

 CC-BY 4.0
CC-BY 4.0
1. Introduction
The use and performance of chromatographic separation in today’s laboratories and industry is greatly impeded by the pressure drop required by available particulates column packings. Chromatography today imposes to force flows through beds of extremely small particles from 50 down to 1.7 μm diameter with extremely high resistance to the percolation of a fluid. This induces high pressure drops of several hundreds of bars [1].
In the analytical field, ultrahigh-performance liquid chromatography (UHPLC) technology is ultimately limited by the extremely high pressure drop of the ultrafine powders used as separating media.
The packing uniformity is of relevance to the chromatographic efficiency. The non-uniformities of the bed will cause irregular flow patterns leading to losses in efficiency. Particulate beds are difficult to stack uniformly [2] due to their size and ultrafine nature and the wall effects of the container, and they suffer from a lack of long-term stability due to their slow deformations when subjected to flow, pressure and cycles. In industrial liquid chromatography (LC), stabilizing and containing large column beds involve levels of complexity and high costs that prohibit the use of this powerful separation method in most chemical processes.
Attempts to solve this problem have been directed toward the use of monolithic packings, manufactured both in polymers [3] and in silica gel [4]. Extrapolation to large sizes has proven a difficult problem for both organic and inorganic monoliths.
The permeability of capillary tubes is much higher. In the field of gas chromatography (GC) the empty monocapillary Golay column [5], 50 to 500 μm diameter, has gained large acceptance. Its low pressure drop allows to make it exceptionally long, and to develop high number of plates or efficiency. This technique has not been used in LC because of the small channels diameters that would be required, 1 to 10 μm. This difference, consequence of the much lower molecular diffusion coefficient in liquids, makes the corresponding LC system unpractical.
Another route has been to use packings composed of a multiplicity of capillary tubes working in parallel [6, 7, 8, 9, 10, 11]. The stationary phase is constituted by or covers the wall material and exchanges mass by diffusion with the fluid in the channel core. The fluid flow path becomes perfectly linear with no obstacles. This geometry could combine the advantage of the low pressure drop of a capillary, and the throughput of existing columns. It could thus be compatible with existing LC instruments.
Multicapillary packings are monolithic in nature and are potentially much easier to extrapolate due to their manufacturing processes.
Unfortunately, the small differences in dimensions between the individual capillaries, the difficulty of coating them evenly with the stationary phase and the differences in their individual aging negatively affect their short- and long-term efficiency and has made this solution very limited in practice [12, 13].
Colocated monolith microstructures with highly interconnected capillary channels have been described and developed for analytical purposes [11].
The field of multicapillary chromatography is nevertheless currently attracting growing interest.
This interest stems from the fact that this approach offers an attractive solution to the pressure drop problem.
Schisla et al. [12], Sidelnikov [13, 14] and Jespers [15] analysed the behaviour of a bunch of individual capillaries.
It has been derived theoretically by Giddings [16] that the band broadening due to mobile phase flow can be affected by the coupling between diffusional transverse flow between fluid elements and the dispersion caused by independent fluid element flowing at different velocities (or so called eddy diffusion).
Their importance has been evaluated [17] for particulate packings. This effect has been studied by several researchers [18, 19, 20, 21, 22].
The simulation of radial diffusional effects in chromatographic columns has been undertaken. The focus of previous studies has been mainly on considering axial diffusion, in one-dimensional column models, because of its direct effect on peak broadening [23, 24, 25, 26, 27]. Some studies [28, 29, 30] examined the effect of a radial transfer phenomenon, heat transfer, on the efficiency of UHPLC columns. Studies did explore by simulation or random walk consideration the effect of radial diffusion through packed beds [18, 19, 20, 31]. Giddings considers that the exchange of mass between different convective flow path can occur by mixing (leading to a partial HETP hf) and by diffusional exchange (leading to a partial HETP hD), both phenomena being coupled. The coupling of both effects must lead to an efficiency better than the one of each of the phenomenon taken separately.
The present multicapillary columns can be considered as limited to the mixing step only. It can thus be expected that the addition of a diffusional exchange between the independant fluid flow paths constituted by the channels core will improve the behaviour of those systems. To the best of our knowledge, the application of this coupling on the theory of multicapillary chromatography and as a way of improvement of this technology has nevertheless never been identified and quantified.
The aim of this study is to estimate the effect of a diffusional flow, passing through bridges of pores between channels causing the eluting bands in the fast or slow channels to discharge by molecular diffusion in channels flowing with an average velocity.
To quantify and check this effect, we thus conducted a short theoretical derivation based on Giddings Random Walk theory. As the quantitative predictions of this coupling were considered by Giddings himself as estimates within an order of magnitude, we verified the results by two independent approaches, by transfer function analysis of the coupled system, and by computer dynamic simulations of a multicapillary array based on the method of lines. The simulations of the residence time distribution (RTD) of a capillary array were performed, recorded, and analyzed, in presence or absence of an interchannel diffusive effect, or diffusional bridging. This first work will focus mainly on the zero-retention case, as the case with solute retention appears to be still the object of discussions [16, 18, 19, 20] and as it describes the case where the resistance to mass transfer of the stationary phase layer is negligible because its thickness is low, its mass diffusivity high, or the solute retention factor is high.
The presented work will be divided in the following sections:
- 1. We will recall some basic findings regarding the pressure drop gain that can be expected using capillary packings as chromatographic beds instead of particulate packings or other existing technologies.
- 2. We will present our results derived from Giddings theories applied to chromatography in multicapillary arrays. The effect of diffusional bridging will be estimated and its effect compared with the performance of present state of the art multicapillary chromatography.
- 3. We will analyze the transfer function of a coupled system constituted of two coupled capillaries.
- 4. We will present our results obtained by the numerical simulation of multicapillary arrays by an ordinary differential equation (ODE) integration and compare them with the theoretical predictions.
- 5. We will conclude on the potential of this new approach for analytical and preparative chromatography.
2. Theoretical results
2.1. Fundamental equations
We adopted for this study the Golay equation [5] written:
| (1) |
Let us recall the expression of the Knox Van Deemter equation:
| (2) |
| (3) |
2.2. Geometry simulated
Detailed morphological structure of the multicapillary packing.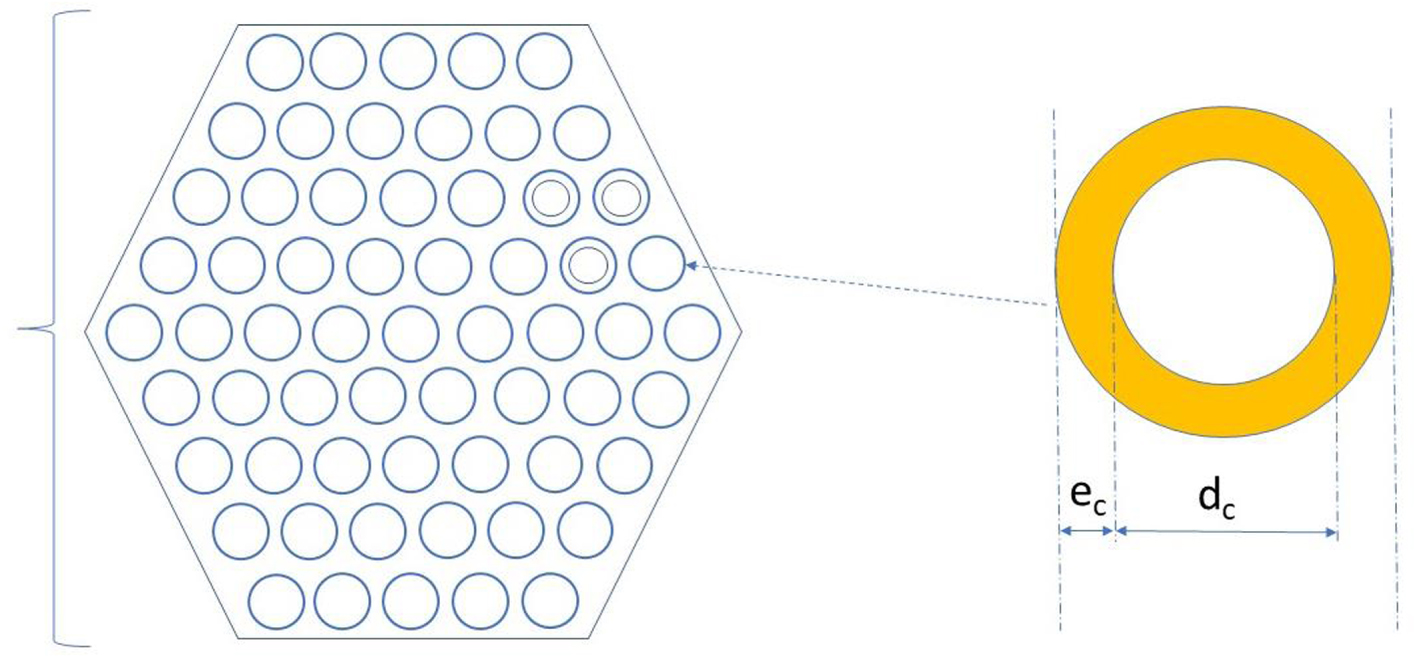
Figure 1 shows the geometry simulated: the packing is composed of a porous mass containing the stationary phase or being constituted by it. This porous mass can be coated with a stationary liquid or gel of thickness ec, or constituted by a mesoporous, high specific surface solid like silica gel or a polymeric, cross linked, gel.
Straight empty channels of diameter dc run through this mass and conduct the convective flow of mobile phase through the packing. The porous mass allows diffusion of sample molecules from each convective channel to its neighbors.
2.3. Pressure drop comparison between capillary and particulate packings
The comparison of pressure drop in capillaries and particulates beds can be established based on Darcy’s law:
| (4) |
For present chromatographic beds made by stacking spherical beads, the permeability coefficient K is comprised between 500 to 800 [1]. For empty capillaries, we derived from Poiseuille’s law:
| (5) |
We found that the ratio Rp of the pressure drops through particulate beds and multicapillary packings at similar diffusional path length, particle diameter and channel diameter is simply given by the ratio of their permeability coefficients and can be expressed as follows:
| (6) |
Within the validity of those assumptions, at equivalent efficiency, the operating pressure of the multicapillary system is from 10 to 30 times lower than that of the particulate packing system depending on the thickness of the stationary phase.
At equivalent HETPs, identical stationary phase, identical void fraction and identical velocities the operating pressure of the multicapillary system is 15 times lower than that of the particulate packing system ΔPp, more than one order of magnitude (ΔPp∕ΔPc = 14.6).
The multicapillary monoliths offers an additional advantage over beds of fully porous particles: the void fraction of the monoliths can be changed by an adjustment of the thickness of the porous layer surrounding the channel. This may give them a kinetic behavior like that of core shell microspheres, or oppositely allow them to carry higher loads of molecules to purify.
The core shell packings have the advantage over fully porous particles of a better kinetic behavior, due to the shortened diffusional path in the outer particle layer and better packing density [34]. With multicapillary packings, the kinetic behavior can be freely improved by making the stationary phase layer coating the tubes as thin as necessary, keeping the advantage of a one order of magnitude gain in pressure drop at equivalent efficiency or speed.
New ordered packings like micropillar arrays [11, 15] have been found efficient but lack in several respects the benefits of radial diffusional dampening, and are subject to wall effects. Micropillar arrays are today limited in resolution to micrometric structures [11] and need specific, microflow equipment. Multicapillary arrays can be manufactured with submicrometric channels with simple processes [35, 36, 37, 38], down to 0.2 μm diameter channels or less. This allows theoretically unsurpassed performances on achievable number of plates and analysis velocity with existing equipment.
2.4. Case of independent channels
As stated previously, the theoretical performance of multicapillary packing has been up to now limited by the difference in individual behavior of the distinct capillaries.
The state of art of multicapillary packings are constituted of a bunch of independent capillaries.
Figure 2 presents a scheme of their behavior. Independent channels behave as independent chromatographic columns with unequal output signals due to differences in channels diameters, stationary phase loading, length, and aging. This unevenness can be considered by an additional variance of the output signal.
Hydrodynamic behavior of an array of independant parallel capillary columns. The different residence times cause a dispersion of the mixed signal at the column outlet.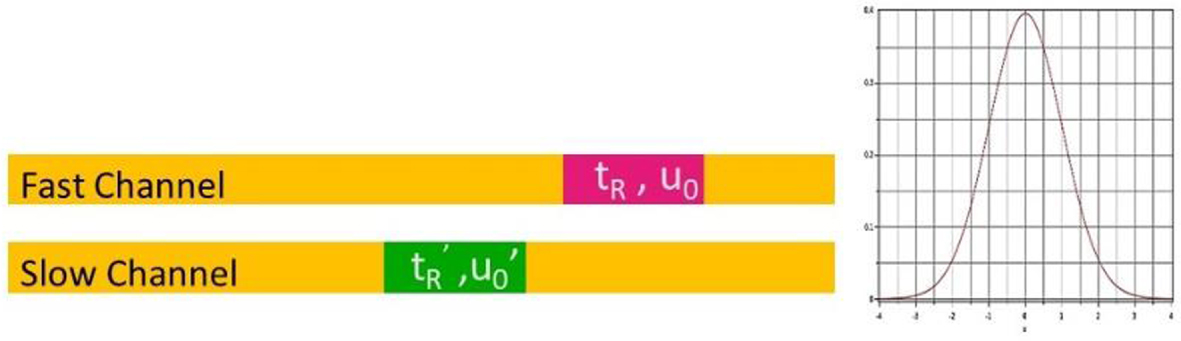
Among those sources of variances, the most sensible is the channel diameter, as the velocity of the mobile phase vary with its square, and the flow rate vary with its fourth power.
Shisla et al. [12] gave an analysis of the chromatographic behavior of a multiplicity of independent channels. This study is based on the Golay equation and considers a normalized distribution of several parallel capillaries of radii distributed according to a function g(R) and of equal lengths.
Writing the Golay equation in partial contributions with a polydispersity term gives:
| (7) |
Shisla equation can be rewritten in terms of partial height contribution , as usual:
Sidelnikov [13] studied the case of a capillary array with a random distribution of diameters. The capillaries are supposed to have a diameter constant along their length, and the diameters are distributed according to a Normal Law of standard deviation . Sidelnikov proposes the following law:
| (8) |
Consequently, the number of equivalent theoretical plates (NETP) increases with length from L∕Hc for short lengths up to a limiting value NETPlimiting that cannot be exceeded.
H is the HETP of the array, Hc is the HETP of the single average capillary, the third term is a HETP Hd attributable to the diameter dispersion of the channels
| (9) |
2.5. Theory of diffusional bridging effect
If the capillaries are not independent but instead communicate by diffusion, the behavior becomes different.
Figure 3 gives a scheme of this disposition. Molecular diffusion is allowed between channels through a porous wall supporting the stationary phase.
Effect of diffusive bridging on an array of parallel capillary columns. The coupling levels the individual differences.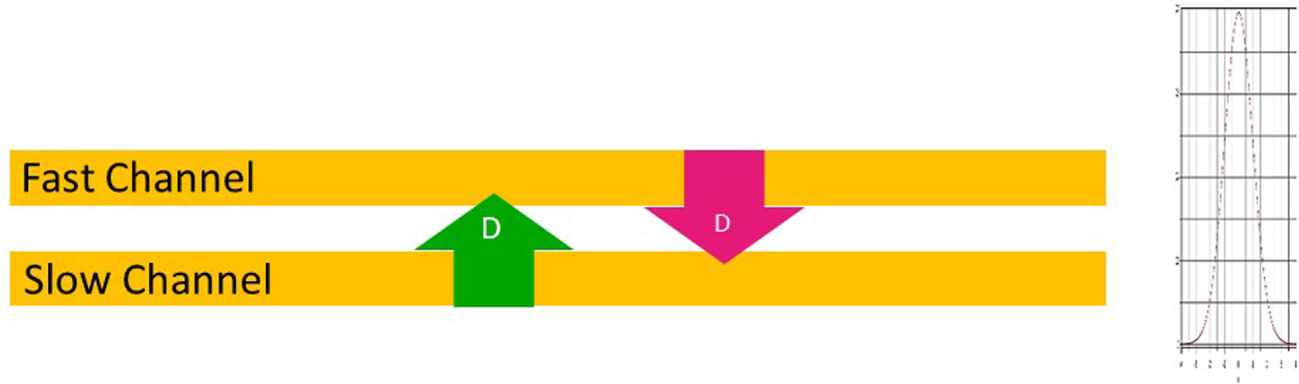
2.5.1. Previous theoretical works: the Giddings random walk considerations
The phenomenon considered in this theoretical work can be considered as a coupling between molecular diffusivity and the diffusion caused by mobile phase velocity inequalities, or eddy diffusivity. Giddings [16] presents in his book theoretical guidelines to consider this aspect of band spreading in particulate beds. Giddings considers that the exchange of mass between different convective flow paths can occur by mixing (leading to a partial HETP ) and by diffusional exchange (leading to a partial HETP hD), both phenomena being coupled. Giddings formulates his reasoning by analyzing the path of a diffusion like a random walker in a column.
The reasoning of Giddings is based on three equations (see also Khirevitch, Tallarek et al. [20]):
- 1. Definition of plate height as the ratio of the variance of the analyte zone to the distance traveled by the center of the band:
(10) - 2. Einstein’s mean square displacement formula
which can be expressed as a characteristic time associated with a characteristic diffusion length l:(11) - 3. The relation for the variance of the displacement of a random walker:
Here, is the column length, l is the average length of the random walker step and n is the number of steps.(12)
Giddings quantifies the effect through the use of three parameters, 𝜔𝛼 is the distance between extreme flow velocities in the packing measured in particles diameters dp units; 𝜔𝛽 is the ratio between one flow extreme and the average flow; 𝜔𝜆 is the persistence-of-velocity distance between which two independent flow paths persist before remixing, measured in dp units.
This derivation gives for the partial HETP term due to this coupling:
| (13) |
Diffusive exchange leads to the plate height expression:
| (14) |
| (15) |
- 1. The transchannel contribution. In the case of Golay columns, this corresponds to the dispersion due to the laminar parabolic velocity profile, or Taylor Aris term.
- 2. The transparticle effect. In the case of a multicapillary packing, this corresponds to a transchannel effect. The source of zone spreading are the inhomogeneities between the individual channels, arising from differences in diameters, in stationary phase loading, in length, and in aging.
- 3. The short range interchannel effect. We will suppose that the capillaries are regularly distributed with only purely random variations in diameter and stationary phase thickness. This effect disappears in this case.
- 4. The long range interchannel effect. Multicapillary structures are monolithic in nature. They are not subject to packing procedures. As such this effect will be supposed to have no validity.
- 5. The transcolumn effect. Multicapillary structures present no defect near column walls, as they have their own fluidic characteristics with their own stationary phase layer. As such this effect will be supposed to have no validity.
Other variables can be possibly affected of a random variation:
- the channels’ averaged ratios of eluent to stationary phase,
- the length of the channels
- the differences in curvatures between channels,
- the channels’ internal surface imperfection or asperities
- other mechanical inhomogeneities or imperfections of the stationary phase.
Those different factors can themselves vary locally along the channel length. We have restricted this first study to the transchannel effect linked to random variations of channels’ diameter at constant retention factor.
𝜔𝛼 is the distance between flow extremes between which molecular diffusion occurs, leading to the dispersion phenomena, measured in this case in dc units. In our case we will assume it equal to 1, distance separating two adjacent channels.
𝜔𝛽 is the ratio in velocity between the extreme and average flow measured in vc units. The velocity in a channel is proportional to the square of its diameter. Neglecting second order terms, we will take 𝜔𝛽 equal to two times the diameter relative standard deviation 2 ∗𝜎rel.
𝜔𝜆 is the persistence-of-velocity distance between which two independent flow paths persist before remixing, measured in dc units. In our case the persistence distance is simply the reduced column length l, so 𝜔𝜆 is equal to l∕dc.
Those values must be taken as simple starting hypothesis, as the underlying phenomenon is in fact constituted of a much more complex arrangement of randomly disposed channels. A complete establishment of their probabilistic relevance has not been done. The problem of a grid of channels having random diameters exchanging mass by diffusion with six neighbors is of a much higher mathematical complexity and a control of the result is not obvious. We have found more complementary to conduct first virtual experiments by computer simulation. In addition, the transfer function of a twin channel has been derived analytically and used to validate the general physical findings and the simulation results.
With those omega values hD and hf are written:
| (16) |
| (17) |
Reintegrating those values in (13), one obtains:
| (18) |
2.5.2. Assessment of packing performance with a stationary phase layer
The influence of the mass transfer in the stationary phase and its coupling with the Giddings eddy-mass transfer theory has been examined by various authors [16, 17, 18, 19, 20].
According to Giddings [16], the eddy dispersion is only slightly influenced by retention factors.
Writing in reduced terms:
| (19) |
| (20) |
| (21) |
2.5.3. Final equation
We suggest writing the modified Golay equation with a term :
| (22) |
3. Transfer function analysis
3.1. Determination of the transfer function
Since the pioneering work of Martin and Synge [39], the transfer function of chromatographic systems has been examined by various authors, Lapidus and Amundson [40], VanDeemter, Zuiderweg and Klinkenberg [41], and others.
The basic method consists in writing the mass balances characteristic of the system in the time domain and transfer them in the Laplace domain to find the expression of the transfer function.
This last expression is inversed in the time domain whenever possible, or its moments are directly computed from the Van Der Laan relationship:
| (23) |
Analytical solutions of the transfer function is in practice possible only for systems limited to two or three differential equations.
We will consider and limit the problem to two adjacent channel exchanging mass by diffusion with an exchange coefficient k12 through a fraction f of their circumference a12. This geometry is represented on Figure 4. The axial diffusion Dm is not considered in the analysis. This limits the ODE order to two and has no impact on the final results as the different contributions are additive in the limit of a high number of plates.
Mass Exchange scheme of the transfer function: twin tubes exchanging mass through an arbitrary fraction f of their circumference.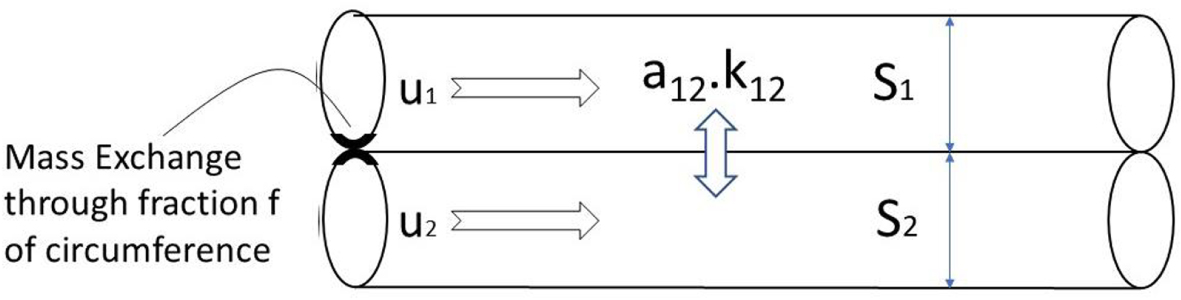
The differential equations to resolve are written:
| (24) |
| (25) |
In the Laplace domain, Equations (24) and (25) become:
| (26) |
| (27) |
| (28) |
| (29) |
| (30) |
| (31) |
The determinant of (30) is:
| (32) |
The roots are written:
| (33) |
We obtain for the transfer function of a column with a length L:
| (34) |
| (35) |
| (36) |
| (37) |
| (38) |
| (39) |
| (40) |
| (41) |
| (42) |
| (43) |
| (44) |
| (45) |
3.2. Determination of the transfer function
in (42) depends on the sum of the mass transfer resistance of the mobile phase in tubes 1 and 2, and of the mass transfer resistance of an eventual stationary phase in between. k12 will approximate the resistance of the mobile phase when the mass transfer resistance of a stationary phase can be neglected. This means that in most practical cases of retention factors, with significant surface diffusion on the stationary phase, liquid–liquid partition chromatography, or small ec∕dc the formula (42) must give a good approximation of the losses due to channel dispersion. This agrees with Gidding’s guesses.
Quantitatively, this can be shortly estimated by the comparison of the transfer coefficient km in the flowing mobile phase of diameter dc and the transfer coefficient ks in the stationary phase layer of thickness ec considering the partition coefficient Ks.
| (46) |
| (47) |
It follows:
| (48) |
| (49) |
It must be underlined that this constitutes an overestimate, as the transfer phenomena to the stationary phase occurs partly in an unsteady state manner. This will be the object of further investigations.
4. Modeling
4.1. Simulation methods and starting hypothesis
The system to model is an infinite array of parallel chromatographic capillary columns with randomly variable diameters.
An abundant literature describes the computation and simulation of chromatographic performance of beds of particles based on either purely mathematical or computational calculations [23, 24, 25, 26, 27, 42]. Computational models are based on different classical schemes, generally propagation through a grid, or an integrated set of differential equations. In this last category of computational models are general rate models, lumped pore diffusion models, and equilibrium dispersive and transport dispersive models. The present modeling approach differs from previous attempts in that it adopts as its starting point a discretized model based on the method of lines [43]. The method of lines consists in writing locally on a grid the partial differential equations to be integrated, separating the space terms and the time dependent terms. The space terms are calculated with the differential terms approximated by algebraic equation linearly interpolated from the grid values, and the integration in time is conducted by an ODE solver.
This allows to rely on a purely physical integration of first principles of chemical engineering, describing basic physical laws, diffusion, fluid dynamics and thermodynamics at a very small scale where they can be assumed linear, in an explicit way easier to understand, correct, and debug. This needs minimal hypotheses. It relies on the discretization to build the integrated result taking account naturally the various effects like Taylor Aris dispersion, dominant mass transfer phenomena, and purely diffusional effects. The result can allow quantitative and precise results with the lowest possible risk due to erroneous preliminaries.
Our aim will be to check the diffusional bridging phenomena limited to the resistance contribution of the channel’s cores. Two resistances can be expected to limit the mass transfer between adjacent channels, the mobile phase flowing in the core channel and the stationary phase separating them. Previous Sections 2.5.2 and 3 did discuss this limitation. We will focus on the analysis of the effect of the mobile phase resistance, that must be the predominant one for practical cases of retention factors, diffusivities, and stationary phase thicknesses. To study the basis of the effect of radial diffusion on column efficiency, the simplest geometry is in this case sufficient, and we will restrict the analysis to the peak at zero retention (residence time distribution) of a bundle of capillaries without stationary phase accumulation and with channels directly exchanging mass.
4.2. Mass balances and initial and boundary conditions
Channels are considered circular with a hexagonal arrangement. Mass is exchanged through one sixth of their circumference with each of the six adjacent channels. The channel diameter is assumed to be uniform along each capillary, and the individual values vary according to a normal law with a standard deviation 𝜎d around a mean value dc. The array of channels is discretized along its length in N equidistant slices defining the cells (Figure 5).
Axial (a) and radial (b) discretization scheme of the multicapillary array.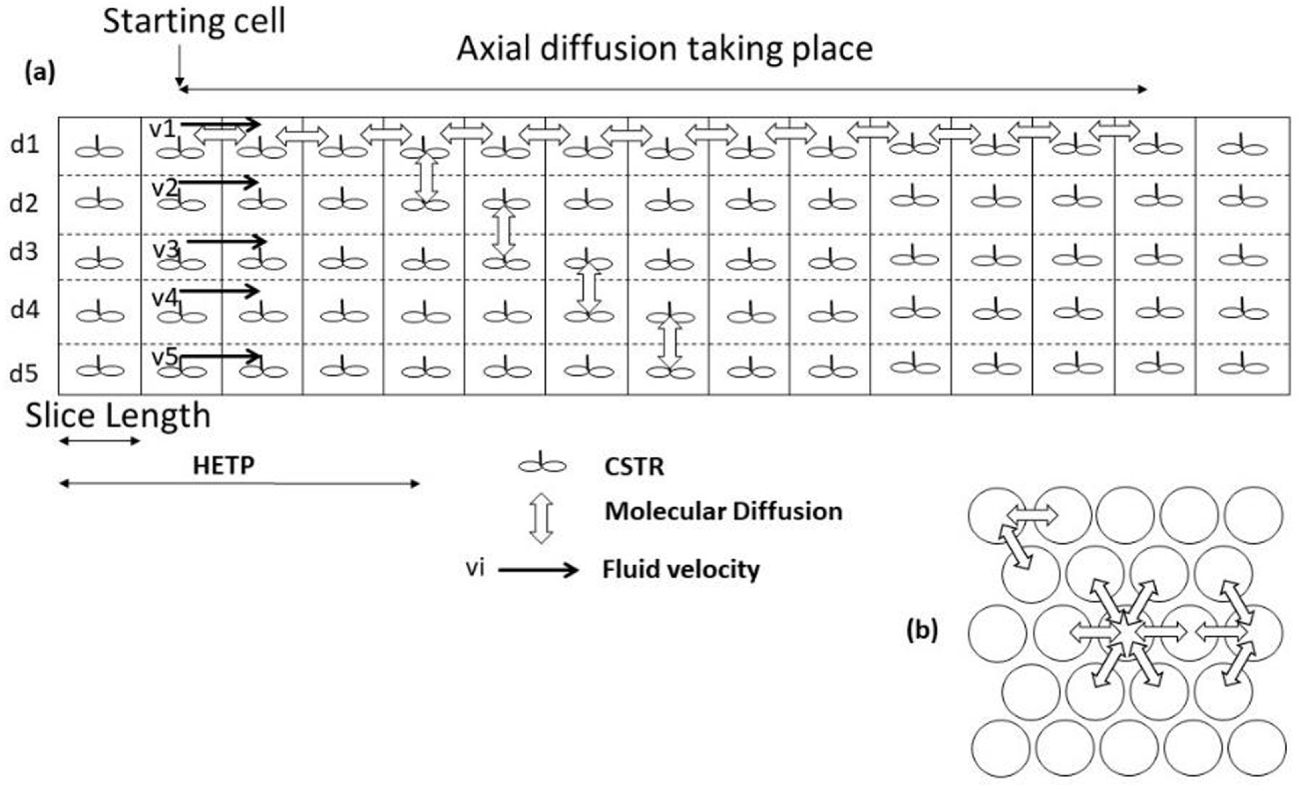
The model is a cascade of continuously stirred tank reactors (CSTRs) (one CSTR of molar accumulation X per cell) (Figure 5 (a)).
The capillary array is arranged in a regular geometry with a hexagonal pattern (Figure 5 (b)). The side effect of a limited size system on corners (two neighbors) and sides (three neighbors) are taken into consideration. The overall simulated system is approximately a square of N × N channels.
N ∗ (ComponentNumber − 1) mass balances (one for each cell) are written in differential form:
Ordinary differential equation set
| (50) |
The initial and boundary conditions are:
| (51) |
is defined by the following equation:
| (52) |
| (53) |
The molar volume of the mixture of solvent and analytes is assumed to be equal to the molar volume of the pure mobile phase, which means that only the case of small molecules (Molar Weight 500 g) are considered in this first study.
4.3. Numerical parameters adjustments
Three main sources of error must be considered.
- The discretization grid
- The time step
- The arrays dimension (number of channels N × N).
The dynamic simulation has been conducted on a meshed model of an array of 41 × 41 capillary columns. This dimension has been chosen after a sensibility study of this parameter. The simulation results are fully stabilized for 41 × 41 grids, with a difference from the asymptotic infinitely wide array of less than 2%.
The elementary cell volume axial thickness is taken to be equal to a fraction of the final HETP (10 to 20%) in order to limit numerical dispersion effects to less than 2% on the final measured asymptotic value. The mobile phase is considered as a molar accumulation in each cell.
The differential equations are integrated with an explicit Runge–Kutta algorithm with a sufficiently small time step to avoid numerical instabilities and inaccuracy, according to the method of lines. In preliminary testing we found the algorithm to be stable if the ratio of the convective flow in one cell over one-time step over cell accumulation is greater than 20, which is like a Courant number. In practice, we used typically time steps of 1 × 10−4 s for simulating 10 μm channels. In this case, the simulation result is overestimated by less than 2% with regard to the limiting case of an infinitely small time step.
4.4. Software testing
Several tests have been done on the final code.
- The unsteady state radial diffusivity from the central channel of the grid has been successfully matched with Fourier heat equation with a difference on the standard deviation of the signal better than 5%.
- The first and second moments of the theoretical transfer function of a single input–single output (SISO) two channels system have been matched with results from a two channels simulation with a difference lower than 4.0%.
4.5. Hardware
The computation was conducted on an Intel Core i7 -6700K with 8 cores and 4 GHz frequency. The average duration of a run was 30 min.
The coding has been realized in object-oriented programming under C++ on Microsoft Visual 2010 platform.
5. Numerical results
5.1. Case of independent channels
The following table (Table 1) gives the evaluation of the number of theoretical plates achievable in the case of independent, unbridged, channels, for random distribution of their individual diameters and for increasing relative standard deviation.
Numerical values of NETPlimiting calculated according to (9)
| RSD =𝜎 r∕r0% | k = 0 | k = 5 |
|---|---|---|
| 0.5 | 10000 | 4981 |
| 1 | 2500 | 1245 |
| 2 | 625 | 311 |
| 5 | 100 | 50 |
| 10 | 25 | 12.5 |
The quantitative results of (8) and (9) are well correlated with the simulation finding reported on Figure 6 as seen from their fit with the theoretical asymptotic values of Table 1.
Simulation of the limitation of NETP due to dispersive phenomena in non diffusive multicapillary array; channel diameter dc, 10 μm; mobile phase velocity v, 940 μm/s.
Theoretical partial height of a multicapillary array with diffudional bridging from Gidding’s RW Theory function of the relative standard deviation of channel diameter
| 𝜎rel (%) | HD (μm) | Hf (μm) | Hdisp (μm) | H (μm) | % Dispersive |
|---|---|---|---|---|---|
| 0 | 0 | 0 | 0 | 3.11 | 0 |
| 1 | 0.0188 | 40.02 | 0.0188 | 3.1288 | 0.601% |
| 2 | 0.0752 | 160.09 | 0.0752 | 3.1852 | 2.36% |
| 5 | 0.47 | 1000.55 | 0.47 | 3.58 | 13.1% |
| 10 | 1.88 | 4002.20 | 1.88 | 4.99 | 37.7% |
| 20 | 7.52 | 16008.81 | 7.52 | 10.63 | 70.8% |
Average channel diameter dc 10 μm; average velocity v 940 μm/s; column length L 100 mm; diffusion coefficient in mobile phase Dm 1 × 10−9 m2/s; 𝛼disp = 1.
A relative standard deviation of 2% can be considered as achievable. It is the standard value of commercial threads of artificial textiles. This maximizes the efficiency at only 625 to 300 plates which is very modest for analytical and preparative purposes, where values from 5000 to 50 000 plates are commonly achieved with present particulate packings. A relative standard deviation of 0.5% is required to reach 5000 theoretical plates, which is the best performance obtained in the monofilament stretching for the optical fiber industry. For a 5 μm channel, this represent 25 nm of average tolerance, in the size range of one nanoparticle. For the smaller channels required to take advantage of the superior characteristics of the multicapillary packings in terms of pressure drop in analytical HPLC, the tolerance would become close to molecular dimensions.
5.2. Effect of diffusional bridging
5.2.1. Theoretical computations from Giddings RW analysis
The Table 2 below reports the values of theoretical plates heights computed by (16) and (17) in the simplest case of virtual channels with separating walls having negligible mass transfer resistance and no retention characteristics.
The main observation is that the loss in efficiency induced by the channels’ diameters dispersion is limited to moderate fractions of the total height for relative square deviation (RSD) lower than 5%.
Correlation of theoretical dispersion height hD ((16) and (17)) of multicapillary packings with diffusional bridging, given by RW Theory with simulated values
| dc (m) | RSD | v (m/s) | Dm (m2/s) | Theor. hd (μm) | Sim. hd (μm) |
|---|---|---|---|---|---|
| 1 × 10−5 | 0.01 | 9.40 × 10−4 | 1 × 10−9 | 0.0188 | 0.0168 |
| 1 × 10−5 | 0.02 | 9.40 × 10−4 | 1 × 10−9 | 0.0752 | 0.0714 |
| 1 × 10−5 | 0.05 | 9.40 × 10−4 | 1 × 10−9 | 0.47 | 0.4636 |
| 1 × 10−5 | 0.1 | 9.40 × 10−4 | 1 × 10−9 | 1.88 | 1.9431 |
| 1 × 10−5 | 0.05 | 4.70 × 10−4 | 1 × 10−9 | 0.235 | 0.1967 |
| 1 × 10−5 | 0.05 | 1.88 × 10−3 | 1 × 10−9 | 0.94 | 0.8997 |
| 5 × 10−6 | 0.1 | 9.40 × 10−4 | 1 × 10−9 | 0.47 | 0.4780 |
| 2 × 10−5 | 0.025 | 9.40 × 10−4 | 1 × 10−9 | 0.47 | 0.4946 |
| 1 × 10−5 | 0.05 | 9.40 × 10−4 | 5 × 10−10 | 0.94 | 0.8997 |
| 1 × 10−5 | 0.05 | 9.40 × 10−4 | 2 × 10−9 | 0.235 | 0.1967 |
5.2.2. Comparison of simulated and random walk (RW) results
Table 3 reports the results of our simulation tools and the corresponding partial height of dispersion computed according to (16) and (17) with 𝛼disp = 1 in different conditions, namely by varying:
- The channel’s diameter RSD at 1, 2, 5, 10%
- The Diffusion coefficient Dm at 0.5 × 10−9, 1.0 × 10−9, 2.0 × 10−9.
- The mobile phase velocity v at 470, 940, 1880 μm/s
- The channel diameter dc at 5, 10, 20 μm.
For the same column length of 1 mm.
Correlation of theoretical dispersion height hD (Equation (44)) from the transfer function of a twin channel geometry with simulated values; Sh = 3.66; f = 1∕6
| dc (m) | RSD | v (m/s) | Dm (m2/s) | Theor. hD (μm) | Sim. hD (μm) |
|---|---|---|---|---|---|
| 0.00001 | 0.01 | 0.00094 | 1 × 10−9 | 0.031 | 0.031 |
| 0.00001 | 0.02 | 0.00094 | 1 × 10−9 | 0.123 | 0.122 |
| 0.00001 | 0.05 | 0.00094 | 1 × 10−9 | 0.770 | 0.747 |
| 0.00001 | 0.05 | 0.00094 | 2 × 10−9 | 0.385 | 0.325 |
| 0.00001 | 0.05 | 0.00047 | 1 × 10−9 | 0.385 | 0.365 |
| 0.00001 | 0.05 | 0.00188 | 1 × 10−9 | 1.541 | 1.59 |
The relation between theoretical and simulated data is linear with a correlation coefficient R2 of 0.9976. The slope shows less than 3% deviation from perfect equality.
5.2.3. Comparison of simulated and transfer function results
Table 4 report the results of the simulation of the twin channel configuration studied in Section 3 and the corresponding partial height of dispersion computed according to (44) in different conditions, namely by varying:
- The channel’s diameter RSD at 1, 2, 5, 10%
- The mobile phase velocity v at 470, 940, 1880 μm/s
- The diffusion coefficient Dm at 1 × 10−9 and 2 × 10−9.
For the same column length of 1 mm.
The relation between theoretical and simulated data is linear with a correlation coefficient R2 of 0.998. The slope shows less than 1% deviation from perfect equality. The simulated twin tube hdisp is in 61% excess over the 41 × 41 channel’s array value, equal to f⋅Sh.
Figure 7 summarizes the fundamental behavior difference between systems with and without diffusional bridging. Diffusional bridging restores the chromatographic functionality of the multicapillary array.
NETP vs length of multicapillary packings with diffusive or non diffusive walls dc = 10 μm; v = 940 μm∕s; Dm = 1 × 10−9 m2∕s; RSD 5%; ec = 0; k = 0.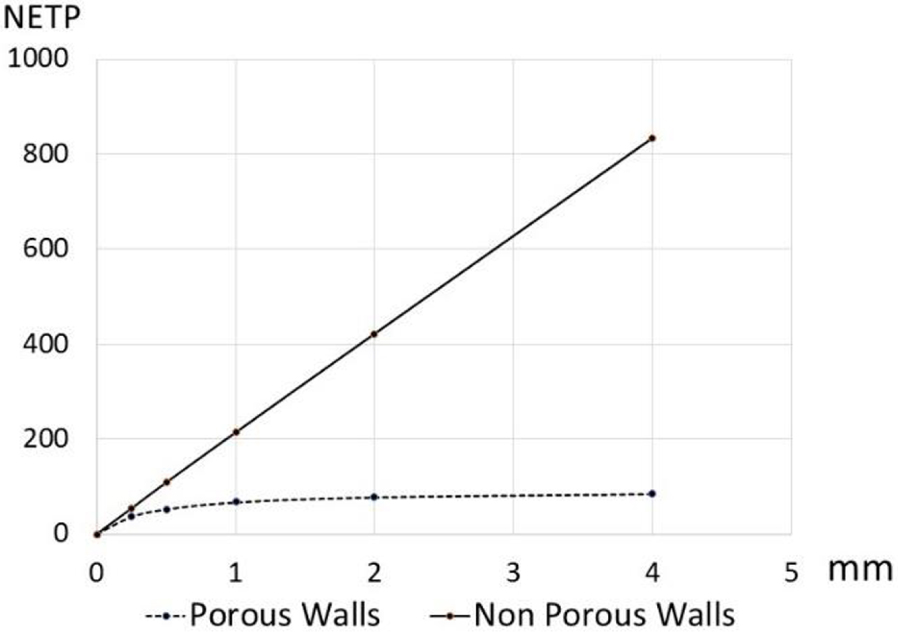
NETP values obtained for channel lengths up to 25 mm with 𝜎 = 5% of the average diameter did show linearity up to at least 5000 plates. No deviation from linearity has been noted.
The agreement of the theoretical results, of the transfer function results, and simulation results is surprisingly good in view of the simplicity of the starting hypothesis of the theoretical interpretation. The agreement between the three models support very efficiently each other. Their very similar numerical predictions lead to a good confidence in the quantitative results of this study.
5.3. Discussion
Several conclusions can be drawn from the analysis of (20) and of this numerical example:
- First, the previous limitation attached to the Shisla–Sidelnikov formula disappears. Due to the coupling between eddy diffusion and transverse molecular diffusivity, the height of a theoretical plate decreases with short column lengths and tends to a constant value for infinite column lengths. The NETP increases linearly with the length and the separative power of chromatography is restored.
- A constant loss of efficiency with respect to the single average column occurs, the polydispersity term on overall variance forecast by Shisla analysis. This correction is strongly dependent on the channel’s diameter dispersity and increases as the square of the RSD.
- The question arises as to what relative dispersity can be reasonably expected. For ordinary textile threads, the usual variance is of 2%. The loss of efficiency attributable to the channel diameter random distribution will in this case be lower than 10%. The counterpart is a slightly higher pressure drop, without changing the order of magnitude of this advantage.
- For very irregular distributions of channels diameters, the loss in NETP can be compensated for by an increase in column length or decrease in channel’s diameter. Due to the large potential gain on pressure drop, the solution remains attractive.
This suggests that provided there is suitable diffusional bridging between capillaries, multicapillary packings allow the throughput capacity and efficiency of conventional particulate packings. Given that multicapillary arrays have a pressure drop that is lower by one order of magnitude, they should be a superior candidate for separation applications.
6. Conclusion
Starting from a short theoretical basis, we have used computer simulation to study quantitatively the behavior of multicapillary arrays with statistical variation in their diameters. In the absence of interchannel radial diffusion, only a slight statistical dispersion between channel dimensions induces a purely hydrodynamic limitation of the efficiency of the packing in terms of NETP. This effect and its magnitude are verified by computer simulation. A 2% standard deviation limits the NETP to a value of 625. This limit cannot be exceeded even with increased packing length. This effect has to date precluded the use of these potentially powerful and very low-pressure-drop systems in analytical and preparative chromatography.
Our theoretical and simulated results show that superimposing a radial diffusive term between adjacent channels, or diffusional bridging, removes this limitation. In this case, the multicapillary array behaves like particulate packing, with an NETP increasing linearly with packing length. In the case where the multicapillary array has comparable effective diffusivity to classical LC porous stationary phases (i.e. silica gel, PS-DVB gels), this effect is strong enough that the efficiency loss due to inhomogeneity in the capillary diameters, which is catastrophic for a non-diffusive array, becomes negligible for typical standard deviations in the capillary diameter. The excellent numerical agreement between the purely mathematical transfer function of a twin tube system and its simulation results without the need of any adjustment parameter constitute a sound proof of the existence and order of magnitude of this diffusional bridging effect.
The 5 to 10% loss in resolving power can be compensated by an increased length of the packing. Due to the large gain on the pressure drop over conventional packings, even very imperfect arrays, easier to manufacture, will still show advantage in operation.
This result could have numerous practical consequences for chromatography and chemical engineering, stemming from the fact that at an identical characteristic dimension (particle diameter versus channel diameter), the pressure drop in multicapillary packing is one order of magnitude lower.
In LC-UHPLC, analytical chromatography has reached its technological limit by decreasing particle sizes to 1.7 μm and working at extremely high pressures, such as 1500 bar. Diffusive multicapillary packing could be an important advance in this field. With multicapillary packing and for a given available pressure drop, the available HETP in LC can increase by one order of magnitude, approaching with standard instruments the 100 000 plates of golay columns. The analysis time can decrease by one order of magnitude making control ultrafast. The high flow rate of the multicapillary structure allows the use of the existing range of detectors, injectors, and pumps.
In industrial separation, this approach will allow chromatography to be conducted with high efficiency and with low-pressure pumps, injectors, lines, and other fluidic appliances. The investment and cost operation will be reduced by an important factor.
The difficulty in stabilizing large particulate beds in columns disappear naturally due to the monolithic and rigid nature of multicapillary packings [28]. Multicapillary packing may be able to be used as modules in parallel or in series for achieving large scale separations.