

1 Introduction
Les métallopolymères présentent l'originalité d'allier les propriétés spécifiques d'un complexe métallique (électroactif, photosensible) aux propriétés électroniques des polymères conjugués, ce qui les rend particulièrement attractifs à la fois pour leurs propriétés intrinsèques fondamentales et pour leurs applications potentielles, par exemple, dans le développement de dispositifs nanoélectroniques et opto-électroniques pour le stockage moléculaire de l'information, l'élaboration de capteurs et pour l'électrocatalyse [1]. Parmi les métallopolymères, les polymétallorotaxanes présentent des propriétés topologiques particulières. Leur stratégie de synthèse repose sur l'effet assembleur [2] d'un ion métallique qui, par son pouvoir complexant, assure l'enfilage d'un ligand possédant des unités polymérisables dans un macrocycle coordinant avant électropolymérisation. Cette stratégie, mise en œuvre avec succès par Swager et al. [3] et par nous-mêmes, a permis l'élaboration de polymétallorotaxanes formés par deux ligands chélatants, respectivement à base de 2,2′-bipyridine ou de 1,10-phénanthroline. Les sites ainsi formés présentent une géométrie tétracoordinante stabilisant les bas degrés d'oxydation des métaux de transition tels que le cuivre ou le cobalt. Selon le mode de greffage des entités polymérisables, deux types de polymères sont élaborés, à savoir les polymères à groupes pendants [4] et les polymères à squelette conjugué [5]. Dans les polymétallorotaxanes à groupes pendants, les motifs pseudorotaxanes sont reliés à l'ossature du polymère (polypyrrole, polythiophène) par l'intermédiaire d'un bras espaceur saturé qui isole électroniquement les deux partenaires, à savoir les complexes métalliques et la matrice conjuguée conductrice. Ces polymères présentent l'additivité des propriétés électrochimiques classiques de la matrice conjuguée et des propriétés redox spécifiques de chaque complexe inclus. En revanche, les fils moléculaires des polymétallorotaxanes conjugués sont constitués d'une alternance d'oligothiophènes et d'unités phénanthroline afin d'assurer une continuité dans la conjugaison. De telles structures entraînent de fortes interactions entre le centre métallique et la matrice [1,6]. Enfin, l'électropolymérisation fige les sites complexants. En conséquence, une propriété tout à fait remarquable de ces matériaux concerne les échanges possibles d'ions métalliques, quelquefois avec l'assistance nécessaire de l'ion lithium pour maintenir la structure du polymère (effet d'échafaudage) [4b,5a].
Récemment, la synthèse d'un nouveau [2]-caténane, constitué de deux macrocycles coordinants, l'un incluant un ligand tridentate et l'autre un ligand bidentate, a été décrite [7]. Cette synthèse est basée sur l'effet de matrice d'un métal pentacoordiné. Ces complexes présentent des propriétés de coordination notablement différentes de celles des caténanes à sites tétracoordinants. En particulier, le potentiel de réduction du complexe de cuivre pentacoordiné subit un déplacement cathodique de 0,7 V en comparaison du complexe tétracoordiné. Par ailleurs, ces différences de propriétés redox ont été mises à profit avec succès pour l'élaboration de moteurs moléculaires [8]. Il nous a donc paru particulièrement intéressant d'inclure ces nouvelles propriétés dans les métallopolymères, d'accéder ainsi à une nouvelle classe de polymétallorotaxanes pentacoordinés pour comparer leurs propriétés à celles des polymétallorotaxanes tétracoordinés.
Nous rapportons ici la synthèse, les propriétés électrochimiques déterminées par voltampérométrie cyclique (VC) et les possibilités d'échange d'ions métalliques dans les polymétallorotaxanes pentacoordinés pendants ou conjugués de cuivre[I] et [II] et de zinc[II] formés à partir d'un ligand bicoordinant incluant une unité phen (phen = 1,10-phénantroline) 1 ou 2 et un macrocycle, incorporant des motifs tridentés de type terpy (terpy = 2,2′,6′,2″-terpyridine) 3 (Fig. 1). Ce choix repose directement sur les résultats de nos précédents travaux : d'une part, le ligand pendant 1 présente la longueur de la chaîne alkyle connue pour être optimale pour l'élaboration de polypyrroles fonctionnalisés [4b] ; d'autre part, le polymétallorotaxane tétracoordiné de cuivre[I] formé à partir du ligand 2 présente d'excellentes propriétés d'échange de métal et une forte interaction métal–ligand [6a]. Dans le cas particulier du poly(2,3,Zn)2+, des mesures de résistance, couplées à la voltampérométrie cyclique in situ, ont pu être réalisées.
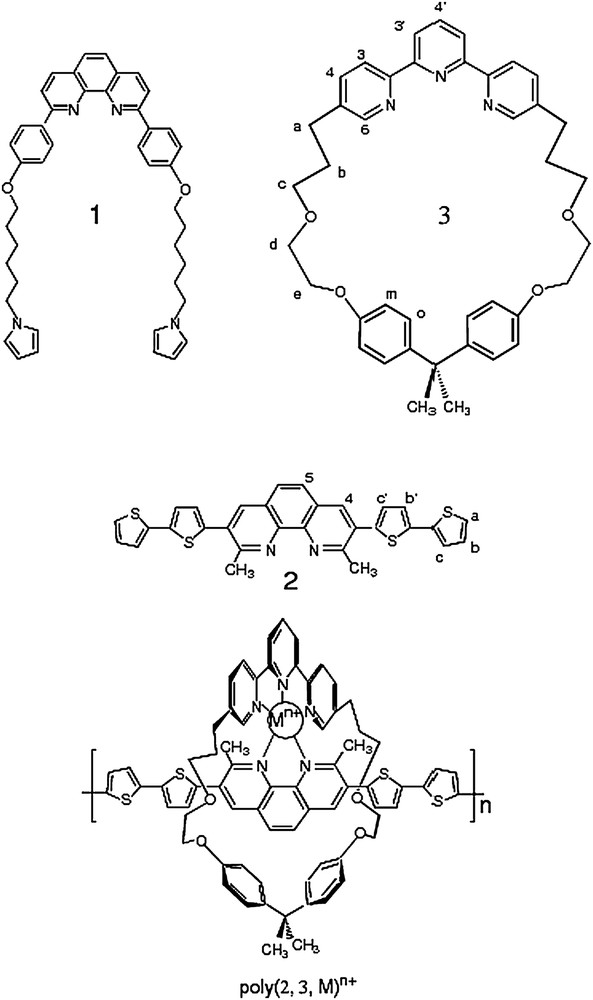
Les fils 1 et 2, et l'anneau 3, éléments constitutifs des pseudorotaxanes poly(1,3,M)n+ et poly(2,3,M)n+.
2 Partie expérimentale
2.1 Synthèses
2.1.1 Synthèse des ligands 1, 2 et 3
Les synthèses des ligands 1 et 3 ont été reproduites [4b,7]. La synthèse du ligand 2 précédemment décrite [6a] a été modifiée selon la procédure suivante.
2.1.1.1 Synthèse de la 3,8-bi(2,2′-bithie´n-5-yle)-1,10-phénanthroline
Une solution de 2,2′-bithiophène (1,016 g, 6,11 mmol) dans le THF anhydre (50 mL) est placée sous argon à −78 °C (bain de carboglace/acétone). Un équivalent de butyllithium (3,8 mL de solution titrée à 2,1 mol/L dans l'hexane, 6,11 mmol) est ajouté, la solution passe de l'incolore au blanc laiteux. Une solution de chlorure de zinc sec (840 mg, 6,16 mmol) dans le THF anhydre (25 mL) est canulée sur le bithiényl-lithium, puis le bain de carboglace est retiré. Une solution d'acétate de palladium (68,5 mg, 308 μmol, 5 % mol) et de triphénylphosphine (320 mg, 1,22 mmol) dans du THF anhydre (30 mL) est portée au reflux sous argon pendant 30 min ; la solution passe du rouge au jaune. La solution de bithiényle zincique lithium est canulée sur une solution de 3,8-dibromo-1,10-phénanthroline (660 mg, 1,95 mmol) dans le THF anhydre (25 mL) ; finalement, le catalyseur au paladium est canulé sur le mélange réactionnel avant d'être chauffé à 70 °C pendant 19 h. Le brut est transféré dans une cartouche de soxlet, la fraction soluble dans le THF étant éliminée. La cartouche est extraite au dichlorométhane et permet d'isoler 750 mg de 3,8-bi(2,2′-bithién-5-yle)-1,10-phénanthroline sous forme d'un solide vermillon (η = 75 %).
UV–vis (CH2Cl2) : λnm (log10 ɛ) = 272 (4,12), 324 (4,09), 441(4,33).
Fluorescence (CH2Cl2) : λex = 440 nm, λem = 525 nm.
Spectroscopie de masse : FAB m/z = 509.
RMN 1H (200 MHz, C2D2Cl4) : δ (ppm) 9,39 (2H, de), 8,37 (2H, d, 4J = 2Hz), 7,87 (2H, s), 7,54 (2H, d, J = 3,8 Hz), 7,34–7,25 (6H, m), 7,08 (2H, dd).
2.1.1.2 Méthylation du 3,8-bi(2,2′-bithién-5-yle)-1,10-phénanthroline
À une suspension de 306 mg de F2 (602 μmol) dans 20 mL de THF anhydre, on ajoute goutte à goutte 2,25 mL de méthyllithium en solution à 1,6 mol/L dans l'hexane (6 équivalents) à température ambiante. Après l'ajout de 2 équivalents, le milieu vire au noir. Après 72 h d'agitation à température ambiante, 1 mL de solution aqueuse saturée de chlorure d'ammonium est ajoutée, puis les solvants sont évaporés. Le brut est dissout 50 mL de CH2Cl2, 500 mg de K2CO3 étant ajoutés avant réoxydation du milieu par l'oxygène de l'air. La solution est chromatographiée sur alumine (CH2Cl2) pour isoler 130 mg de 2,9-diméthyl-3,8-bi(2,2′-bithién-5-yle)-1,10-phénanthroline sous forme d'un solide orange (η = 40%).
UV–vis (CH2Cl2) : λnm (log10 ɛ) = 310 (4,66), 372 (4,78).
Fluorescence (CH2Cl2) : λex = 384, épaul(365) nm ; λem = 451, épaul(488) nm.
Spectroscopie de masse (MALDI) : m/z = 536,77.
RMN 1H (200 MHz, C2D2Cl4) : δ (ppm) 8,25 (2H, s), 7,75 (2H, s), 7,29–7,23 (8H, m), 7,09–7,04 (2H, dd), 3,12 (6H, s).
2.1.2 Synthèse du complexe enfilé (1,3,Cu)BF4
On dissout 4,04 mg de 3 (M = 629,8 g/mol, 6,4 μmol) dans 500 μL de dichlorométhane-d2 dans un tube RMN. On pèse en boîte à gants 14,5 mg de tétraquis acétonitrile de cuivre[I], qui sont ensuite dissous dans 750 μL d'acétonitrile-d3. Par la méthode des ajouts dosés par RMN, le volume de solution de tétraquis acétonitrile de cuivre[I] à ajouter est déterminé, soit 105 μL. Finalement, 4,25 mg de 1 (M = 662,8 g/mol, 6,4 μmol) sont ajoutés en solution dans le dichlorométhane-d2.
RMN : 1H (200 MHz, CD2Cl2) : δ (ppm) 8,78–8,82 (3H, m), 8,58 (2H, d, 3J = 1,2 Hz), 8,37–8,41 (4H, m), 8,20 (2H, d, 3J = 8,8 Hz), 8,12 (2H, s), 7,19 (6H, d), 6,88 (4H, d, 3J = 8,8 Hz), 6,60–6,66 (12H, m), 6,02 (4H, t, 3J = 2,1 Hz), 4,08 (4H, t, 3J = 6,4 Hz), 3,95 (4H, dd), 3,86 (4H, t, 3J = 7,0 Hz), 3,66 (4H, dd), 3,55 (4H, t, 3J = 5,6 Hz), 3,04 (8H, m), 1,94–1,50 (16H, m).
2.1.3 Synthèse du complexe enfilé (2,3,Cu) (BF4)2
On introduit dans une boîte à gants 2,31 mg de 2 (M = 536,7 g/mol, 4,3 μmol) et 2,71 mg de 3 (M = 629,8 g/mol, 4,3 μmol), qui sont ensuite dissous dans 5 mL environ de dichlorométhane. On ajoute alors 430 μL d'une solution à 10−2 mol/L de Cu(BF4)2 dans l'acétonitrile. La solution passe du jaune au brun quasi instantanément. Le complexe est utilisé sans autre purification.
UV–vis (CH2Cl2) : λnm (log10 ɛ) : 327 (4,32), 352 épaul(4,23) et 415(4,11).
VC dans CH2Cl2 + 0,2 mol/L de nBu4NPF6, E1/2 = −308 mV vs Fc, quasi réversible (Cu[II]/Cu[I]]).
2.1.4 Synthèse du complexe enfilé (2,3,Zn)(BF4)2
À une solution de 5,2 mg de 3 (M = 629,8 g/mol, 8,26 μmol) dans 500 μL de dichlorométhane-d2 sont ajoutés un équivalent de Zn(BF4)2 en solution dans l'acétonitrile-d3 par la méthode des ajouts dosés en RMN. On ajoute alors 4,43 mg de 2 (M = 536,7 g/mol, 8,26 μmol). La solution passe du jaune clair à un orange plus foncé. Après enregistrement du spectre RMN, le solvant est évaporé et le complexe utilisé sans autre purification.
RMN : 1H (200 MHz, CD2Cl2, 300 K) : δ (ppm) : 8,64–8,70 (5H, m), 8,52 (2H, d), 8,06 (2H, dd), 7,80 (2H, s), 7,43–7,03 (16H, m), 6,54 (4H, d), 3,66 (4H, t), 3,36 (4H, t), 3,11 (4H, t), 2,42–2,54 (14H, m).
Spectroscopie de masse (MALDI) : 630 (6000) 3, 691 (1000) (3, Zn), 1136 (3000) (22, Zn), 1230 (1200) (2,3,Zn).
2.2 Électrochimie
Toutes les analyses et synthèses électrochimiques ont été effectuées en boîte à gants sous atmosphère d'argon à l'aide d'un potentiostat EG&G Princeton Applied Research PAR273 dans une cellule électrochimique constituée d'un montage classique à trois électrodes. L'électrode de référence est une électrode Ag+/Ag constituée d'un fil d'argent plongeant dans une solution de nitrate d'argent AgNO3 10−2 mol/L dans un électrolyte CH3CN + 0,1 mol/L de n-Bu4NPF6. Tous les potentiels donnés sont calculés par rapport au couple ferricinium/ferrocène (Fc) déterminé par voltamétrie cyclique (solution à 10−3 mol/L dans l'électrolyte) par rapport à cette électrode de référence. Les électrolytes sont des solutions à 0,1 mol/L de n-Bu4NPF6 dans CH3CN et 0,2 mol/L de n-Bu4NPF6 dans CH2Cl2. L'électrode de travail est un disque de platine de 7 mm2. Les électrosynthèses des polymétallorotaxanes ont été réalisées à partir de solutions à 10−3 mol/L de monomère par cyclages itératifs entre −0,1 V ou −0,2 V et 0,8 V ou 1 V à la vitesse de 50 mV/s. Les films ainsi obtenus sont copieusement rincés dans du CH2Cl2 avant analyse. Pour les expériences de démétallation, les solution de cyanure de lithium ont été obtenues par dissolution de quantité stœchiométriques de n-Bu4NCN et de LiPF6 dans CH3CN. Les films sont trempés dans cette solution pendant 15 min avant d'être rincés dans CH3CN, puis dans CH2Cl2. La remétallation par le cuivre[II] a été effectuée par immersion du film pendant 72 h dans une solution saturée d'hydrate de Cu(BF4)2 dans CH3CN, puis rinçage important dans CH3CN, puis CH2Cl2.
3 Résultats expérimentaux et discussion
La stratégie de synthèse des polymétallorotaxanes pentacoordinés est analogue à celle mise en jeu pour les polymétallorotaxanes tétracoordinés. Elle repose sur la formation, par effet de matrice, des précurseurs (1,3,M)n+ et (2,3,M)n+ (Mn+ : Cu+ ou Cu2+ et Zn2+) avant électropolymérisation.
3.1 Les complexes précurseurs
Les complexes précurseurs (1,3,M)n+ et (2,3,M)n+ (Mn+ : Cu+ ou Cu2+ et Zn2+) ont été préparés de la manière classique par addition, en quantité légèrement sous-stœchiométrique, du sel métallique correspondant (Cu(CH3CN)4BF4, Cu(BF4)2 ou Zn(BF4)2) au macrocycle 3, suivie par l'addition d'un équivalent de phen substituée 1 ou 2. Ces additions sont suivies par RMN dans le cas des ions métalliques non paramagnétiques. Cette méthode, particulièrement informative, démontre sans ambiguïté l'enfilage du ligand linéaire dans le macrocycle. En effet, cet enfilage provoque un très fort déplacement vers les champs forts des protons des phényles du ligand 1 (Ho (Δδ = −1,9 ppm) et Hm (Δδ = −0,6 ppm) dans le cas de (1,3,Zn)2+) ou bien des substituants méthyles du ligand 2 (Δδ = −0,3 ppm dans le cas de (2,3,Zn)2+) (Fig. 1). Ces blindages sont la conséquence de la proximité spatiale des substituants en C2 et C9 de la phénanthroline avec le cône d'anisotropie de la terpyridine du macrocycle 3. De même, les protons en C6 du cœur terpyridine du macrocycle 3 subissent un déplacement vers les champs forts (Δδ = −1,3 ppm pour (1,3,Zn)2+ et Δδ = −1,2 ppm pour (2,3,Zn)2+) du fait des cônes d'anisotropie des groupes phényles et thiényles des ligands enfilés 1 et 2. Ces effets de blindage sont caractéristiques des complexes à ligands encastrés 2,9-diarylphénanthroline. Un exemple de ce suivi par RMN est montré dans le cas de la formation du complexe (2,3,Zn)2+ (Fig. 2). Les solutions se colorent de manière attendue et contiennent essentiellement les précurseurs purs correspondants (1,3,M)n+ ou (2,3,M)n+ (Mn+ : Cu+ ou Cu2+, Zn2+). Les complexes sont caractérisés par voltampérométrie cyclique (Tableau 1). La réponse des solutions des complexes de cuivre[I] ainsi préparées est constituée, dans le domaine cathodique, d'un pic quasi réversible, caractéristique d'un transfert monoélectronique lent centré à E1/2 = −0,32 V pour (1,3,Cu)+ et −0,31 V pour (2,3,Cu)+, où E1/2 est défini comme la demi-somme des potentiels de pic anodique et cathodique (E1/2 = (Epa + Epc)/2). Ces valeurs sont en accord avec celles rapportées précédemment pour le couple formel Cu[II]/Cu[I] pentacoordiné et présentent un important décalage cathodique, respectivement −0,60 V et −0,69 V, par rapport aux complexes tétracoordinés correspondants, dont le macrocycle inclut une 2,9-diaryl-phénanthroline. Les complexes de Cu[I] sont stabilisés par un environnement tétraédrique, alors que les complexes de Cu[II] sont plus stables dans un environnement pentaédrique [8a,9]. La nature du ligand enfilé 1 ou 2 a peu d'influence sur la valeur de E1/2 du couple formel Cu[II]/Cu[I], indiquant clairement que l'échange électronique concerne les orbitales redox du métal. En revanche, les voltampérogrammes des complexes de zinc sont caractérisés, dans le domaine cathodique, par deux vagues monoélectroniques quasi réversibles, centrées, d'une part, à −1,59 V et −1,90 V pour le complexe (1,3,Zn)2+et, d'autre part, à −1,47 V et −1,73 V pour (2,3,Zn)2+, associées respectivement aux couples formels Zn[II]/Zn[I] et Zn[I]/Zn[0]. À la différence des complexes de cuivre, ces transferts électroniques mettent en jeu les orbitales redox des ligands. Ainsi, on observe un déplacement anodique des E1/2 entre les voltampérogrammes des deux complexes, mettant en évidence un effet donneur moins prononcé du ligand 2 en comparaison du ligand 1.
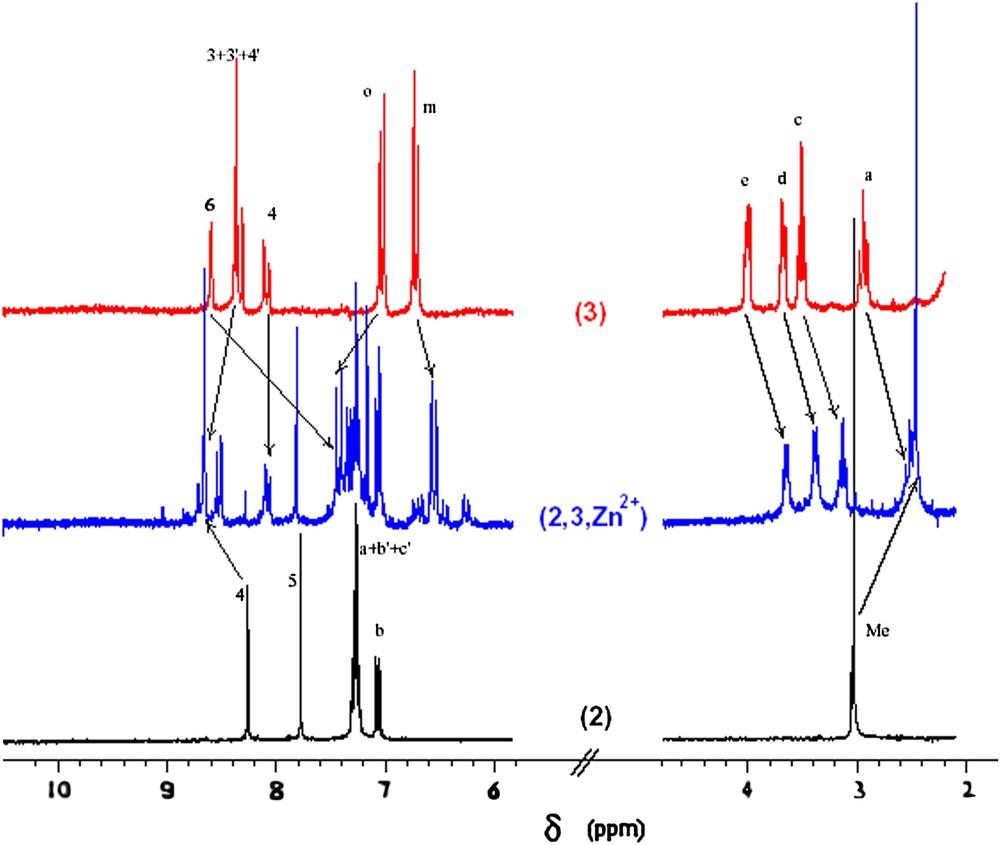
Suivi par RMN de la synthèse de (2,3,Zn)2+.
Valeurs des E1/2a (V) et ΔEb (mV) déterminées par voltampérométrie cyclique, v = 50 mV/s, dans CH2Cl2, 0,1 mol/L de nBu4NPF6, associées aux couples redox formels des métaux des complexes pentacoordinés
| Complexes | M[II]/M[I] | M[I]/M[0] | ||
| E1/2 | ΔE | E1/2 | ΔE | |
| (1,3,Cu)+ | −0,32 | 300c | ||
| (2,3,Cu)+ | −0,31 | 116 | ||
| (1,3,Zn)2+ | −1,59 | 160 | −1,90 | 125 |
| (2,3,Zn)2+ | −1,47 | 95 | −1,73 | 100 |
a E1/2 = (Epa + Epc)/2 en V vs Fc+/Fc.
b ΔE = Epa − Epc.
c Ce complexe présente un pic de désorption à −0,460 V et une voltampérométrie irréversible.
3.2 Les polymétallorotaxanes « pendants » : poly(1,3,M)n+ (Mn+ : Cu+, Zn2+)
Alors que les tentatives d'électropolymérisation du complexe de cuivre (1,3,Cu)+ ont échoué, l'électropolymérisation du complexe de zinc (1,3,Zn)2+ conduit bien à la formation d'un film très fin, jaune pâle, lors d'un balayage lent (v = 5 mV/s) entre 0 V et 0,9 V, correspondant à une charge de synthèse de Qsynthèse = 1,3 mC/cm2. Le voltampérogramme de ce film (Fig. 3) montre, dans le domaine anodique, une réponse caractéristique de l'électroactivité de la matrice N-polypyrrole et, dans le domaine cathodique, deux vagues, associées au centre métallique, caractéristiques de systèmes redox attachés à l'électrode à −1,5 V et −1,75 V. Les balayages suivants indiquent clairement une diminution progressive du signal du centre métallique, sans modification de la réponse de la matrice. Ce polymère présente bien l'additivité des propriétés de la matrice et du centre métallique. Enfin, la faible stabilité du centre métallique dans ce film a interdit toute expérience de métallation–démétallation.
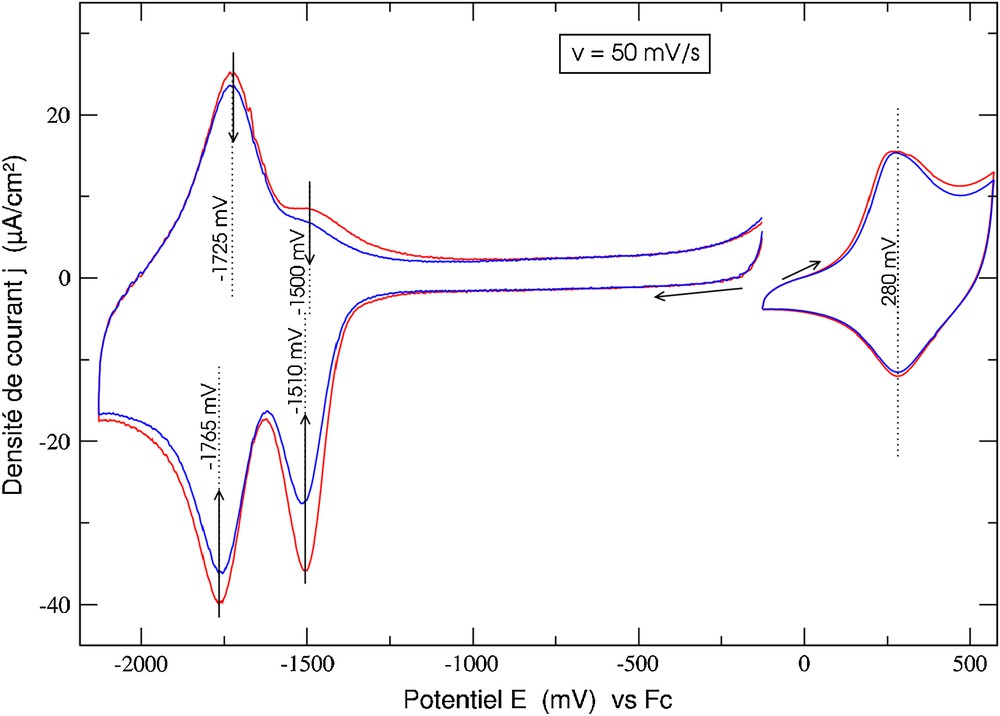
Voltampérogramme d'un film de poly(1,3,Zn)2+ (Qsynthèse = 1,3 mC/cm2) dans CH3CN + 0,1 mol/L de nBu4NPF6.
3.3 Les polymétallorotaxanes conjugués : poly(2,3,M)n+ (Mn+ : Cu+, Zn2+)
L'électropolymérisation des monomères conjugués (2,3,Cu)+ et (2,3,Cu)2+ s'accompagne de la décomplexation du site métallique. Ainsi, le voltampérogramme du film synthétisé à partir du monomère de cuivre[I] confirme l'absence de réponse du centre métallique dans le domaine cathodique. En revanche, il présente deux vagues rédox réversibles, bien résolues, situées à E1/2 = 0,70 V et 0,92 V, caractéristiques d'oligothiophènes à quatre unités thiényles, dont les extrémités sont substituées par des groupes non électroactifs [10].
Devant cette absence de réponse du cuivre pentacoordiné dans le polymère précédent, il nous a paru intéressant d'essayer une autre stratégie de synthèse du polymétallorotaxane de cuivre, mettant à profit les propriétés des polymétallorotaxanes vis-à-vis des échanges d'ions [5a]. L'électrosynthèse précédente fait intervenir un changement du degré d'oxydation du cuivre[I] lors de la polymérisation de (2,3,Cu)+, donc un changement de sa sphère de coordination avant électropolymérisation, pouvant induire une certaine instabilité du complexe. Le zinc[II] dans un environnement pentacoordinant ne présente pas cet inconvénient. En effet, cet état redox est stable jusqu'à des potentiels très supérieurs à l'oxydation des unités bithiényles. L'électropolymérisation du métallorotaxane de zinc[II] peut donc conduire avec succès à la synthèse d'un polymétallorotaxane de zinc[II] puis par démétallation-métallation au polymétallorotaxane conjugué de cuivre[I] ou [II].
À la différence des complexes de cuivre, une voltampérométrie itérative de (2,3,Zn)2+ en solution dans le dichlorométhane (Tableau 1) ou l'acétonitrile conduit bien à la formation d'un film de poly(2,3,Zn)2+, dont le voltampérogramme dans l'acétonitrile est présenté sur la Fig. 4. Dans le domaine anodique, on observe une seule vague peu structurée associée à l'électroactivité de la matrice conjuguée, indiquant que l'ion métallique n'induit pas de coopérativité dans le matériau, c'est-à-dire n'induit pas une localisation organisée des charges dans les chaînes conjuguées. Un phénomène semblable avait été observé pour le polymétallorotaxane de zinc tétracoordiné correspondant [7]. Dans le domaine cathodique, les trois vagues successives centrées à −1,45, −1,70 et −2,00 V sont attribuées respectivement aux réductions successives des ligands complexant le zinc[II], puis aux ligands libres, indiquant qu'une décomplexation se produit lors de la synthèse. Les balayages suivants montrent une stabilité de la réponse de la matrice. En revanche, on constate la disparition de la vague à −1,45 V et une forte diminution des intensités des vagues plus cathodiques, confirmant une faible stabilité du zinc dans un site complexant pentacoordinant immobilisé. Après une dizaine de cycles dans le domaine cathodique, les vagues relatives au complexe de zinc ont disparu. Il est alors impossible de remétaller avec quelque ion métallique que ce soit : cuivre[I], cuivre[II], zinc[II], cobalt[II], nickel[II]. La démétallation par balayages cathodiques entraîne donc une destruction irréversible du site complexant.
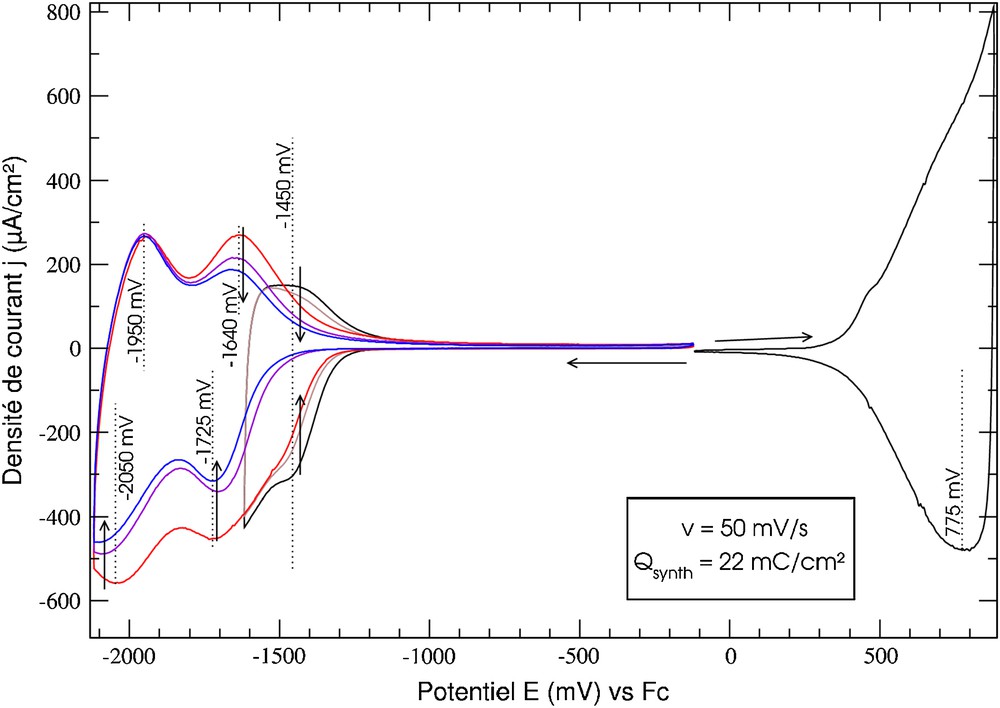
Voltampérogramme d'un film de poly(2,3,Zn)2+ déposé sur du platine (Qsynthèse = 22 mC/cm2) dans CH3CN + 0,1 mol/L de nBu4NPF6.
Si la démétallation est effectuée, d'une manière classique [5a], par action d'un agent décomplexant tel que le cyanure de lithium en solution dans l'acétonitrile pendant 15 min, la métallation par le cuivre peut être réalisée par immersion de l'électrode pendant 72 h dans une solution de Cu2+·2BF4− saturée dans l'acétonitrile. La Fig. 5 montre bien, dans le domaine cathodique, la réponse électrochimique réversible caractéristique du cuivre pentacoordiné et, dans le domaine anodique, la réponse caractéristique du premier système redox de la matrice, centré à E1/2 = 0,68 V. À des potentiels plus anodiques, se trouve une autre vague liée à la matrice polymère. Le système électrochimique du cuivre pentacoordiné apparaît comme une vague faiblement réversible à E1/2 = −0,41 V. Ce système n'est pas stable aux cyclages, puisqu'en une dizaine de cycles l'intensité des pics d'oxydation a diminué de 30%. La structuration de la réponse électrochimique de la matrice peut être liée à la coopérativité entre le complexe métallique et la matrice polymère. La charge du complexe provoque la localisation des charges sur la matrice. Enfin, nous remarquons une diminution sensible de l'intensité de la réponse électrochimique du polymétallorotaxane de cuivre en comparaison de celle du polymétallorotaxane de zinc avant échange. Nous attribuons cette diminution à une dégradation du film pendant les différentes étapes de la démétallation–métallation, en particulier à l'ion cuivre[II], susceptible d'oxyder la matrice polymère.
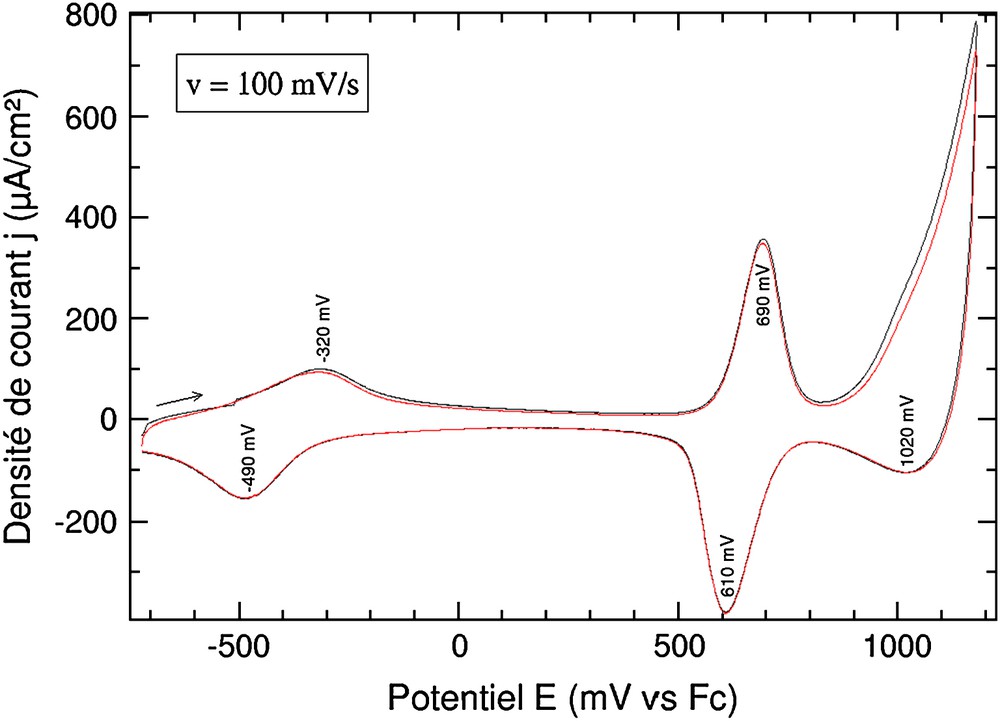
Voltampérogramme d'un film de poly(2,3,Zn)2+ (Qsynthèse = 22 mC/cm2) dans CH3CN + 0,1 mol/L de nBu4NPF6 après trempages successifs dans LiCN (15 min) puis dans une solution de cuivre[II] (72 h).
Des mesures de résistance couplées à la voltampérométrie ont pu être réalisées sur le poly(2,3,Zn)2+ et sont montrées sur la Fig. 6. La résistance chute lors du dopage de la matrice. La valeur de cette résistance est alors de 800 kΩ et correspond à une conductivité de σ = 1,3 × 10−5 cm−1 Ω−1, qui est du même ordre de grandeur que celle mesurée précédemment pour le polymétallorotaxane tétracoordiné correspondant. Le polymère est isolant dans tout le domaine cathodique. Ceci provient vraisemblablement de l'instabilité des zincs[I] et [0] dans l'environnement pentacoordinant du polymère. En effet, la comparaison des deux voltampérométries réalisées avant et après la mesure de résistance couplée montre la disparition de la première vague associée au centre métallique.
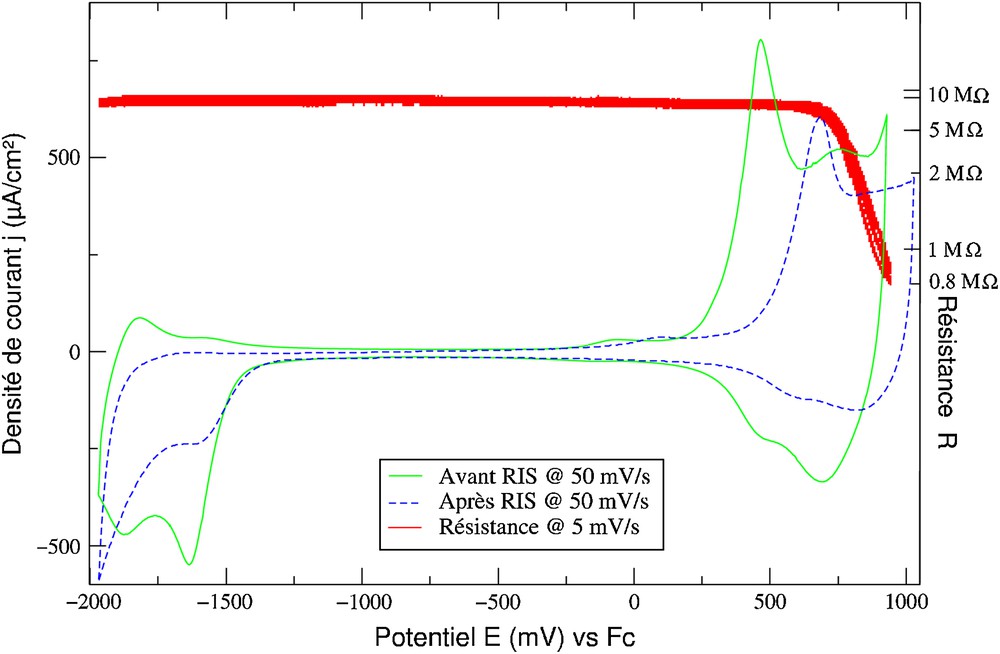
Mesure de résistance couplée à la voltamétrie cyclique du poly(2,3,Zn)2+ déposé en film (Qsynthèse = 23 mC/cm2 à v = 5 mV/s). Les voltampérogrammes sont enregistrés à une vitesse de balayage de 50 mV/s.
4 Conclusion
La synthèse par effet de matrice des cations cuivre[I] et [II] et zinc[II] a permis d'enfiler deux ligands linéaires 1 et 2 dans un macrocycle terpyridinique 3 conduisant à quatre pseudorotaxanes pentacoordinés différents, potentiellement électropolymérisables.
Les pseudorotaxanes basés sur le ligand linéaire conjugué 2 ont permis l'électropolymérisation et le dépôt en couche épaisse de polymère. Néanmoins, la présence du cuivre dans les sites complexants n'a pu être mise en évidence après polymérisation des complexes (2,3,Cu)+/2+. En revanche le polymétallorotaxane de zinc[II], poly(2,3,Zn)2+, a bien conservé le métal après la polymérisation, et l'ion Zn2+ a pu être échangé avec du Cu2+ après démétallation au cyanure de lithium, conduisant ainsi au poly(2,3,Cu)2+ par une voie chimique. Le polymétallorotaxane de zinc présente une faible conductivité, de l'ordre de 10−5 Ω−1 cm−1, dans le domaine d'électroactivité de la matrice. Les réponses en voltamétrie cyclique des polymétallorotaxanes pentacoordinés indiquent une interaction métal–ossature conjuguée plus importante dans le cas du cuivre[II] que dans celui du zinc[II], en accord avec les interactions métal–ligand précédemment mises en évidence sur les polymétallorotaxanes analogues tétracoordinés.