

1 Introduction
The coordination chemistry of organic compounds has attracted considerable attention with the development of asymmetric synthesis [1] and biomedical inorganic chemistry [2]. Phosphines ligands are the most often encountered in enantioselective reactions mediated by palladium complexes [3]. However, some phosphine free palladium ligands have been developed [4]. In example, a cationic palladium–sparteine complex was synthesized in order to get a chiral pocket around the metal. It was shown to be efficient in asymmetric alkylation of allylic acetates [5a,b]. Other palladium/sparteine-like diamine complexes catalyzed the oxidative kinetic resolution of 1-indanol [5c]. Inorganic elements are also involved in many biological processes [6] or used, when complexed to organic substrates, as therapeutic or diagnostic agents [7] ([8] for magnetic resonance imaging agents). Platinum complexes [9], including the most known cisplatin, are now currently used against resistant tumors. Recently, a cytotoxic activity similar to cisplatin or carboplatin was obtained with the palladium(II) complex formed with naturally occurring alkaloid harmine [10].
With the aim of synthesizing new radiotracers for the in vivo imaging of nicotinic receptors, we [11] and others [12] recently prepared pyridone-substituted derivatives of (–)-cytisine 1. This alkaloid, extracted from many leguminosae [13], is a potent and selective agonist of nicotinic cholinergic receptors of subtype α4β2. It is generally used as a reference for the study of nicotinic receptors. Moreover, it has been shown that cytisine, probably via an iron complex, reduces hydroxy radical formation in vitro and is able to protect dopaminergic neurons against the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) induced toxicity in vivo [14]. The low cost, the original chiral structure close to sparteine, make (–)-cytisine a new attracting tool both for the development of novel metal biomolecules or chiral complexes for asymmetric synthesis [15]. In order to progress in understanding the coordination chemistry and biochemistry of (–)-cytisine complexes, we have studied the reactivity of this ligand and two of its derivatives towards various palladium salts. A palladium–cytisine complex from palladium acetate and (–)-cytisine has been prepared, characterized by its spectroscopic properties and its X-ray structure. The recent report on the synthesis of PdCl2(cytisine)2 complex which has been characterized solely by its spectroscopic properties [16] urges us to present our results.
2 Experimental section
Solvents were dried and distilled under argon before use (dichloromethane over CaCl2 and diethyl ether over sodium/benzophenone) and kept over molecular sieves. 1H and 13C NMR spectra were recorded on an AC 250 Bruker in CDCl3 as the solvent with TMS as reference for 1H spectra and CDCl3 (δ 77.0) for 13C spectra. The infrared spectra were recorded with Spectrafile IRTM Plus MIDAC. C, H and N analyses were performed on a Perkin Elmer 2400 CHN.
2.1 Characterization data of the free cytisine [17]
M.p. 153–154 °C (heptane–EtOH). [α]22D = –150° (c 1, CHCl3).
EI-MS 190 (M+, 78%), 186 (100%).
IR (KBr, cm−1): 791, 1140, 1472, 1542, 1646, 2800, 2935, 3278.
1H NMR (CDCl3): δ = 1.60 (br s, N–H), 1.96 (s, 2 H-8), 2.33 (br s, H-9), 2.90–3.12 (m, 2 H-11, 2 H-13, H-7), 3.90 (dd, J = 15.0, 6.9 Hz, Hax-10), 4.13 (d, J = 15.0 Hz, Heq-10), 6.00 (dd, J = 6.9, 1.2 Hz, H-5), 6.9 (dd, J = 9.1, 7.31 Hz, l. 180… H-4) failed.
13C NMR (CDCl3): δ = 25.9 (C-8), 27.4 (C-9), 35.2 (C-7), 49.4 (C-10), 52.7 (C-13), 53.6 (C-11), 104.7 (C-5), 116.3 (C-3), 138.5 (C-4), 150.9 (C-6), 163.3 (C-2).
Anal. Calcd for C11H14N2O (190 g mol−1): C, 69.45; H, 7.45; N, 14.72. Found: C, 69.12; H, 7.45; N, 14.52.
2.2 Syntheses of compounds 2b and 3
2.2.1 Synthesis of PdCl2(-cytisine)2 2b
(–)-Cytisine 1 (0.035 g, 0.18 mmol) dissolved in 8 ml of CH2Cl2 was added to a solution of PdCl2(MeCN)2 (0.23 g, 0.09 mmol) in 10 ml of CH2Cl2. After 24-h stirring at r.t., the orange–yellow solution was evaporated under reduced pressure. The resulting orange powder was washed with two portions of 5 ml of diethyl ether (0.030 g, 75%).
M.p. 211 °C Dec.
[α]22D = –460° (c 2, CHCl3).
IR (KBr, cm−1): 237, 279, 329, 506, 562, 805, 1147, 1548, 1574, 1649, 2940, 3214.
1H NMR (CDCl3): δ = 1.85 (d, J = 12.4 Hz, H-8), 2.02 (d, J = 12.4 Hz, H-8′), 2.33 (sl, H-9), 3.25 (m, 2 H-11, 2 H-13, NH), 2.92 (sl, H-7), 3.82 (dd, J = 15.6, 6.2 Hz, Hax-10), 4.12 (d, J = 15.6 Hz, Heq-10), 6.00 (d, J = 6.2 Hz, H-5), 6.47 (d, J = 9.4 Hz, H-3), 7.32 (dd, J = 9.4, 6.2 Hz, H-4).
13C NMR (CDCl3): δ = 25.0 (C-8), 27.9 (C-9), 35.2 (C-7), 48.5 (C-10), 56.1 (C-13), 56.8 (C-11), 105.8 (C-5), 118.2 (C-3), 139.0 (C-4), 146.8 (C-6), 163.1 (C-2).
Anal. Calcd for C22H28N4O4Cl2Pd·H2O (607.4 g mol−1): C, 45.88; H, 5.21; N, 9.73. Found: C, 45.92; H, 5.01; N, 9.28.
2.2.2 Synthesis of Pd(OAc)2(C11H14N2O)2 3 and of its hydrate
Palladium acetate (0.35 g, 1.6 mmol) was dissolved in CH2Cl2 (20 ml). To this solution, (–)-cytisine 1 (0.59 g, 3.1 mmol) in CH2Cl2 (10 ml) was slowly added. The mixture was stirred at r.t. for 24 h. After evaporation of the solvent, the yellow–orange oil was treated with diethyl ether to afford an orange powder (0.94 g) in 97% yield.
M.p. 174 °C Dec.
[α]22D = –230° (c 3, CHCl3).
IR (KBr, cm−1): 810, 1179, 1315, 1418, 1546, 1570, 1614, 1650, 2856, 2924, 3300.
1H NMR (CDCl3): δ = 1.77 (s, 3H, CH3, Ac), 1.81–2.03 (m, N–H, 2 H-8), 2.30 (br s, H-9), 2.84–3.22 (m, 2 H-11, 2 H-13, H-7), 3.82 (dd, J = 15.6, 6.2 Hz, Hax-10), 4.12 (d, J = 15.6 Hz, Heq-10), 5.98 (dd, J = 6.2, 1.2 Hz, H-5), 6.49 (dd, J = 9.4, 1.2 Hz, H-3), 7.32 (dd, J = 9.4, 6.2 Hz, H-4).
13C NMR (CDCl3): δ = 23.7 (CH3, Ac), 25.6 (C-8), 27.7 (C-9), 34.9 (C-7), 48.6 (C-10), 54.3 (C-13), 55.0 (C-11), 105.4 (C-5), 118.1 (C-3), 138.7 (C-4), 147.6 (C-6), 163.5 (C-2), 180.4 (CO, Ac).
Anal. Calcd for C26H34N4O6Pd (604.4 g mol−1): C, 51.62; H, 5.62; N, 9.26. Found: C, 52.02; H, 5.90; N, 9.26.
Solubilization of the powder in CH2Cl2 then addition of a minimum amount of Et2O afforded suitable crystals for X-ray analysis after evaporation of part of the solvent mixture.
M.p. 137–138 °C Dec.
IR, 1H NMR and 13C NMR spectra identical to the above spectra.
Anal. Calcd. for C26H34N4O6Pd·H2O (622.4 g mol−1): C, 50.12; H, 5.78; N, 8.99. Found: C, 49.89; H, 5.28; N, 8.78.
2.3 X-ray crystallographic studies
2.3.1 X-ray analysis of Pd(OAc)2(C11H14N2O)2
The data were collected on a CAD4 Enraf–Nonius diffractometer using the molybdenum Kα radiation isolated with a graphite monochromator (λ = 0.71073 Å) until θ = 90°. The reflections were corrected for Lorentz, polarization, absorption effects. The crystal structure was solved using the heavy atom method and subsequent Fourier synthesis. The pseudocentrosymmetric character of the structure did not allow the refinement to work efficiently. In a first step, all atoms except the ones of the two terminal pyridone rings were supposed to be related by a symmetry center located on the palladium atom. All the anisotropic thermal factors were considered as associated through the pseudo center. With this strategy the results of the refinements were sufficiently good (R = 0.051 and Rw = 0.064) to determine accurately the palladium coordination and to show that the complex is a palladium diacetate dicytisine hydrate. However, this approach led to very short interatomic distances (about 1.18 Å) between atoms belonging to the boundaries of the ‘free refined cycle’ and the ‘restrained part’ of the molecule. A second strategy was thus used for the refinement. As the size and the bond lengths of cytisine molecule [18,19] are known, we used one of the facilities of the Xtal 3.7 package [19]: the possibility to constrain the distance between two atoms.
2.3.2 X-ray analysis of N-benzylcytisine
The X-ray intensity data were collected at room temperature with a MAR345 image plate detector using MoKα (λ = 0.71069 Å) monochromatized radiation from a RU200 rotating anode. Hundred images, with Δφ = 3°, at a crystal to detector distance of 160 mm were recorded. After processing and merging, there was 1991 unique reflections (Rint = 0.070). The reflections were corrected for Lorentz polarization, not for absorption. The unit cell parameters were refined using all the collected spots after the integration process. The structure was solved by direct methods with SHELXS97 [20] and refined by full-matrix least-squares on F2 using SHELXL97 [21]. All the non-hydrogen atoms were refined with anisotropic temperature factors. The hydrogen atoms were calculated with AFIX and included in the refinement with a common isotropic temperature factor. The details of the refinement and the final R indices are presented in Table 1.
Summary of crystal data, intensity measurement and structure refinement for palladium diacetate dicytisine hydrate 3 and N-benzylcytisine 5
| Chemical formula | PdN4O7C26H30 | C18H24N2O |
| Formula weight | 617.02D | 284.39 |
| Crystal system | Triclinic | Orthorhombic |
| Space group | P1 (1) | P212121 |
| Cell dimensions | a = 6.6064(9) Å | a = 7.183(2) Å |
| b = 7.5159(7) Å | b = 8.591(3) Å | |
| c = 14.169(2) Å | c = 24.994(8) Å | |
| α = 84.039(9)° | α = 90° | |
| β = 76.64(1)° | β = 90° | |
| γ = 88.11(1) | γ = 90° | |
| Cell volume V | 680.7(2) Å3 | 1542.4(8) |
| Z | 1 | 4 |
| Density Dcalcd. (g cm−3) | 1.505 | 1.225 |
| μ (cm−1) | 7.32 | 0.76 |
| Measured reflections | 11,460 | 11,757 |
| Independent reflections (Rint) | 1991 (0.070) | |
| 2 Θmax (°) | 45 | |
| Tmin, Tmax | 0.9101, 0.9391 | No correction |
| hmin, hmax | –12, 13 | –7, 7 |
| kmin, kmax | –14, 14 | –9, 9 |
| lmin, lmax | 0, 28 | –26, 26 |
| Reflections with I > 3σ(I) | 2910 | I > 2σ(I) 1891 |
| Temperature of the data collections T (°C) | 21 | 25 |
| Number of variables | 217 | 192 |
| R (Fo) a | 0.0453 | 0.0544 |
| RW | 0.0544 | 0.1546 |
3 Results and discussion
3.1 Syntheses of compounds 2b and 3
In 2000, Khisamutdinov et al. [16] described the formation of PdCl2((–)cytisine)2 from Na2PdCl4. On the basis of the spectroscopic data of the complex, they proposed around the palladium atom a cis arrangement, respectively, of the chlorides and the two cytisines. We studied the coordination of the cytisine with other palladium chlorides precursors (PdCl2 or PdCl2MeCN2). With PdCl2, problems of solubility of the precursor occurred, so we decided to use PdCl2MeCN2, a soluble form of PdCl2. The resulting complex showed similar NMR spectroscopic data as the cis complex described by Baikova what is relatively surprisingly when trans geometry around a PdII center is commonly accepted [22]. In contrary in IR, we observed in the area 200–800 cm−1, only one band for Pd–N antisymmetric stretching at 506 cm−1 and only one band for Pd–Cl antisymmetric stretching at 329 cm−1. These results are in accord with a trans geometry around the palladium center; as a matter of fact, Perry et al. [23] showed that Pd–N antisymmetric stretching led, respectively, to two bands at 495 and 474 cm−1 for cis-Pd(NH3)2Cl2 and one band at 494 cm−1 for trans-Pd(NH3)2Cl2. In the same way, they reported for Pd–Cl antisymmetric stretching two strong bands at 327 and 306 cm–1 for cis-Pd(NH3)2Cl2 and one strong band at 333 cm−1 for trans-Pd(NH3)2Cl2. Next, the melting point of 2b measured at 211 °C is also different that the melting point of 2a proposed at 238 °C [16].
Furthermore, in the presence of K2CO3 as a base, no difference concerning the resulting complex was detected. Employment of stronger bases (NaOMe, NaNH2) led to degradation products. We were so enable to deprotonate the secondary amine of cytisine and to obtain covalent coordination of the amine (Scheme 1).
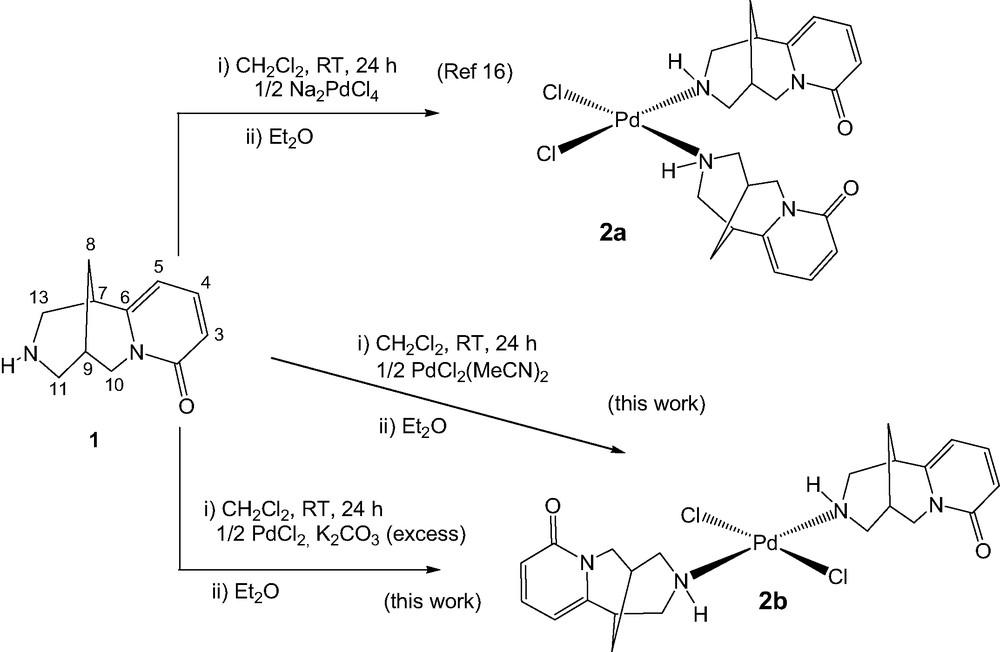
Complexation of (–)-cytisine with palladium dichloride.
These results led us to employ another PdII source as palladium acetate. As described in Scheme 2, coordination of the cytisine occurred and the complex 3 was obtained in quantitative yield, the trans geometry of the complex around the palladium center being confirmed by X-ray analysis.
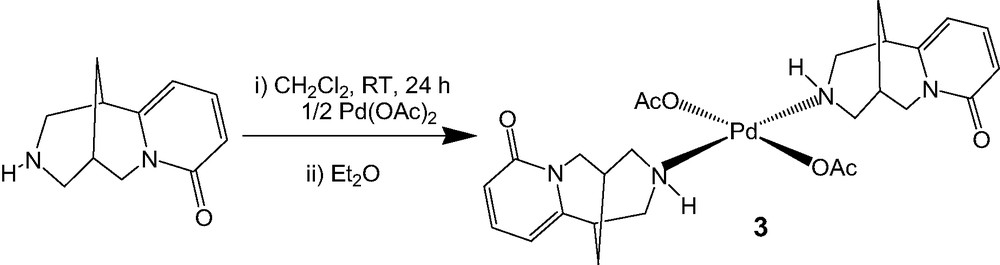
Complexation of (–)-cytisine with palladium diacetate.
The comparison of 1H NMR spectra of cytisine 1 and its Pd(OAc)2-complex 3 shows a few modifications in terms of chemical shift and multiplicity of the signals. In the complex, the exchangeable proton of the secondary amine is shifted downfield by 0.2–0.3 ppm inside a multiplet at 1.81–2.03 ppm including the H8 protons. These protons at the C8 atom (bridging methylene) also become non-equivalent and the corresponding signal changes into a multiplet (singlet in the cytisine spectrum). The other major difference from the spectrum of the free cytisine arises from protons at the C7, C11 and C13 atoms. The original signal, a somehow narrow multiplet at 2.90–3.12 ppm, is broaden into a wide multiplet at 2.84–3.22 ppm. Two other signals experience a slight shift for one of the protons at C10 atom (0.09 ppm upfield) and for the H-3 proton (0.06 ppm downfield).
In the 13C NMR spectrum of complex 3, some carbons are shifted downfield (more than 1 ppm) compared to the free ligand 1. These are C11, C13, the carbons adjacent to the amino group, by 1.4 and 1.6 ppm, respectively, as well as C3 and C5 (in the pyridone ring) by 1.8 and 0.7 ppm, respectively. On the other hand, the signals of two carbons bonded to the nitrogen atom of the pyridone ring, C6 and C10, are shifted upfield by 3.3 and 0.8 ppm, respectively. As for the 1H NMR spectrum, all these values compare well with the previously described complex of cytisine, PdCl2(cytisine)2 [16].
Since cytisine is capable to coordinate metal ions by several sites, all these spectroscopic observations are consistent with X-ray analysis which shows a coordination of palladium(II) only through the nitrogen atom of secondary amino group: all the NMR signals of carbons or hydrogens adjacent to this nitrogen atom are clearly affected by the coordination and are mainly shifted downfield. At the same time, the NMR signals of atoms surrounding the two other heteroatoms remained unchanged or clearly shifted upfield.
The coordination N-methyl-cytisine and N-benzylcytisine [24] towards palladium acetate have been also studied. In both cases, using strictly anaerobic conditions and distilled solvents, black palladium appeared relatively quickly and, respectively, a part of the ligands is recovered. The reduction of PdII into Pd0 is probably due to the presence of the tertiary amine function present in the two structures, such functional group being able to reduce palladium(II) salts in black palladium as reported by Vollmüller et al. [25a], following a mechanism proposed by Mc Crindle et al. [25b,c] (Scheme 3).

(–)-Cytisine derivatives.
Considering the use of (–)-sparteine in palladium-catalyzed oxidative kinetic resolution of secondary alcohols with molecular oxygen [26], cytisine and N-methylcytisine have been combinated to Pd(OAc)2 or PdCl2(MeCN)2 to catalyze the oxidative kinetic resolution of phenethyl alcohol (Scheme 4). Preliminary results are quite disappointing, but improving experiments are still in course by varying the nature of the metallic precursor and sources of oxygen.

Oxidative kinetic resolution.
3.2 X-ray analysis of Pd(OAc)2(C11H14N2O)2
The cell parameters reported in Table 1 are determined and refined by diffractometric technique at 21 °C with a least-squares refinement based upon 25 reflections with 18 < θ < 22°.
A colorless plate crystal with dimensions 0.210 × 0.122 × 0.084 mm was selected for the structure determination. Its quality was tested with film techniques on a Weissenberg camera. The triclinic unit cell parameters are: a = 6.6064(9) Å, b = 7.5159(7) Å, c = 14.169(2) Å, α = 84.038(9)°, β = 76.64(1)°, γ = 88.11(1)°. Two thousand nine hundred and nine data with I ≥ 3σ(I) were used to solve and to refine the crystal structure (see the data collection and refinement conditions in Table 1). The space group may be either non-centrosymmetric P1 or centrosymmetric P. The distributions and the statistics over the Ehkl values are favorable to the centrosymmetric P space group but (–)-cytisine is a molecule with stereogenic carbon atoms. Therefore the space group must be non-centrosymmetric i.e. P1. This contradiction can be explained by the fact that a symmetry center located on the Pd atom generates correctly the acetate groups and the N1, N2, C6–C13 atoms of the cytisine but not the pyridone ring, so that about 75% of the atoms may be centrosymmetric.
The coordination of the palladium atom is square planar with two nitrogen atoms, one from each cytisine molecule, and two oxygen atoms, one from each acetate group (Fig. 1) with Pd–O = 2.019(4) Å and Pd–N = 2.067(3) Å. As observed previously for cytisine 1 [27] the pyridone rings in the complex 2 are essentially planar with maximum deviations of 0.024(9) and 0.05(1) Å from the least-square plane of the two pyridone rings, respectively. The planarity of the latter ones confers an envelope conformation on the adjacent rings with the bridge-head atoms C(8) and C(28) out of their respective plane by 0.76(2) and –0.71(2) Å. Note that the pyridone rings are almost coplanar with their adjacent rings, the angles between the two least-squares planes being about 176°. The rings near the palladium atom adopt a conventional chair conformation. Fig. 2 shows the stacking of the palladium diacetate dicytisine and water molecules.
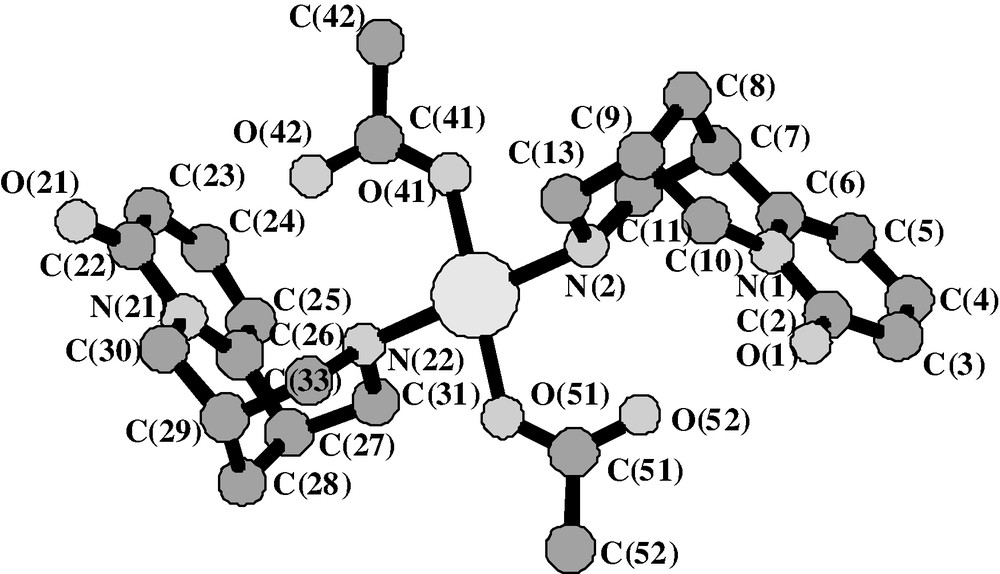
The palladium diacetate di-(–)-cytisine molecule 3 with the square planar coordination of the palladium atom.

The projection of the structure along a of palladium diacetate di-(–)-cytisine hydrate showing the stacking of the molecules.
3.3 X-ray analysis of N-benzylcytisine
Slow evaporation of the reacting mixture (PdCl2(MeCN)2/N-benzylcytisine (2 eq.) in CH2Cl2) led to the formation of suitable crystals for X-ray analysis.
A colorless crystal of approximate dimensions 0.30 × 0.15 × 0.15 mm was chosen and glued on a glass fiber. The crystal data and the data collection parameters are summarized in Table 1. The geometry of the molecule is very similar to that of (–)-cytisine and (–)-N-methylcytisine [27] (Fig. 3).

ORTEP style view of N-benzylcytisine [21].
4 Conclusion
A new complex palladium/cytisine has been synthetized from palladium acetate and characterized by X-ray analysis showing a trans geometry. A palladium/cytisine complex generated from Pd(MeCN)2Cl2 is also assigned as a trans complex based on strong spectral similarities with the newly synthetized palladium complex of this study. N-substituted cytisine derivatives do not coordinate to the palladium and reduce PdII to Pd0. X-ray analysis of the N-benzylcytisine completes the characterization of this derivative. Experiments about the contribution of the complex Pd(OAc)2(cytisine)2 in oxidative kinetic resolution of secondary alcohols are still in course.
Acknowledgements
We thank Dr. Philippe Bazin for technical support. This work was supported by the‘Pôle universitaire normand’.