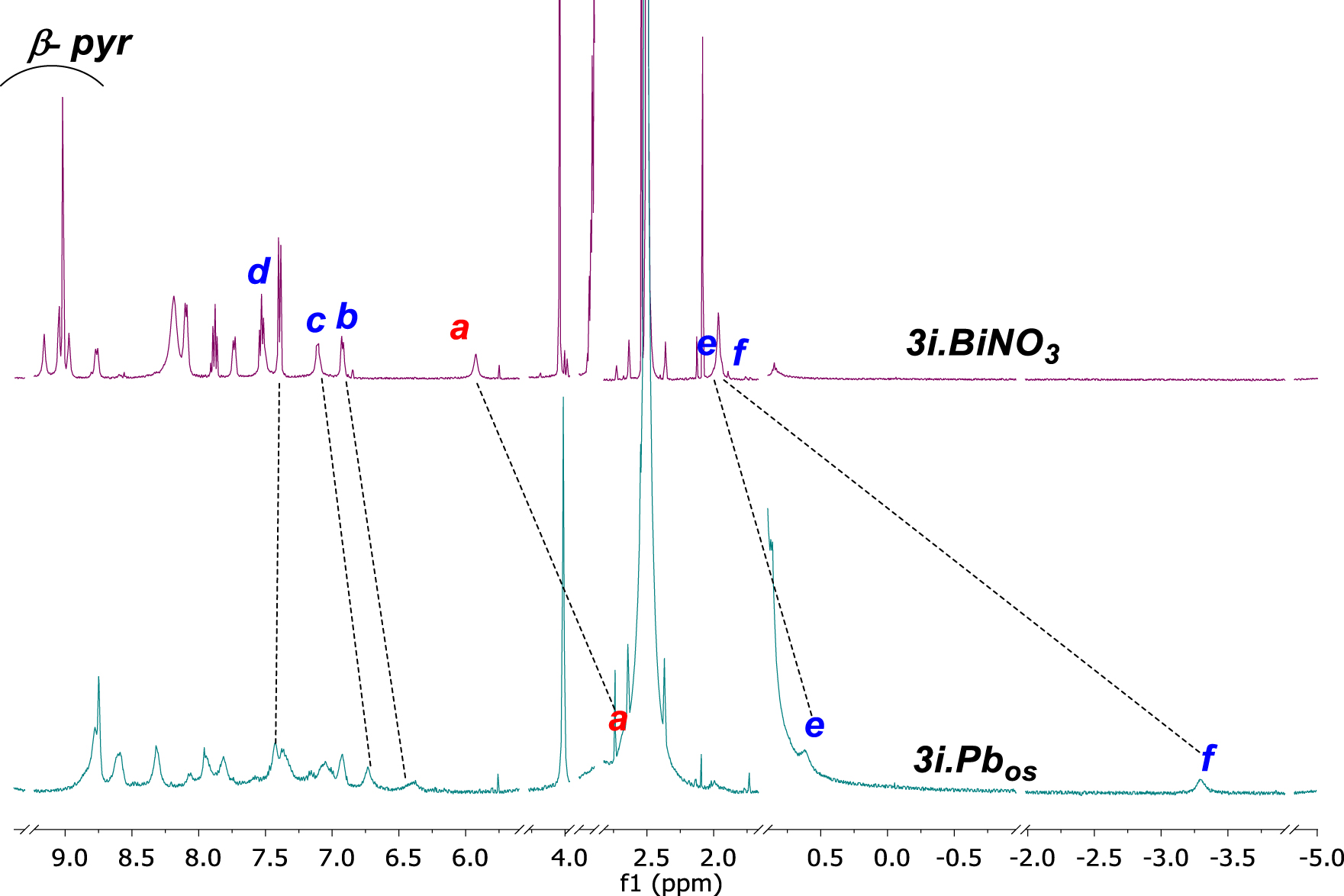
It was also interesting to investigate bismuth(III) insertion in these new ligands. The formation of a bismuth complex with a single strapped porphyrin (2o and 2i) requires a long time of reaction (4–16 h at 85 °C) in the case of bismuth nitrate [19]. To increase the kinetics of Bi(III) insertion, a transmetalation of Pb(II) by Bi(III) was envisaged as it was found efficient with related porphyrins [28,29]. The transmetalation process was achieved by the addition of bismuth nitrate on the two lead complexes, 2i.Pb and 2o.Pb. In both cases, the formation of the bismuth complex was observed quasi instantaneously as attested by the 1H NMR spectra (Figure 4a, d). As an example, in the case of 2i, the addition of Bi(NO3)3 bismuth salt to the mixture of Pb(II) complexes 2i.Pbss and 2i.Pbos led to the formation of a single bismuth complex, in which the metal coordinates from the side of the strap (Scheme 3b and Figure 4a). Likely, in this complex, the bismuth nitrato counter-ion is stabilized by two hydrogen bonds with the amide functions of the strap, which stands in a straight-up position to accommodate the “BiNO3” moiety in the cavity, in an inverted W-shape conformation. This was deduced by analogy with the previously reported X-ray structure of the 2o.BiNO3 bismuth complex obtained directly from the free-base (Scheme 1, bottom) [19]. Therefore, owing to the specific arrangement of this strap, it becomes possible to form instantaneously a bismuth complex at room temperature by transmetalation instead of heating 4–16 h at 85 °C.
We applied with success this strategy to the coordination of Bi(III) with 3i, difficult to achieve at room temperature as no coordination was observed after the addition of 3 eq of Bi(NO3)3 to the free base. Furthermore, heating the NMR tube up to 353 K to insert Bi(III) into the N-core led to decarboxylation. Thus, the bismuth complex was formed instantaneously through transmetalation of lead complex 3i.Pbos with 3 eq of bismuth salt (Scheme 3c). The 1H NMR spectrum recorded at 298 K evidenced the coordination of Bi(III) with changes in the conformation of the strap from bent-over to straight-up (Figure 5) as revealed by the variation of the chemical shifts of Ha, He and Hf. For instance, Ha which initially resonated at 2.66 ppm was shifted to 5.93 ppm during the transmetalation process (𝛿𝛥Ha = 3.27 ppm). In the same line, He and Hf shifted from 0.59 ppm and −3.30 ppm in 3i.Pbos to 1.96 ppm and 1.93 ppm, respectively after Bi(III) insertion. Therefore, bismuth likely coordinates from the strap side and could be stabilized by an exogenous nitrate molecule, likely linked to the strap by two hydrogen bonds as in the case of 2i.BiNO3. With this type of complex, the strap is likely arranged in an inverted W-shape straight-up conformation with the Ha protons lightly oriented outward the center of the macrocycle. This conformation remains consistent with its chemical shift at 5.93 ppm (instead of Ha = 7.32 ppm in case of 2i.BiNO3). However, only the resolution of the X-ray structure of this bismuth complex would allow a definite structural validation of its conformation.
3. Conclusions
We have designed new strapped porphyrins bearing a cyano group in α position of the overhanging carboxylic acid group. Coordination studies of Pb(II) and Bi(III) cations with/without cyano group beside the carboxylic acid have shown different conformational behaviors of the strap leading to unexpected metal complexes. The coordination of Pb(II) with compound 2 lacking the cyano group led to the formation of two metal complexes in which Pb(II) coordinates either from the side of the strap or from the naked side of the macrocycle with different steroselectivity depending on the considered stereoisomer 2i or 2o. For the former, no stereoselectivity was observed as both complexes (2i.Pbss and 2i.Pbos) were formed. In contrast, for the latter, a significant stereoselectivity was achieved as 2o.Pbss was formed with a 9:1 ratio. Conversely, the steric hindrance generated by the presence of the cyano group in compound 3i resulted in the stereoselective formation of a single metal complex with Pb(II) coordinated on the naked side. Moreover, transmetalation of Pb(II) by Bi(III) was shown to be an efficient method for the instantaneous and stereoselective formation of the bismuth complex, which forms exclusively from the side of the strap with 3i. Coordination of these isomers at the level of the strap with different metals, targeting supramolecular coordination assemblies, is currently being under investigation by our group.
4. Experimental section
4.1. General
1H and 13C NMR spectra were recorded at respectively 500 MHz and 125 MHz and referenced to the residual protonated solvents. THF was distilled over Na/benzophenone according to a standard procedure. Other chemicals were used as received without any further purification. All reactions were performed under argon and monitored by TLC (silica, CH2Cl2/CH3OH). Column flash chromatography was performed on silica gel (Merck TLC-Kieselgel 60 H, 15 μm).
Typical procedure for the porphyrin synthesis here detailed in case of 3i: sodium (50 eq) in EtOH was stirred at room temperature for 30 min in a two-neck round bottom flask and ethyl-2-cyanoacetate (50 eq) was added. After 1 h, the resulting mixture was added to a solution of porphyrin 5 (1 eq) in CH2Cl2. The reaction mixture was stirred for 2 h at RT, then water was added. The organic layer was separated from water and evaporated under vacuum. Finally, the two isomers 4o and 4i were separated on a silica gel chromatography column eluted with 0.1% MeOH/CH2Cl2with a yield of 10% and 75%, respectively. In the next step, a 100 ml two-neck round bottom flask equipped with stir bar was charged with KOH (30 eq) in distilled ethanol. After 30 min, porphyrin 4i (38 μmol, 40 mg, 1 eq) was dissolved in 80 mL THF and slowly added to the reaction mixture, which was stirred for 2 h at room temperature. The reaction was quenched by adding distilled water, neutralized by 1 M HCl, and filtrated. The resulted product was recovered by MeOH/CH2Cl2. The solvent was removed under vacuum. The product was obtained in quantitative yield.
α-5,10-bis{2,2′-[3,3′-(2-(cyano)-2-(ethoxycarbonyl)propane-1,3-diyl)benzoylamino]phenyl}-15,20-bis(4-methoxyphenyl)porphyrin: 4o.1H NMR (CDCl3, 298 K, 500 MHz): 𝛿H, ppm 8.96 (2H, s, H5), 8.93 (2H, s, H6), 8.67 (2H, d, J = 4.2 Hz, H7), 8.65 (2H, d, J = 8.3 Hz, H1), 8.61 (2H, d, J = 4.2 Hz, H8), 8.52 (2H, d, J = 7.5 Hz, H4), 8.21 (2H, d, J = 8.3 Hz, Hi), 7.95 (2H, t, J = 7.7 Hz, H2), 7.75 (2H, t, J = 8 Hz, H3), 7.72 (2H, d, J = 8 Hz, Hd), 7.30 (2H, d, J = 7.5 Hz, Hl), 7.27 (2H, d, J = 8.2 Hz, Hk), 7.09 (2H, t, J = 7.6 Hz, Hc), 7.07 (2H, m, Hj), 7.06 (2H, s, NHCO), 6.74 (2H, d, J = 6.8 Hz, Hb), 4.07 (6H, s, O–CH3), 3.29 (2H, s, Ha), 1.64 (2H, q, J = 7 Hz, CH2ester), 0.89 (2H, d, J = 12.4 Hz, He), −0.5 (3H, t, J = 7 Hz, CH3 ester), −1.97 (2H, d, J = 12.6 Hz, Hf), −2.06 (2H, s, NH pyr).HSQC (CDCl3, 298 K, 500 MHz): 𝛿c, ppm, 135.82 (Ck), 135.64 (Ci), 135.22 (C8), 134.18 (C4), 132.54 (C7), 131.95 (Cb), 129.99 (C2), 129.15 (Cc), 129.11 (C5), 128.98 (C6), 127.90 (Cd), 124.42 (Ca), 123.67 (C3), 121.85 (C1), 112.33 (Cj), 112.57 (Cl), 59.77 (Cester), 56.11 (COMe), 39.98 (Ce), 39.98 (Cf), 12.33 (Cester). UV-vis (CHCl3): λ/nm (𝜀, dm3⋅mol−1⋅cm−1): 426 (272120), 520 (26710), 556 (14080), 595 (5000), 652 (6800). ESI-HRMS: m/z calcd. 1050.39734 [M + H]+ for C67H52N7O6, found 1050.3968 (0 ppm).
α-5,10-bis{2,2′-[3,3′-(2-(cyano)-2-(ethoxycarbonyl)propane-1,3-diyl)benzoylamino]phenyl}-15,20-bis(4-methoxyphenyl)porphyrin: 4i.1H NMR (CDCl3, 298 K, 500 MHz): 𝛿H, ppm 8.98 (2H, s, H5), 8.97 (2H, s, H6), 8.71 (2H, d, J = 4.3 Hz, H7), 8.61 (2H, d, J = 4.3 Hz, H8), 8.56 (2H, d, J = 7.5 Hz, H1), 8.54 (2H, d, J = 8.7 Hz, H4), 8.22 (2H, d, J = 7.9 Hz, Hi), 7.95 (2H, t, J = 8 Hz, H2), 7.78 (2H, t, J = 7.4 Hz, H3), 7.67 (2H, d, J = 7.7 Hz, Hd), 7.31 (2H, d, J = 7.9 Hz, Hl), 7.24 (2H, d, J = 8.2 Hz, Hk), 7.05 (2H, d, J = 7.6 Hz, Hj), 6.94 (2H, s, NHCO), 6.92 (2H, t, J = 7.8 Hz, Hc), 6.27 (2H, d, J = 7.5 Hz, Hb), 4.06 (6H, s, O–CH3), 3.50 (2H, s, Ha), 2.97 (2H, q, J = 7 Hz, CH2 ester), 1.06 (2H, d, J = 12.5 Hz, He), 0.11 (3H, t, J = 7 Hz, CH3 ester), −2.15 (2H, s, NH pyr), −2.44 (2H, d, J = 13.2 Hz, Hf). HSQC (CDCl3, 298 K, 500 MHz): 𝛿c, ppm, 136.12 (Ck), 135.53 (Ci), 135.28 (C8), 133.44 (C1), 132.72 (C7), 130.95 (Cb), 129.83 (C2), 128.51 (Cc), 128.35 (C6), 127.94 (C5), 127.71 (Cd), 125.54 (Ca), 123.98 (C3), 123.43 (C4), 112.80 (Cl), 112.21 (Cj), 61.32 (Cester), 56.06 (COMe), 39.44 (Ce), 39.44 (Cf), 12.77 (Cester). UV-vis (CHCl3): λ/nm (𝜀, dm3⋅mol−1⋅cm−1): 426 (264950), 520 (41900), 557 (20200), 593 (8400), 650 (3800). ESI-HRMS: m/z calcd. 1050.39736 [M + H]+ for C67H52N7O6, found 1050.3970 (0 ppm).
α-5,10-bis{2,2′-[3,3′-(2-(cyano)-2-(carboxyl)propane-1,3-diyl)benzoylamino]phenyl}-15,20-bis(4-methoxyphenyl)porphyrin: 3o.1H NMR (DMSO, 298 K, 500 MHz): 𝛿H, ppm 8.92 (2H, s, H5), 8.81 (2H, s, H6), 8.55 (2H, broad, H7), 8.52 (2H, d, J = 4.3 Hz, H8), 8.35 (2H, d, J = 7.7 Hz, H1), 8.27 (2H, d, J = 8.1 Hz, H4), 8.19 (2H, s, NHCO), 8.11 (2H, d, J = 8.1 Hz, Hi), 7.91 (2H, t, J = 8 Hz, H3), 7.73 (2H, d, J = 7.9 Hz, H2), 7.51 (2H, t, J = 8.2 Hz, Hk), 7.33 (2H, broad, Hd), 7.32 (2H, broad, Hl), 7.17 (2H, d, J = 7.8 Hz, Hj), 6.94 (2H, t, J = 7.6 Hz, Hc), 6.69 (2H, d, J = 7.6 Hz, Hb), 4.31 (2H, s, Ha), 3.99 (6H, s, O–CH3), 0.72 (2H, d, J = 12.8 Hz, He), −0.63 (2H, broad, Hf), −2.48 (2H, s, NH pyr), 1H of carboxylic acid was not observed. HSQC (CDCl3, 298 K, 500 MHz): 𝛿c, ppm, 135.73 (Ck), 135.48 (Ci), 134.80 (C1), 132.59 (C6), 132.11 (Cb), 129.81 (C3), 128.02 (Cc), 127.77 (Ca), 126.10 (Cd), 124.95 (C2), 123.36 (C4), 112.74 (Cj), 112.98 (Cl), 40.45 (Ce), 40.45 (Cf). UV-vis (DMSO): λ/nm (𝜀, dm3⋅mol−1⋅cm−1): 426 (218250), 520 (24600), 560 (15440), 592 (10080), 649 (7810). ESI-HRMS: m/z calcd. 1022.36606 [M + H]+ for C65H48N7O6, found 1022.36660 (0 ppm).
α-5,10-bis{2,2′-[3,3′-(2-(cyano)-2-(carboxyl)propane-1,3-diyl)benzoylamino]phenyl}-15,20-bis(4-methoxyphenyl)porphyrin: 3i.1H NMR (DMSO, 298 K, 500 MHz): 𝛿H, ppm 9 (2H, s, H5), 8.97 (2H, s, H6), 8.55 (2H, broad, H7), 8.6 (2H, d, J = 7.6 Hz, H1), 8.55 (2H, broad, H8), 8.40 (2H, d, J = 8 Hz, H4), 8.23 (2H, d, J = 7.9 Hz, Hi), 7.98 (2H, t, J = 8.5 Hz, H3), 7.9 (2H, d, J = 7.1 Hz, Hj), 7.84 (2H, t, J = 7.2 Hz, H2), 7.50 (2H, d, J = 8.8 Hz, Hd), 7.37 (2H, d, J = 8.8 Hz, Hl), 7.21 (2H, d, J = 7.4 Hz, Hk), 6.92 (2H, s, NHCO), 7.05 (2H, t, J = 7.6 Hz, Hc), 6.43 (2H, d, J = 7.7 Hz, Hb), 3.98 (6H, s, O–CH3), 3.35 (2H, s, Ha), 1.05 (2H, d, J = 12.5 Hz, He), −2.32 (2H, s, NH pyr), −2.47 (2H, d, J = 13.2 Hz, Hf), 1H of carboxylic acid was not observed. HSQC (CDCl3, 298 K, 500 MHz): 𝛿c, ppm, 135.70 (Ck), 135.49 (C8), 135.37 (Ci), 133.60 (C1), 132.40 (C7), 131.88 (Cb), 130.25 (C3), 129.51 (C5), 129.34 (Cc), 128.35 (C6), 127.33 (Cd), 124.93 (C2), 124.56 (Ca), 123.36 (C4), 113.10 (Cj), 113.03 (Cl), 55.61 (COMe), 37.70 (Ce), 37.70 (Cf), UV-vis (DMSO): λ/nm (𝜀, dm3⋅mol−1⋅cm−1): 427 (167810), 519 (22270), 556 (9050), 592 (5640), 650 (3760). ESI-HRMS: m/z calcd. 1022.36606 [M + H]+ for C65H48N7O6, found 1022.36662 (0 ppm).
4.2. Typical procedure for metal insertion
A solution (S1) of Pb(NO3)2 (37.6 μmol, 12.4 mg, 12.5 eq) in DMSO-d6 (500 μL) was prepared. Pb(II) insertion was performed in an NMR tube by mixing either 2o or 2i (3.012 μmol, 3 mg, 1 eq), DMSO-d6 (500 μL), DIPEA (17.2 μmol, 3 μL, 5.7 eq), and S1 (200 μL, 3 eq). Leaving the NMR tube at room temperature for respectively a few minutes and 4 hours led quantitatively to 2o.Pb and 2i.Pb, as attested by 1H NMR spectra recorded at 333 K and 298 K, respectively.
A solution (S2) of Bi(NO3)3 (37.6 μmol, 18.2 mg, 12.5 eq) in DMSO-d6 (500 μL) was prepared. The complexes 2o.BiNO3 and 2i.BiNO3 were formed instantaneously by adding 3 eq from S2 (200 μL) to each NMR tube (2o.Pb and 2i.Pb). The proton NMR spectrum was recorded at 298 K.
A solution (S3) of PbNO3 (36.6 μmol, 11.9 mg, 12.5 eq) in DMSO-d6 (500 μL) was prepared. The complex 3i.Pbos was formed by mixing 3i (2.93 μmol, 3 mg, 1 eq), DMSO-d6 (500 μL), DIPEA (16.7 μmol, 2.9 μL, 5.7 eq), and S3 (120 μL, 3 eq) for 5 h at room temperature. The proton NMR spectrum was recorded at 353 K.
A solution (S4) of Bi(NO3)3 (36.6 μmol, 17.8 mg, 12.5 eq) in DMSO-d6 (500 μL) was prepared. The complex 3i.BiNO3 was formed instantaneously by transmetalation through mixing 3i.Pbos and S4 (120 μL, 3 eq). The proton NMR spectrum was recorded at 298 K.
2o.Pbss: 1H NMR (DMSO-d6, 333 K, 500 MHz): 𝛿H, ppm 8.84 (2H, s, H5), 8.82 (2H, s, H6), 8.75 (2H, broad, H7), 8.73 (2H, broad, H8), 8.43 (2H, broad, H1), 8.05 (2H, broad, Hk), 8.05 (2H, broad, Hi), 7.87 (2H, broad, H4), 7.85 (2H, t, J = 7.7 Hz, H2), 7.58 (2H, broad, H3), 7.36 (2H, broad, Hd), 7.33 (2H, d, J = 8.5 Hz, Hj), 7.33 (2H, d, J = 8.5 Hz, Hl), 6.92 (2H, broad, Hc), 6.69 (2H, broad, Hb), 5.59 (2H, s, Ha), 4.04 (6H, s, O–CH3), 1.45 (2H, broad, He), 1.32 (2H, broad, Hf), 0.72 (1H, s, Hα), 2H of NHCO and 1H of carboxylic acid were not observed. HSQC (DMSO-d6, 333 K, 500 MHz): 𝛿c, ppm 135.97 (Ck), 135.97 (Ci), 134.95 (C4), 132.67 (C6), 132.39 (C5), 132.52 (Cb), 132.30 (C8), 131.82 (C7), 129.30 (C2), 128.04 (Cc), 126.40 (Ca), 126.19 (Cd), 124.23 (C1), 123.67 (C3), 112.38 (Cj), 112.38 (Cl), 56.13 (COMe), 50.87 (Cα), 36.16 (Ce), 35.97 (Cf).
2o.Pbos: 1H NMR (DMSO-d6, 333 K, 500 MHz): 𝛿H, ppm 8.37 (2H, d, J = 8.1 Hz, H1), 8.31 (2H, d, J = 7 Hz, H4), 7.85 (2H, broad, H2), 7.67 (2H, d, J = 7.9 Hz, H3), 7.21 (2H, d, J = 7.9 Hz, Hd), 6.81 (2H, d, J = 8.2 Hz Hc), 6.52 (2H, d, J = 7.1 Hz, Hb), 4.37 (2H, s, Ha), 4.01 (6H, s, O–CH3), 0.92 (1H, s, Hα), 0.14 (2H, broad, He), −0.24 (2H, broad, Hf), 2H of H5, 2H of H6, 2H of H7, 2H of H8, 2H of Hi, 2H of Hj, 2H of Hk, 2H of Hl, 2H of NHCO and 1H of carboxylic acid were not observed. UV-vis (DMSO): 𝜆max, nm (rel. abs). 439 (0.38), 471 (1), 661 (0.02).
2i.Pbss: 1H NMR (DMSO-d6, 298 K, 500 MHz): 𝛿H, ppm 9.08 (2H, s, H5′), 9.08 (2H, s, H6′), 9.06 (2H, s, NH′CO), 8.69 (2H, d, J = 4.3 Hz, H7′), 8.67 (2H, d, J = 4.7 Hz, H8′), 8.51 (2H, d, J = 7 Hz, H1′), 8.27 (2H, d, J = 7.7 Hz, H4′), 7.93 (2H, m, H3′), 7.76 (2H, m, H2′), 7.28 (2H, d, J = 7.6 Hz, Hd′), 6.86 (2H, d, J = 7.5 Hz, Hc′), 6.64 (2H, d, J = 7.1 Hz, Hb′), 5.37 (2H, s, Ha′), 4.55 (6H, s, O–CH3′), 1.7 (2H, broad, He′), 1.25 (1H, s, Hα′), 0.5 (2H, broad, Hf′), 2H of Hi, 2H of Hj, 2H of Hk, 2H of Hl, and 1H of carboxylic acid were not observed.
2i.Pbos: 1H NMR (DMSO-d6, 298 K, 500 MHz): 𝛿H, ppm 8.87 (2H, s, H5), 8.87 (2H, s, H6), 8.77 (2H, d, J = 3.2 Hz, H7), 8.75 (2H, d, J = 3.8 Hz, H8), 8.22 (2H, d, J = 7.7 Hz, H1), 8.16 (2H, d, J = 7.7 Hz, H4), 8.05 (2H, broad, Hk), 8.05 (2H, broad, Hi), 7.93 (2H, broad, H3), 7.82 (2H, s, NHCO), 7.75 (2H, m, H2), 7.33 (2H, d, J = 8.5 Hz, Hj), 7.33 (2H, d, J = 8.5 Hz, Hl), 7.18 (2H, d, J = 8 Hz, Hd), 6.85 (2H, d, J = 6.9 Hz, Hc), 6.5 (2H, d, J = 6.6 Hz, Hb), 4.03 (6H, s, O–CH3), 3.65 (2H, s, Ha), 0.85 (1H, s, Hα), −0.32 (2H, broad, He), −2.06 (2H, broad, Hf), 1H of carboxylic acid was not observed. UV-vis (DMSO): λ/nm: 438 (0.41), 472 (1), 664 (0.02).
3i.Pbos: 1H NMR (DMSO-d6, 353 K, 500 MHz): 𝛿H, ppm 8.80 (2H, s, H5), 8.75 (2H, s, H6), 8.75 (2H, broad, H7), 8.75 (2H, broad, H8), 8.66 (2H, broad, H1), 8.41 (2H, broad, H4), 7.9 (2H, t, J = 7.9 Hz, H2), 7.72 (2H, broad, H3), 7.45 (2H, d, J = 7.7 Hz, Hd), 7.25 (2H, broad, Hl), 7.25 (2H, broad, Hj), 6.95 (2H, t, J = 7.7 Hz, Hc), 6.56 (2H, broad, Hb), 4.02 (6H, s, O–CH3), 2.68 (2H, s, Ha), 0.88 (2H, broad, He), −3.31 (2H, broad, Hf), 2H of Hk, 2H of Hi, 2H of NHCO, 1H of carboxylic acid was not observed. HSQC (DMSO-d6, 353 K, 500 MHz): 𝛿c, ppm 132.20 (C5), 132.20 (Cb), 131.75 (C6), 131.75 (C7), 131.75 (C8), 131.25 (C1), 129.34 (C2), 128.74 (Cc), 126.75 (Cd), 124.24 (C3), 123.41 (C4), 112.82 (Cj), 112.82 (Cl), 55.93 (COMe). UV-vis (DMSO): 𝜆max, nm (rel. abs). 472 (1), 615 (0.02), 666 (0.05).
3i.BiNO3: 1H NMR (DMSO-d6, 298 K, 500 MHz): 𝛿H, ppm 9.16 (2H, s, H5), 9.05 (2H, d, J = 4.6 Hz, H7), 9.02 (2H, s, NHCO), 9.01 (2H, d, J = 4.5 Hz, H8), 8.97 (2H, s, H6), 8.77 (2H, d, J = 8.4 Hz, H1), 8.10 (2H, d, J = 7.6 Hz, Hk), 8.10 (2H, d, J = 7.6 Hz, Hi), 7.88 (2H, t, J = 8.2 Hz, H2), 7.73 (2H, J = 7.7 Hz, H4), 7.53 (2H, m, H3), 7.53 (2H, m, Hd), 7.39 (2H, d, J = 8.6 Hz, Hl), 7.39 (2H, d, J = 8.6 Hz, Hj), 7.10 (2H, broad, Hc), 6.92 (2H, d, J = 7 Hz, Hb), 5.93 (2H, s, Ha), 4.04 (6H, s, O–CH3), 1.96 (2H, broad, He), 1.93 (2H, broad, Hf), 1H of carboxylic acid was not observed. HSQC (DMSO-d6, 298 K, 500 MHz): 𝛿c, ppm 136.28 (Ck), 136.28 (Ci), 135.68 (C4), 134.72 (C7), 134.61 (Cb), 133.30 (C6), 133.12 (C5), 132.72 (C8), 130.24 (C2), 128.81 (Cc), 127.62 (Ca), 127.26 (Cd), 123.75 (C3), 123.21 (C1), 113 (Cj), 113 (Cl), 55.98 (COMe), 40.45 (Ce), 40.45 (Cf). UV-vis (DMSO): 𝜆max, nm (rel. abs). 354 (0.26), 476 (1), 606 (0.045), 649 (0.05).
4.3. X-ray crystallographic studies
Data were collected with a D8 VENTURE Bruker AXS diffractometer (founded by FEDER) equipped with a (CMOS) PHOTON 100 detector, Mo-Kα radiation (𝜆 = 0.71073 Å, multilayer monochromator), T = 150 K. The structure was resolved by dual-space algorithm using the SHELXT program [30], and then refined with full-matrix least-squares methods based on F2 (SHELXL) [31]. The contribution of the disordered solvents to the calculated structure factors was estimated following the BYPASS algorithm [32] implemented as the SQUEEZE option in PLATON [33]. A new data set, free of solvent contribution, was then used in the final refinement. All non-hydrogen atoms were refined with anisotropic atomic displacement parameters. Except nitrogen-linked hydrogen atoms that were introduced in the structural model through Fourier difference maps analysis, H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. The crystals of both complexes were crystallized by the same method. Typically, the samples were dissolved in the mixture of two (chloroform, methanol) or three solvents (dimethyl sulfoxide, methanol, chloroform) in a vial fitted with a needle to allow very slow evaporation. 5–6 drops of water were gently added to the mixture before closing the vial. The vials were stored in the dark at room temperature for approximately one month, until single crystals were obtained.
3i: CCDC 1971401, crystals grown from chloroform, methanol, and water, (C65H47N7O6, CHCl3, CH4O); monoclinic P 21/n (I.T.#14), a = 14.1676(17), b = 16.033(2), c = 24.954(4) Å, 𝛽 = 93.491(5)°, V = 5657.9(14) Å3, Z = 4, d = 1.378 g⋅cm−3, 𝜇 = 0.226 mm−1. Except nitrogen and oxygen linked hydrogen atoms that were introduced in the structural model through Fourier difference maps analysis, H atoms were finally included in their calculated positions and treated as riding on their parent atom with constrained thermal parameters. A final refinement on F2 with 12658 unique intensities and 789 parameters converged at 𝜔R(F2) = 0.2357 (R(F) = 0.1006) for 6618 observed reflections with I > 2𝜎(I).
3i.Pbos: CCDC 1971367, single crystals grown from dimethyl sulfoxide, methanol, chloroform and water (C65H44N7O6Pb); triclinic P -1 (I.T.#2), a = 13.462(2), b = 14.764(2), c = 21.115(3) Å, 𝛼 = 93.891(6), 𝛽 = 99.656(5), 𝛾 = 94.702(6)°, V = 4108.9(10) Å3, Z = 2, d = 0.991 g⋅cm−3, 𝜇 = 2.093 mm−1. The contribution of the disordered solvents to the calculated structure factors was estimated following the BYPASS algorithm, implemented as the SQUEEZE option in PLATON. A final refinement on F2 with 18330 unique intensities and 516 parameters converged at (RF = 0.0383) for 16483 observed reflections with I > 2𝜎(I).
Acknowledgment
Région Bretagne is acknowledged for financial support to WB.
Supplementary data
Supporting information for this article is available on the journal’s website under https://doi.org/10.5802/crchim.94 or from the author.
1Note that the “in” and “out” labeling was initially chosen with the strap in a broken-shape conformation before we realized that most of these ligands adopt a W-shape conformation; see Ref. [18].
2ss: metal coordinates from the same side of the strap. os: metal coordinates from the opposite side of the strap.