

1 Introduction
Metallic materials including stainless steel, titanium alloys, and cobalt-based alloys represent, due to their high strength, ductibility, and good corrosion resistance, an important class of materials in hard tissue replacement, especially load-bearing implants for the repair or replacement of diseased or damaged bone tissues. However, these metallic materials are not biodegradable in the human body and can cause long-term complication (infection). So a second surgery may be necessary after the tissues have healed. Thus a new domain of research in metallic implants focuses on new biodegradable implants, which dissolve in biological environment after a certain time of functional use. Biodegradable implants represent an appropriate solution because of cost, convenience and aesthetic reasons favourable to patients. Magnesium as a degradable implant material provides both biocompatibility and suitable mechanical properties. Mg2+ is present in large amount in the human body, involved in many metabolism reactions and biological mechanisms. Previous in vivo studies have shown that magnesium is suitable as degradable biomaterial for use in medical implant [1,2]. Compared to current implant materials, magnesium alloys have a lower elastic modulus (about 45 GPa which is closed to that of natural bones (10–40 GPa)) and higher yield strength, that provide to these alloys the potential for avoiding the “stress shield” [3–5]. Their use as a permanent metallic implant is seriously limited because of their poor corrosion resistance (10.5–210 mm per year for Mg with purity of 99.9% in 3% NaCl solution, relevant for biomedical applications) [6]. The fact that they corrode relatively fast is a good starting point to consider them for potential applications as bioabsorbable implant, but the corrosion performance still has to be improved. Additionally, studies concerning the corrosion behaviour of magnesium in biological environment and cytotoxicity of magnesium are lacking.
From a corrosion perspective, magnesium is one of the most active elements and will, as well as its alloys, corrode rapidly in water-containing media except in the alkaline pH range. Furthermore, rapid production of hydrogen gas during the corrosion process may not be tolerated by host tissues [7,8]. As a result of the high corrosion susceptibility of Mg alloys, temporary protection of the surface is necessary when this material is used in aqueous and saline media such as the environment of the human organism. Recently, a number of studies have been carried out to investigate the corrosion behaviour of magnesium alloy in artificial physiological fluids. However, most of them were considering an Al containing Mg alloy [9–12]. Most alloying elements will dissolve into the human body during the magnesium alloy degradation. If, for example, the AZ91D magnesium alloy is used, the Al present in the alloy would get into the human body, which might be hazardous, from a health point of view.
The purpose of this study is to investigate the in vitro corrosion behaviour in contact with different simulated biological environments of a model Mg–Y–RE alloy (untreated and anodised) representing the actual trend in alloys considered for biodegradable implants. Because of the potential use of Mg alloys for different types of implants, two types of physiological solutions were used depending on the envisaged implant location: artificial plasma (AP) for an implant in contact with blood and a buffered simulated body fluid (SBF K9) for the other applications where blood is absent. Protection of Mg alloy by using plasma–electrochemical anodising processes has been extensively studied in the past but mainly to obtain a very thick (in order of 10 μm) and porous coating with long-term corrosion protection [13–20]. This is typically not a protection strategy that can be applied to the surface considered for a biodegradable implant purpose: a biodegradable implant needs to corrode at a controlled rate after a certain delay in the corrosion initiation. The final objective is to modify the composition and/or the surface of the alloy with an anodic layer to obtain a suitable initial stability during the initial period where mechanical strength is required, followed after a couple of weeks by a full degradation of the implant. So, as a temporary corrosion protection, a thin (hydr)oxide film has been considered in this work using the galvanostatic anodising treatment, and then the corrosion behaviour of this coating was documented. Characterization of the oxide film structure and composition for optimized conditions was performed by different electrochemical impedance spectroscopy (EIS), optical microscopy and Auger Electron Spectroscopy (AES) measurements. The AES investigation of the surface composition has evidenced the different oxidation state. In order to take in account this fact, the term of (hydr)oxide is used in this paper, indicating the possible presence of both structure.
2 Experimental
Test specimens were prepared from an Mg–Y–RE alloy. Its composition in weight is the following: 3.9% yttrium, 2.1% neodymium, 0.51% zirconium, and other elements in trace amount. The samples were polished using abrasive paper up to grit 4000 with ethanol as lubricant and then polished with diamond paste (particle size < 0.25 μm). Fig. 1 shows the SEM image of the complex microstructure of the alloy.

SEM image of the freshly polished Mg–Y–RE alloy surface (Scale in microns).
For the corrosion investigation, two types of physiological solutions were used. Artificial plasma (composition according to ISO 10993-15) [21] and simulated body fluid (SBF K9) [4] were both prepared by dissolving the analytical grade reagents in deionised water. The ion concentrations of SBF K9 and AP are listed in Table 1. Two main differences need to be mentioned. First of all, the SBF K9 contains buffer agent (Tris–(CH2OH)3CNH2) that induces good pH stabilization for SBF K9 contrary to the AP media. Secondly, AP is about six times more concentrated in carbonate than in SBF, and the carbonate is well known as a good corrosion inhibitor in certain conditions for Mg alloys [22].
Ion concentrations (mM) in artificial plasma (AP) and simulated body fluid (SBF) K9
| Artificial plasma | SBF K9 | |
| Na+ | 144.5 | 142.0 |
| K+ | 5.4 | 5.0 |
| Mg2+ | 0.8 | 1.5 |
| Ca2+ | 1.8 | 2.5 |
| Cl− | 125.3 | 148.8 |
| (HCO3)− | 26.2 | 4.2 |
| (HPO4)2− | 3.0 | 1.0 |
| SO42− | 0.8 | 0.5 |
2.1 Anodising
The anodising bath solution was 0.1 M NaOH + 0.05 M Na3PO4. Potassium phosphate is a commonly used chemical in anodising solutions for magnesium alloys [16–19]. It has to be mentioned that in pure NaOH solutions, high voltage anodising is very difficult. This is due to the fact that in high pH solution, the thin Mg hydroxide layer is semiconducting, so it is necessary to add ions like phosphate in the anodising bath, in order for them to integrate in the Mg hydroxide, preventing water dissociation [23,24]. Phosphate ions are essential in order to get an insulating surface oxide at the beginning of the anodising. Surface film modification such as phosphate integration in the Mg hydroxide can also help to reduce the subsequent uncontrolled growth of the surface oxide in neutral electrolytes. Anodising was performed using a galvanostatically controlled anodising current provided by a KEITHLEY 2400 power supply system with the current/voltage source whose maximum current and voltage can be preset. The specimen was the anode and a platinum electrode was used as a cathode. The anodising current density was 15 mA/cm2 applied for 5 min, with a maximum voltage limit set to 200 V (limitation of the potentiostat is 250 V).
2.2 Electrochemical impedance spectroscopy (EIS)
In order to identify and investigate the electrochemical reaction mechanisms (uniform versus localized corrosion susceptibility), the electrochemical behaviour of untreated and treated samples (anodised) was examined by electrochemical impedance spectroscopy (EIS). An Autolab PGSTAT 30 system from Eco Chemie B.V. (Utrecht, the Netherlands) was used and both artificial plasma and simulated body fluid have been tested as electrolytes. The EIS spectra were measured at 37 ± 2 °C. The measured area was 1 cm2, smaller than the anodised region (diameter 2.6 cm). The AC potential amplitude applied for EIS measurement was 10 mV and the frequency was varied from 100 kHz to 10 mHz. The electrochemical cell consisted of a classical three-electrode cell. A saturated calomel electrode (SCE) was used as a reference electrode and a Pt counter electrode.
For every EIS measurements, the whole frequency spectra from 100 kHz down to 10 mHz are recorded. Afterwards, often the value at 10 mHz is used to characterize the charge transfer resistance related to uniform processes. It is clearly an approximation because, depending on the measured value that is ranging from 10 to 250 kΩ/cm2, this impedance corresponds to different processes. However, it is a good indication of how good the surface protection is as a function of the used electrolytes and conditions.
2.3 Surface characterization by Auger electron spectroscopy (AES)
Characterization of the surface oxide formed and its influence on the stability in aqueous media is a key aspect of the study. A PHI/Perkin Elmer 4300 system equipped with an LaB6 filament was used for scanning electron microscopy, surface analysis and sputter depth profiling measurements. The in-depth elemental composition of the oxide formed on the magnesium alloys was obtained by combining Auger electron spectroscopy (AES) with controlled sputtering of the sample surface using a 4 keV argon ion beam. The sputtered area was 2 × 2 mm and after each data acquisition cycle, the sputter duration was 1 min. The sputtering rate was around 20 nm/min, as determined by calibration using a 100 nm SiO2/Si reference sample. Quantitative analysis was based on standard atomic sensitivity factors obtained from PHI reference handbook [15].
3 Results and discussion
3.1 Alloy degradation
3.1.1 Electrochemical analysis
The first aspect of the study was to monitor the corrosion reaction rates as a function of time in simulated body fluids and get a first picture of the uniform versus localized corrosion attack. The electrochemical impedance spectroscopy (EIS) measurements with its frequency depending information allow distinguishing between different corrosion reactions and to assess the protecting ability of surface. The EIS measurements are performed in the different body fluids mentioned in Section 2. The denomination untreated samples refer to freshly polished surfaces before the measurement.
Fig. 2 shows the Bode representation of an impedance measurement performed on a polished Mg–Y–RE alloy after immediate immersion in AP. Depending on the frequency domain considered, the Z modulus (later called impedance) and the phase shift curves give information concerning the type of electrochemical processes occurring during the immersion. In the high frequency domain (around 100 kHz), there should not be a capacitive charging of the electrochemical double layer and the system resistance can be defined as the solution resistance Rs only. In the low frequency domain (<1 Hz in our case) the system resistance depends mostly on the polarisation resistance of the oxidized surface and is related to corrosion resistance. The (hydr)oxide film formed during the immersion can act as a more or less efficient electric barrier for the charge transfer depending on its homogeneity and stability. The higher the charge transfer resistance of the formed surface film is, the higher would be the uniform corrosion resistance of this (hydr)oxide layer, and accordingly the higher would be the impedance value measured at low frequency. It has to be mentioned at this point that because of the complexity of the surface processes with competing uniform and localized corrosion processes as well as oxide film formation on an alloy with heterogeneous microstructure, it is not possible and reasonable to formulate equivalent circuit models for the impedance measurements.
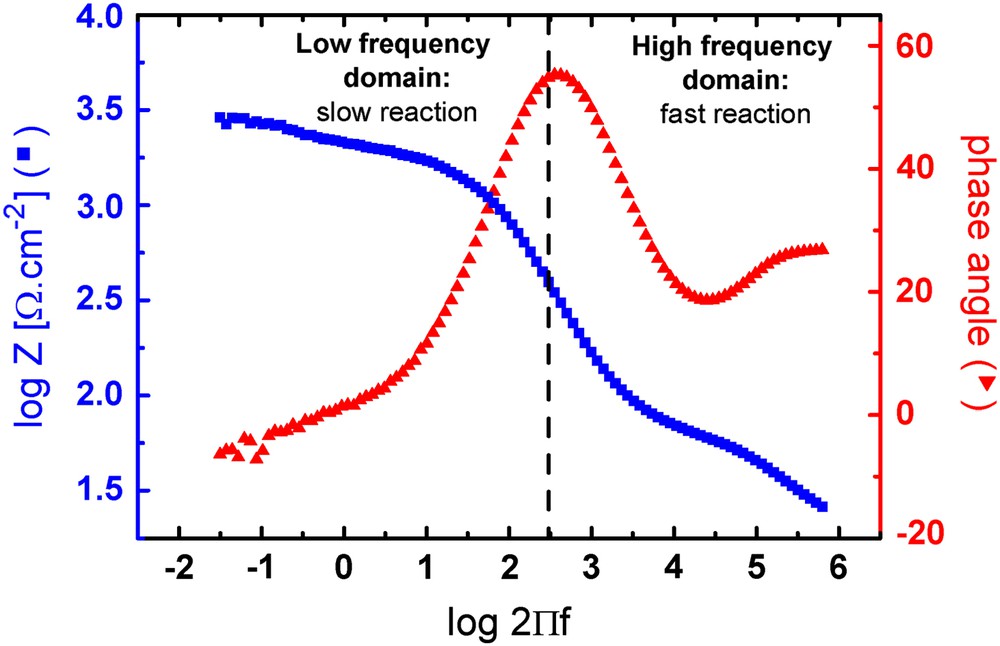
Bode representation of the modulus and phase shift for a bare surface after immediate immersion in AP.
In order to study the uniform corrosion resistance of the alloy in the different body fluids, attention was turned to the impedance results in the low frequency domain. The impedance values for the Z modulus (obtained at f = 0.01 Hz) measured in AP and SBF K9 at open circuit potential increase as a function of immersion time during the first 24 h. In these experimental conditions considered (pH, buffer ability…), the initial air-formed (hydr)oxide layer is not expected to be protective.
Large differences between the impedance values for each solution are obtained. Concerning the immersion in SBF K9, even if an increase of the impedance value is observed at longer immersion time (Fig. 3), impedance range values are 20–30 times lower than that for AP immersion. This discrepancy represents a problem for an accurate implant life prediction which is necessary when a specific application is envisaged. AP solution is significantly less aggressive than SBF solution for the untreated surface shown by the charge transfer through the surface being more hindered than in SBF. Degradation is critically dependant on the surface pH and the buffering ability of the media. The buffering ability of SBF induces a low corrosion resistance: the increase in pH at the electrode surface is not high enough to reach a value where thermodynamics predicts the formation of a stable and protecting Mg oxide or hydroxide layer.

Impedance values measured at f = 0.01 Hz of a bare surface Mg–Y–RE alloy as a function of immersion time in AP compared to SBF.
Concerning the immersion in AP, the high impedance values found after few hours of immersion indicated that the surface film becomes increasingly resistant and insulating as a function of immersion time. Afterwards, the corrosion resistance slightly decreases after 4 h of immersion and seems to stabilize. The result of this (hydr)oxide film stabilization is the initiation of localized corrosion attack and formation of pits at defects in the surface film. Fig. 4 shows deep localized attacks (pits) visible after 24 h of AP immersion and with a size estimated at 50–100 μm, investigated using optical microscopy observations. There are no corrosion products present in these pits, so that the defect is completely open to the attacks. The presence of this defect is detrimental to the global corrosion resistance and especially to mechanical failure of the implants.
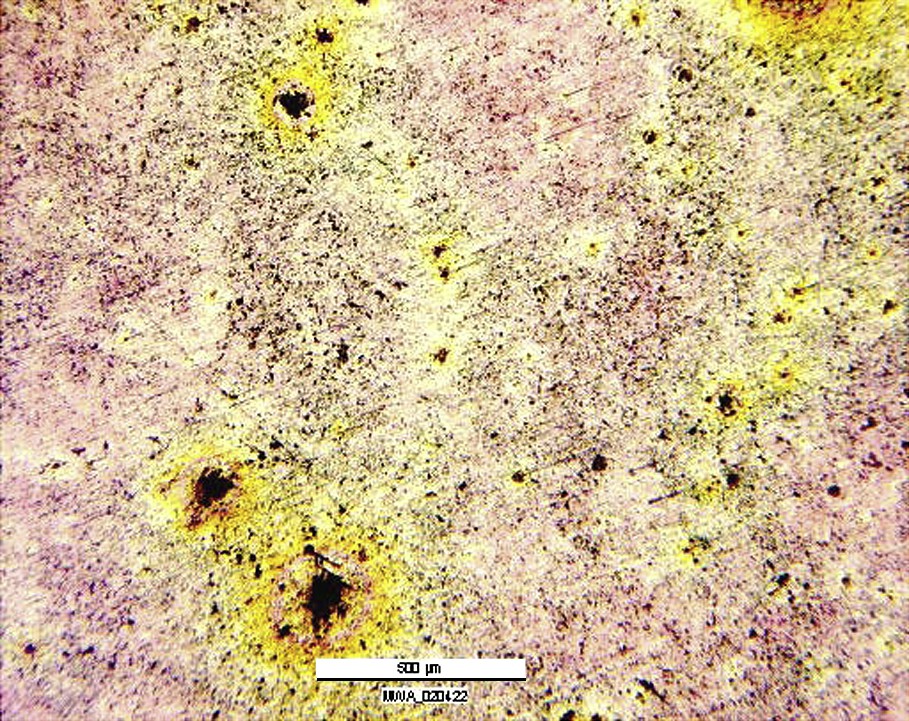
Optical micrograph of corroded surface after 24 h immersion in AP at 37 °C for a polished sample of Mg–Y–RE alloy.
The pH of the environment that is in contact with the (hydr)oxide film surface is one of the most important parameters concerning the corrosion of magnesium. In a previous publication [23], it was shown that the Mg and Mg alloy corrode relatively quickly in aggressive electrolytes (for example, chloride solutions) as long as the pH of the surface does not reach a value high enough. The pH 12 provides a good protection with the formation of a passive oxide. Due to the low electrochemical potential of the surface, the anodic Mg dissolution is accompanied by a very high rate of cathodic hydrogen evolution guaranteeing the charge balance of the corrosion process. As a result of this H+ consumption, the pH at the surface rises quickly inducing a spontaneous improvement of the corrosion resistance.
Fig. 5 shows the pH increase measured in small amount (5 ml/1 cm2) in AP, SBF K9 and DI water during alloy corrosion. The measurements are performed with a micro-pH sensor positioned 1 mm above the surface (needle tip micro-pH electrode by Thermo Orion). In contact with distilled water, the pH at the surface increases very quickly up to a value of around 9.5–10. In this range of surface pH, the chemical equilibrium exists for this solution volume. The alkalinization of the surface in the DI water occurs also in the absence of aggressive ions because of the thermodynamic instability of Mg resulting in uniform dissolution. It had been evidenced that the rate of pH increase depends obviously on the solution volume to exposed area ratio [25].

pH evolution as a function of immersion time of Mg–Y–RE alloy surface in AP, SBF and distilled water measured at 37 °C.
In contact with the AP, the pH value measured at the surface increases quickly during the first few minutes of immersion but reaches a slightly lower pH value limit than for distilled water immersion. Some buffering ability of carbonates can explain this trend. When the surface is in contact with SBF K9, important pH transients are visible induced by the competition between the important H+ consumption during localized attack which increases the pH and the buffering ability of the solution. The pH of the buffered SBF is 7.4, but after these severe corrosion attacks, it can clearly not be maintained over longer period of time.
3.1.2 Influence of the carbonate amount and the buffering ability on the corrosion rate of the Mg–Y–RE alloy
The difference in corrosion behaviour between immersion in AP and SBF K9 might also be related to the difference of the ionic species content of these solutions (Table 1). One main difference is the amount of carbonate which is six times less concentrated in SBF. In order to study the influence of the carbonate ions on the corrosion mechanisms, two kinds of AP solutions referred as AP and AP low (HCO3−) were prepared. In the “usual” AP solution, the carbonate ion concentration is very similar to that of human plasma. The amount of carbonate ions in the AP low (HCO3−) solution is adjusted to be the same concentration than the one present in the SBF K9 (e.g. Table 1). The second difference is the buffering ability of SBF K9. This means that the composition of SBF K9 without buffer agent corresponds to AP low (HCO3−). Fig. 6(a) displays impedance spectra showing the influence of the buffering ability of the media directly after immersion of the alloy surface in SBF K9 and SBF K9 without buffer agent. As described before, the impedance behaviour of the alloy at low frequency may be related to the uniform corrosion resistance of the surface (hydr)oxide film. A large difference in the corrosion rate is observed as a function of the buffering capacity of the media. Although a good buffering ability decreases strongly the uniform corrosion resistance of the alloy, when the pH slightly increases, susceptibility to localized corrosion is also immediately higher. This is visible in Fig. 6(a) by the fast reaction present in the high frequency domain (104–105 Hz) of the spectrum for the immersion in SBF without buffer agent. An additional phase shift peak as well as a shoulder in the impedance graph is a clear indication of a fast electrochemical process which is the case of localized attack. In order to model the localized breakdown in an oxide layer due to corrosive attack, previous publications have developed different pitting models for the case of Al alloys [26–31]. Different authors had evidenced that the phase angle is a very sensitive indicator of the localized corrosion. In these models, artificial pitting was generated on the coating. When pits had grown to a certain size, a new time constant appeared at higher frequency [28]. In an another active pit model, it had been shown that even a small amount of pitting lead to a pronounced change of the spectra in the high frequency region [26,30].

(a) Bode representation of the impedance modulus diagram of an untreated surface after immediate immersion in SBF K9 and SBF K9 without buffer agent at 37 °C. (b) Impedance evolution as a function of immersion time of the alloy in SBF K9, SBF K9 without buffer agent and AP.
Fig. 6(b) shows the low frequency impedance evolution as a function of immersion time in SBF K9, SBF K9 without buffer agent and AP for the Mg–Y–RE alloy. The high amount of carbonate in AP seems to speed up the initial surface protection and induces a faster stabilization of the surface in the first 4 h of immersion. But after few hours of immersion in AP, the dissolution rate of the alloy decreases and the impedance values measured during immersion in SBF without buffer agent are very similar to the one measured during immersion in AP low (HCO3−). In summary, it can be stated that no long-term protecting effect is observed for high amount of bicarbonate that could be related to the presence of a continuous protecting MgCO3 layer. A positive effect of carbonate in reducing the corrosion rates is only observed in the early stage of the corrosion process.
3.1.3 Corrosion behaviour of magnesium sample in AP and AP without Cl−
The formulation of the corrosion mechanisms of Mg–Y–RE alloys in body fluids is complicated because of the presence of a large number of species in the solution. Moreover, we are not in the presence of the direct active dissolution of the alloy because in this case there would not be an increase of the impedance values as a function of immersion time. The direct passivation of the alloy with formation of a protecting oxide layer is not possible in this pH range as well. Usually, a critical parameter influencing the corrosion resistance is the amount of chloride ions in the solution. Chloride ions are aggressive for the magnesium alloy not from a passivation breakdown perspective, but because the absorption of chloride ions on the oxide films of the magnesium surface transforms Mg(OH)2 to easily soluble MgCl2 [32]. In the buffered chloride solution, the corrosion rates of magnesium alloys are determined by the pH and the chloride ion concentration [33]. In fact, in physiological solution, some corrosion products are present at the interface, and the stability of these corrosion products in solution can be increased by the integration of different anions like Cl− or HCO3−. The situation is totally different for passive systems where the chloride ions are part of the most detrimental elements for the corrosion resistance.
AP solutions with and without chloride ions were used to investigate the influence of this ionic species on the corrosion of untreated samples. In the chloride free AP solution, all the chemicals containing chloride ions have been removed, inducing also small changes in cationic species concentration. Since the amount of ions present in solution is less important, the solution resistance will also be higher. The Bode plot of impedance modulus diagrams measured at 37 °C in AP and AP without Cl− directly after immersion and after 24 h of immersion is given in Fig. 7(a) and (b), respectively. After immediate immersion, the resistance in the low frequency domain associated to the film formation (corrosion product layer) obtained for AP is five times higher than the one observed in AP without Cl−. For low impedance values, subtraction of the solution resistance (RS value measured at high frequency) is necessary. The difference in corrosion rate (which is evaluated by the difference RP − RS) is then even more evident. AP without Cl− shows higher corrosion rate than conventional AP for the Mg–Y–RE alloy. This indicates that the presence of chloride in the solution has a beneficial effect on the overall surface stability inducing better corrosion product stability. This observation clearly demonstrates that it is not possible to think in terms of oxide breakdown in this type of corrosion processes. Then, after a longer immersion time, a fast electrochemical reaction is observed in the high frequency domain of the spectrum in Fig. 7(b) for the alloy immersion in chloride containing AP. The negative effect of chloride is now visible in terms of the initiation of a fast localized attack as soon as the surface stability is increasing (visible as well in Fig. 3).
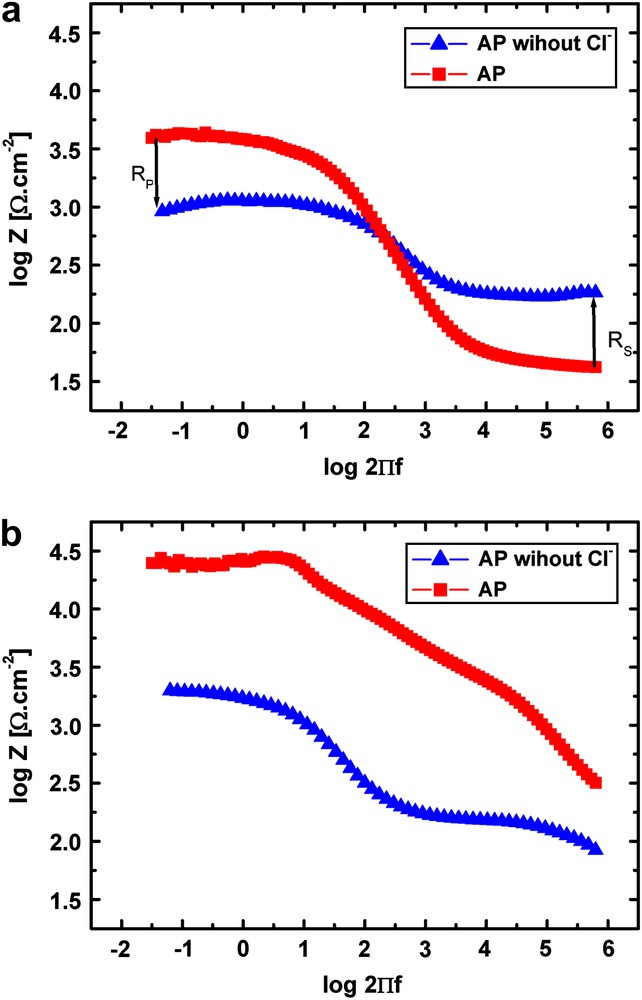
Bode plot of the impedance modulus diagram of an untreated surface after (a) immediate and (b) 24 h immersion in AP and AP without (Cl−) at 37 °C.
In summary, the stability of the corrosion products in body fluids depends also on the ions present in the solution and their integration in the corrosion products. This observation is typical for hindered active corrosion processes with the formation of stable reaction products on the surface and localized corrosion susceptibility in the case of metals like Cu, Zn or Ni. In the case of these metals, the corrosion susceptibility is influenced by the ion diffusions and the solubility of dissolution products in solution. Chloride alone in the solution will not stabilize the corrosion product layer [32], but in combination with bicarbonate and/or phosphate, it has a beneficial effect for uniform corrosion protection of Mg surfaces.
3.2 Protection by anodic surface oxides
3.2.1 Galvanostatic electrochemical anodising
To obtain an initial delay in the corrosion processes, a temporary protecting surface layer is necessary on the implant surface. An initial corrosion rate in physiological solution still too fast has been mentioned previously. Electrochemical anodising in high pH solution is one of the ways to grow “thick” oxides and has been envisaged for the initial protection of the alloy surface. Fig. 8 shows a typical potential evolution curve during galvanostatic anodising of Mg–Y–RE alloy in pH 13, 0.05 M Na3PO4 solution. For an applied current density of 20 mA/cm2 in the galvanostatic mode, the potential increases very quickly to reach the 200 V value that represents the limit set to the Keithley current source. This behaviour for the potential evolution indicates the formation of a dense protecting oxide layer. As indicated previously, phosphate ions are necessary to suppress the semiconducting nature of the Mg hydroxide and to produce a stable surface layer where no water dissociation reaction can occur. This mode of anodising allows controlling accurately the film thickness which is around 350–400 nm in this case. When the limiting voltage of 200 V in our case is reached, then the current drops automatically because the film formation also stops.
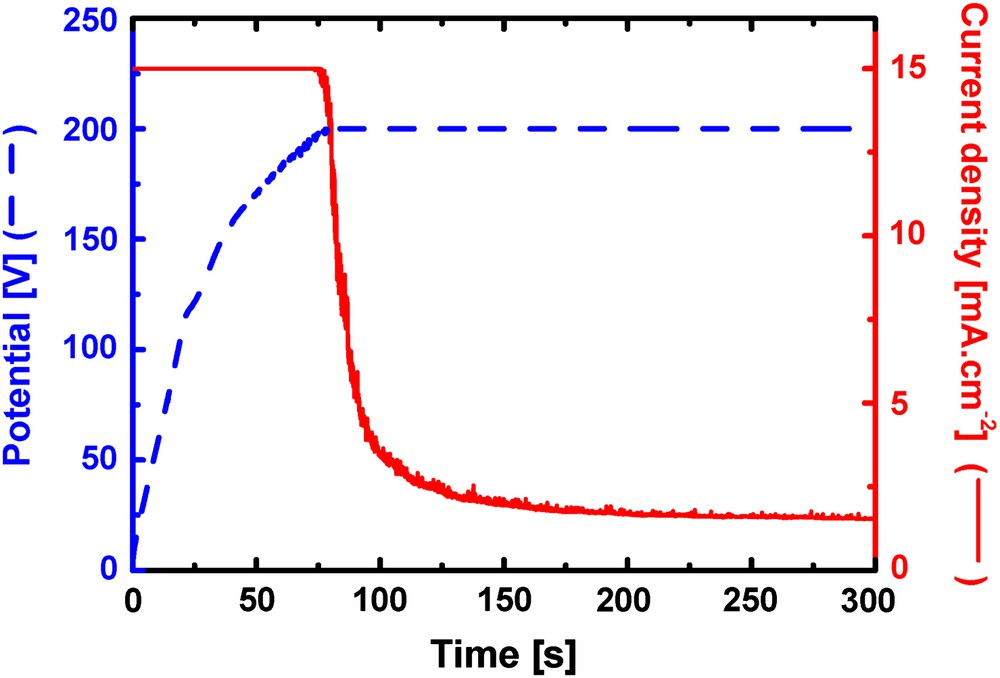
Potential evolution during galvanostatic anodising of an Mg–Y–RE alloy in pH 13 phosphate solution.
3.2.2 Characterization of the (hydr)oxide surface
From the potential evolution during anodising, it can be assumed that a “thick” and dense oxide is produced. Auger electron spectroscopy (AES) was used to obtain additional information on surface composition and, together with sputter depth profiling, on the in-depth composition and thickness of the (hydr)oxide layer grown by the anodising process. A depth profile was performed on a sample previously anodised for 5 min (potential reached 200 V) at 20 mA/cm2 current density, and is presented in Fig. 9(a). It is possible to use the O KLL transition to characterize the oxidation process. The thickness of the hydroxide film was determined from the depth corresponding to the half-maximum of the O KLL intensity. This profile shows that the (hydr)oxide film on an as anodised sample was relatively thick, and estimated from the sputtering time (removal rate of 20 nm/min) to be approximately 350–400 nm for the (hydr)-oxide layer. Focused ion beam sectioning confirmed the thickness determined by AES [26]. The (hydr)oxide contains mainly magnesium and oxygen. At the (hydr)oxide metal interface, a slight yttrium enrichment is observed which maybe related to the faster oxidation of Mg. In the substrate, only magnesium is detected; this observation is expected due to the very small amount of yttrium (4%) present in the alloy and mainly present in the intermetallic phases. A further observation when considering in detail the Mg and O spectra as a function of depth is that the anodised (hydr)oxide can be divided into two sublayers: a very thin surface layer and a much thicker inner layer. Fig. 9(b) shows the AES spectra measured at different depths. The spectra show a clear shift between the two different oxidized species of Mg in the (hydr)oxide layer. A peak is visible at an energy of 1178 eV for the outer layer and 1170 eV for the inner layer. A comparable shift is observed for the O peaks. The kinetic energy of the oxidized species of Mg and oxygen for the different magnesium oxide and hydroxide is referenced in Table 2, and allows us to also have some additional information on the type of oxide structure formed in the anodic layer consisting mainly of Mg(OH)2 with a native and thin outer layer of MgO. A more detailed analysis of the surface (hydr-)oxide formed on Mg alloys including comparison of AES and XPS data can be found in detail in an another study [34].
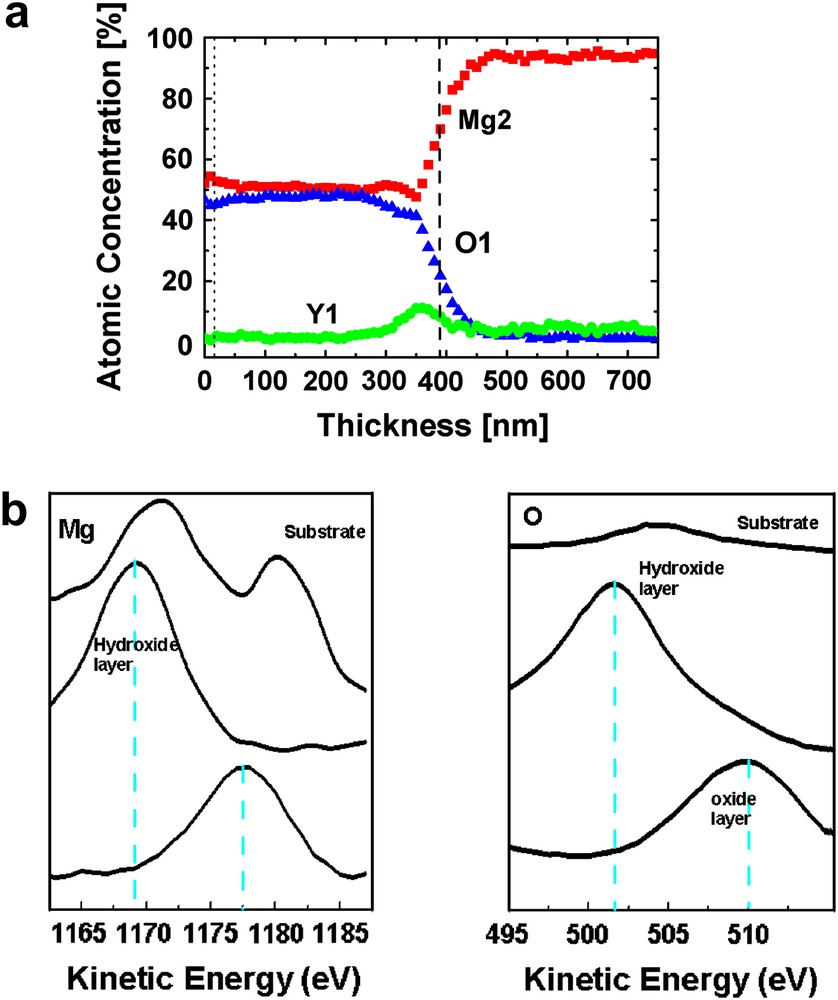
(a) AES composition depth profiles of Mg–Y–RE alloy after the galvanostatic polarization. (b) AES spectra (magnesium, oxygen) as a function of sputtered depth in the oxide and alloy substrate.
Specific kinetic energy of Mg and O peaks for Mg(OH)2 and MgO
| E (eV) | Mg | O |
| Mg(OH)2 | 1170–1172 | 502–504 |
| MgO | 1176–1178 | 508–510 |
3.2.3 Surface film corrosion resistance during immersion in AP and SBF
Even in the presence of a 400 nm thick and homogeneous hydroxide, localized “filiform like” corrosion occurs during the immersion in AP solution, as can be observed in Fig. 10(a). This type of corrosion attack seems to be the result of a combination of the better corrosion resistance obtained but with poor mechanical properties of the hydroxide film. It is not the classical undermining of an organic coating, but an open to solution attack which then propagates laterally like the classical filiform attack. A similar type of “filiform” corrosive attack under a hydroxide film has already been evidenced on pure Mg immersed in NaCl solutions containing dichromate [35]. The first step of the corrosion propagation is a local breakdown of the (hydr)oxide which induces an easier solution access to the filament head, and then allows further lateral propagation of the corrosion. Although in-depth growth of this type of attack is slow because of the local alkalizing occurring, it can be very detrimental to the protection of the surface, because considerable damage is produced, followed by new attacks so that the final result is a uniformly corroded surface. It is necessary to modify the anodising bath by adding carbonate in the solution to obtain a coating that will not break open after the corrosion initiation as it is shown in Fig. 10(b). In this second case, corrosion attack does not propagate laterally after initiation. This difference in the filiform propagation is probably due to the presence of a mechanically stronger film combined with high pH conditions which hindered the propagation of the attack under the anodised layer.

Optical micrograph of the attack immersed in AP at 37 °C for anodised (hydr)oxide: (a) phosphate anodising; (b) mixed phosphate–carbonate anodising.
The low frequency values (f = 0.01 Hz) of the impedance obtained after different immersion times in AP at 37 °C for an untreated and an anodised surface are shown in Fig. 11. It is clear that the oxi-hydroxide has better corrosion resistance properties, evidenced by the increasing impedance value obtained as a function of immersion time in AP. But, as shown before in Fig. 10(a), a local “filiform corrosion” attack is observed for this phosphate coating after 24 h of immersion. Localized corrosion can be detrimental on the long term for the general corrosion resistance properties. The high impedance values measured during immersion do not always reflect correctly this fact especially if the size and amount of the localized attack stay small. The homogeneity of the dissolution is a very important point when optimized biodegradable surfaces have to be developed. EIS curves of the anodised (hydr)oxide after immediate immersion and 24 h of immersion are shown in Fig. 12. After immediate immersion of the anodised sample, only one time constant (one peak in the phase shift curve) is observed. But after few hours of immersion in AP, a second peak in the phase shift curves appears corresponding to a fast localized reaction visible in the high frequency domain of the spectrum. From this time, two distinct electrochemical processes are proceeding on the surface, whereas only one process is detected at the beginning of the immersion. The second peak corresponds to the deep localized attacked corrosion observed in Fig. 10(a). An important comment related to data interpretation in the case of uniform and localized corrosion occurring simultaneously and in parallel is that the impedance modulus at low frequency is a convolution of the impedance of the two processes. In the processes presented in this paper, the impedance value measured at low frequency is the sum of the contribution of the uniform corrosion resistance improvement of the (hydr)oxide and the localized corrosion attacks which decrease the impedance values. This behaviour is typical for a defective oxide or coating [36] where the intrinsic corrosion resistance of the oxide is always higher, but the lower impedance value measured is due to the additional contribution of the defect of the oxide or coating. To assess intrinsic corrosion resistance of the anodised layer that is certainly exceeding the MΩ/cm2 range in this case, it would be necessary to either perform local impedance spectroscopy measurements or to characterize very precisely the effective surface of the localized attack. These characterizations are certainly a next step in the current investigation, but not a high priority in this first phase of development of protecting oxide.
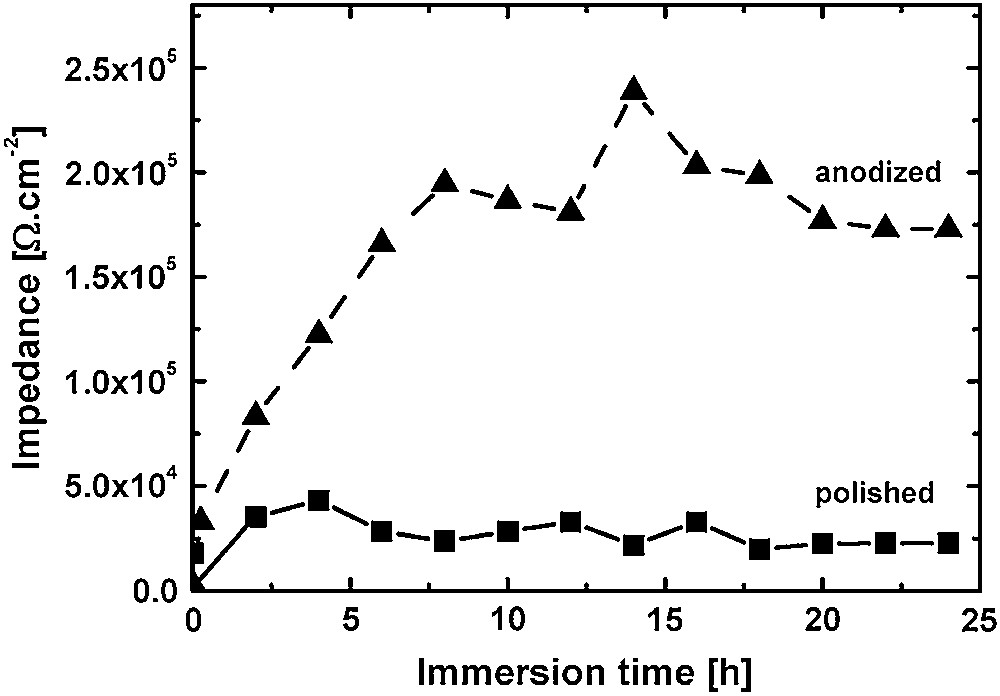
Impedance values obtained at f = 0.01 Hz for an anodised and an untreated Mg–Y–RE surface as a function of immersion time in AP at 37 °C.

Impedance (square) and phase shift (triangle) curves of anodised layer after immediate and 24 h immersion in AP at 37 °C.
3.2.4 Influence on the corrosion processes of the carbonate content of the physiology solutions
The low frequency (f = 0.01 Hz) impedance values obtained after different immersion times in AP and AP low (HCO3−) at 37 °C for an anodised surface are shown in Fig. 13. It is clear that the HCO3− ion content in the solution plays an important role in the uniform corrosion resistance of the surface. As described previously for an untreated surface, the presence of a high amount of carbonate ions allows one to decrease the initial dissolution rate, but do not induce the formation of a protecting (hydr)oxide (Fig. 6). The beneficial protecting effect of the carbonate ions is here more visible in the case of localised corrosion attack of an already formed anodised oxide. In the presence of low amount of carbonate in AP, the corrosion resistance of an anodised oxide is not better than that of an untreated sample, because localized attack can initiate and proceed at a very high dissolution rate. When higher amounts of carbonate ions are present, they react locally to slow down the corrosion processes, as observed in Fig. 6 on the untreated surfaces. The most obvious interpretation is that the high carbonate amount produces more stable corrosion products into the pits as is shown in Fig. 14. In the case of already anodised surfaces, this effect is combined with the already existing protecting oxi-hydroxide. When only part of the surface needs to be protected by the carbonate, the fact that a uniform protection could not be obtained for the untreated surface is less a problem on anodised surfaces.

Impedance values obtained at f = 0.01 Hz for a phosphate anodised surface as a function of immersion time in AP and AP with low [HCO3−] at 37 °C.
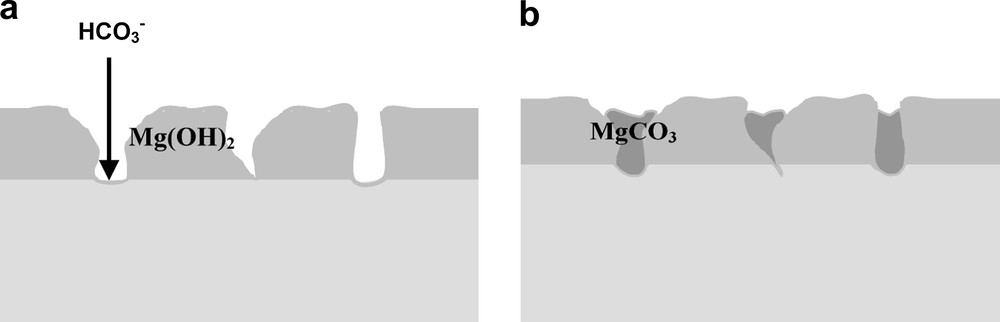
Schematic description of corrosion process on anodised oxides: (a) at short immersion times; (b) at longer immersion times.
4 Conclusions
The investigations presented in this paper allow the description of the concept of the positive use of corrosion processes to obtain tailored biodegradable metallic implants. The electrochemical data are only treated qualitatively because it is first necessary to identify the corrosion mechanism of this very complex degradation process with temporary surface protection. Some of the key issues related to the dissolution of this type of metallic implants are discussed and can be summarized in the following way:
- - A main problem related to the use of Mg alloys in physiological solution is the very large difference in the corrosion rate (a factor of 20–30) and mechanisms obtained between AP and SBF used for in vitro characterization experiments.
- - The exact prediction of the implant life is relatively difficult and the correlation with in vivo results is still investigated. The media used in this study should simulate the different environments that an implant can be in contact with different medical application fields. Artificial plasma (AP) is related to blood contact and simulated body fluids (SBF), for implants where blood contact is less probable.
- - A better uniform corrosion is furthermore often related to higher localized corrosion susceptibility. This can be monitored and investigated by electrochemical impedance spectroscopy (EIS). The Bode plot representation allows identifying the presence of the high frequency electrochemical process only present in the case of fast localized attack and the slower uniform dissolution processes measured at low frequency.
- - A second aspect related to biodegradable implants is clearly to avoid corrosion in a first period in order to guarantee the mechanical integrity and the functionality of the implants. This is difficult to achieve with a bare alloy surface and electrochemical anodising has been used to grow an approximately 350–400 nm phosphate–carbonate containing Mg hydroxide.
- - An initial corrosion protection is obtained, but localized attack cannot be hindered. The susceptibility and type of attack are, however, strongly reduced with the phosphate/carbonate combination in the high pH anodising bath compared to the use of pure phosphate. On the long term, localized attacks are necessary in order to have degradation of the implants!