

1 Introduction
Les apatites sont des matériaux inorganiques. Elles correspondent à une large famille de composés de formule générale Me10(XO4)6Y2, avec Me un cation divalent (Sr2+, Ca2+, Ba2+…), XO4 un groupement trivalent (PO43−, VO43−…) et Y− un anion monovalent (F−, Br−, Cl−…). Généralement, l'apatite cristallise dans le système hexagonal (groupe d'espace P63/m) [1]. Les cations Me2+ se repartissent entre deux sites cristallographiques différents : quatre sont localisés dans le site Me(1), de coordinence 9, et les six restants se placent dans le site Me(2), de coordinence 7. En raison de la stabilité et de la flexibilité de leur structure, les apatites offrent de nombreuses possibilités de substitutions [2–4]. Ces sites peuvent être occupés par des cations mono-, bi-, tri-ou tétravalents. Les groupements XO4 peuvent être substitués par des groupements bi-, tri-ou tétravalents. Quant aux ions Y−, ils peuvent être remplacés par des ions divalents. Les britholites sont des dérivés apatitiques obtenus par substitution couplée du cation divalent par une terre rare trivalente et du groupement trivalent XO4 par le groupement tétravalent silicaté, SiO4 [5–7].
Les matériaux apatitiques sont utilisés dans divers domaines, tels que la médecine [8,9], la catalyse [10], la luminescence [11], l'optique [12,13] ou la conductivité ionique [14,15]. Ils constituent aussi des matériaux pour laser [16]. Plusieurs études sur la possibilité de confinement d'éléments radioactifs au sein de la structure apatitique ont été suscitées par des observations réalisées sur des réacteurs naturels, tels que celui d'Oklo, au Gabon [17], ou du Hoggar en Algérie [18]. Ces études ont montré que les britholites sont capables de piéger des radionucléides, comme le césium ou les actinides [19–23].
Nous nous sommes intéressés dans ce travail à la synthèse de britholites renfermant du césium, de formule générale Sr8−xLa1+xCs(PO4)6−x(SiO4)xF2, avec x = 0, 1 et 6. Les produits obtenus ont été caractérisés par diffraction des rayons X, par spectroscopies infrarouge et Raman, et par microscopie électronique à balayage. De même, les échantillons ont été analysés par thermogravimétrie, microanalyse X à dispersion en énergie et par fluorescence X.
2 Protocole expérimental
2.1 Préparation des poudres
Les britholites à base de césium ont été synthétisées par voie sèche en deux étapes à partir de SrCO3 (>96% Riedel de Haen), La2O3 (>99,5% Prolabo), SiO2 (>99,5% Alfa), SrF2 (>99,5% Prolabo), Cs2CO3 (>99% Fluka), NH4F (> 95% Merck ) et Sr2P2O7.
2.1.1 Préparation de Sr2P2O7
Le di-phosphate de strontium a été obtenu selon la réaction suivante :
| 2SrCO3 + 2(NH4)2HPO4 → Sr2P2O7 + 4NH3(↑) + 2CO2(↑) + 3H2O(↑) | (1) |
Après avoir été broyé et homogénéisé dans un mortier d'agate, le mélange de carbonate de strontium et de di-ammonium-hydrogénophosphate a été mis en forme par pressage uniaxial à froid, puis calciné à 900 °C pendant 10 h. La montée en température et le refroidissement ont été effectués à une vitesse de 10 °C/min.
2.1.2 Préparation des britholites lacunaires
Des britholites lacunaires de formule générale Sr8−xLax(PO4)6−x(SiO4)xF ont été préparées selon l'équation suivante :
| 1/2SrF2 + 3/2SrCO3 + (6 − x)/2Sr2P2O7 + x/2La2O3 + xSiO2 → Sr8−xLa1+x(PO4)6−x(SiO4)xF + 3/2CO2(↑) | (2) |
Après broyage manuel et homogénéisation prolongés des réactifs dans un mortier d'agate, les mélanges ont été mis en forme par pressage uniaxial à froid. Les pastilles obtenues ont été traitées sous atmosphère dynamique d'argon à 900 °C pendant 12 h pour effectuer leur dégazage, puis, après une nouvelle étape de broyage/homogénéisation, un traitement thermique de 12 h à 1400 °C a été appliqué.
2.1.3 Préparation des britholites dopées au césium
Les britholites lacunaires obtenues ont été mélangées avec du carbonate de césium (Cs2CO3) et du fluorure d'ammonium (NH4F), broyées et compactées selon le protocole opératoire précédent. Après un premier traitement à 800 °C pendant 2 h, les pastilles obtenues ont été de nouveau broyées, homogénéisées et mises en forme. Afin de limiter la volatilisation du césium [24,25], les pastilles ont été placées dans une nacelle munie d'un couvercle, enrobées par une poudre de même nature, mais enrichie en carbonate de césium (20% en masse), pour créer une atmosphère autostabilisante. Elles ont été ensuite calcinées à 1100 °C pendant 4 h. La réaction est la suivante :
| Sr8−xLa1+x(PO4)6−x(SiO4)xF + 1/2Cs2CO3 + NH4F → Sr8−xLa1+xCs(PO4)6−x(SiO4)xF2 + NH3(↑) + 1/2H2O(↑) + 1/2CO2(↑) | (3) |
2.1.4 Nomenclature des composés
Dans l'étude qui suit, les britholites lacunaires seront désignées par Sr8La, Sr7La2 et Sr2La7, respectivement, pour x = 0, 1 et 6, alors que les britholites à base de césium seront dénommées respectivement Sr8LaCs, Sr7La2Cs et Sr2La7Cs.
2.2 Caractérisation des poudres
L'examen par DRX des échantillons a été effectué au moyen d'un diffractomètre Philips PW 1800, en utilisant la radiation Kα du cuivre (λ = 1.5406 Å). Les données ont été collectées dans l'intervalle 2θ 5–90° avec un pas de 0,02° et un temps de comptage égal à 1 s. L'identification des phases cristallines a été réalisée par comparaison avec les fichiers JCPDS (Joint Commitee Powder Diffraction Standard).
Les spectres infrarouge ont été acquis dans le domaine 400–4000 cm−1 à l'aide d'un spectromètre de type PerkinElmer 1283 à transformée de Fourier. Les échantillons dispersés dans du bromure de potassium (KBr) pur et sec ont été mis en forme par pressage uniaxial.
Les spectres Raman ont été enregistrés à la température ambiante dans l'intervalle 100–1200 cm−1 à l'aide d'un spectromètre Renishaw inVia équipé d'un détecteur CCD Renishaw RenCam, refroidi par effet Peltier. Un microscope Leica (objectif 50 ×) complète ce dispositif. La source excitatrice est un laser à argon de longueur d'onde λ = 514,5 nm.
Les échantillons ont été observés et caractérisés à l'aide d'un microscope électronique à balayage (MEB) Philips XL 30 équipé d'un détecteur électronique à dispersion en énergie (EDS).
Les échantillons ont été aussi analysés à l'aide d'un spectromètre à fluorescence X Bruker AXS S4 Explorer. L'appareil utilisé pour l'analyse thermogravimétrique est de type Setaram DTA-TG 92. La montée en température a été effectuée avec une vitesse de 10 °C/min.
3 Résultats et discussion
L'analyse par DRX des échantillons lacunaires révèle la formation d'une phase apatitique majoritaire indexée à partir de la fiche JCPDS n° 17-609. Dans le cas des compositions Sr8La (Fig. 1a) et Sr7La2 (Fig. 1b), on note la présence des phases secondaires Sr3La(PO4)3 et Sr3(PO4)2, indexées respectivement à partir des fiches nos 85-905 et 32-493. Dans le cas de la composition Sr2La7 (Fig. 1c), les phases secondaires identifiées sont La2Si2O7 et La2SiO5. Elles correspondent respectivement aux fiches nos 44-346 et 40-234. Il est bien connu que les apatites non stœchiométriques ne sont pas stables à haute température [25] ; les phases secondaires seraient alors respectivement formées selon les réactions suivantes :
| 3SrCO3 + SrF2 + 6Sr2P2O7 + La2O3 → Sr10(PO4)6F2 + Sr3La(PO4)3 + Sr3(PO4)2 + LaPO4 + 3CO2(↑) | (4) |
| 3SrCO3 + SrF2 + 5Sr2P2O7 + 2La2O3 + 2SiO2 → Sr8La2(PO4)4(SiO4)2F2 + Sr3La(PO4)3 + Sr3(PO4)2 + LaPO4 + 3CO2(↑) | (5) |
| SrF2 + 3SrCO3 + 12SiO2 + 7La2O3 → Sr4La6(SiO4)6F2 + 2La2Si2O7 + 2La2SiO5 + 3CO2(↑) | (6) |
| Sr3(PO4)2 + LaPO4 → Sr3La(PO4)3 | (7) |
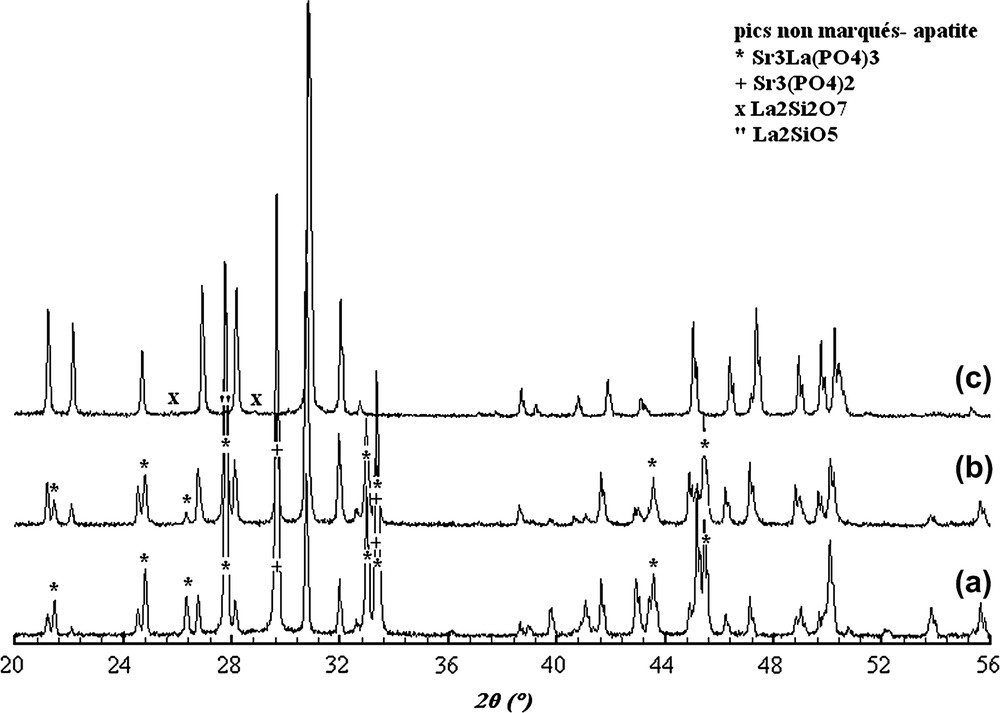
Diagrammes DRX des britholites lacunaires de compositions Sr8La(PO4)6F (a), Sr7La2(PO4)5(SiO4)F (b) et Sr2La7(SiO4)6F (c).
Paramètres cristallographiques des britholites lacunaires et dopées au césium
| Compositions | a (Å) | c (Å) |
| Sr8La(PO4)6F | 9715(5) | 7282(4) |
| Sr7La2(PO4)5(SiO4)F | 9725(3) | 7275(4) |
| Sr2La7(SiO4)6F | 9740(2) | 7260(1) |
| Sr8LaCs(PO4)6F2 | 9729(5) | 7290(5) |
| Sr7La2Cs(PO4)5(SiO4)F2 | 9746(1) | 7289(2) |
| Sr2La7Cs(SiO4)6F2 | 9752(5) | 7270(3) |
Les diagrammes DRX des britholites dopées au césium sont présentés sur la Fig. 2. Après incorporation du césium, parmi les trois compositions intermédiaires, seule la composition Sr7La2 permet l'obtention d'une phase apatitique pure (Fig. 2b), de formule Sr7La2Cs(PO4)5(SiO4)F2. Pour la composition Sr8La, il y a disparition des phases secondaires Sr3La(PO4)3 et Sr3(PO4)2 et apparition d'une nouvelle phase SrLaCs(PO4)2 (cf. fiche n° 35-426 et Fig. 2a). Dans le cas de la phase lacunaire purement silicatée traitée au césium, le diagramme DRX (Fig. 2c) montre la présence d'une phase apatitique majoritaire de formule Sr2La7Cs(SiO4)6F2 et de deux nouvelles phases secondaires CsLaSiO4 et Sr2SiO4, identifiées respectivement à partir des fiches nos 49-663 et 38-271. Aussi, l'incorporation du césium [28] dans les sites cationiques et des ions F− dans les lacunes anioniques des tunnels parallèles à l'axe c s'accompagnse d'une augmentation des paramètres a et c (Tableau 1). En effet, d'après une étude de Fourier réalisée par Senamaud sur des britholites substituées au néodyme, l'incorporation du césium a lieu dans les sites cationiques de l'apatite. Cependant, sa localisation sur le site Me(1) ou Me(2) n'a pas été possible [25]. Ainsi, parmi les trois compositions lacunaires étudiées, la composition Sr7La2 apparaît comme celle permettant une meilleure rétention du césium dans la structure apatitique, puisqu'aucune phase secondaire renfermant cet élément n'a été détectée. À la même température, son analogue au calcium et néodyme a été obtenu mélangé à la phase CaNdCs(PO4)2, dont la formation serait due à la non-stœchiométrie de la britholite [25]. D'après Campayo et al. [24,29], cette phase CaNdCs(PO4)2 ne semble pas être rédhibitoire pour le conditionnement du césium, alors que les phases secondaires formées avec les pôles phosphaté et silicaté sont suffisamment solubles pour entraver leur utilisation pour le confinement de cet élément.
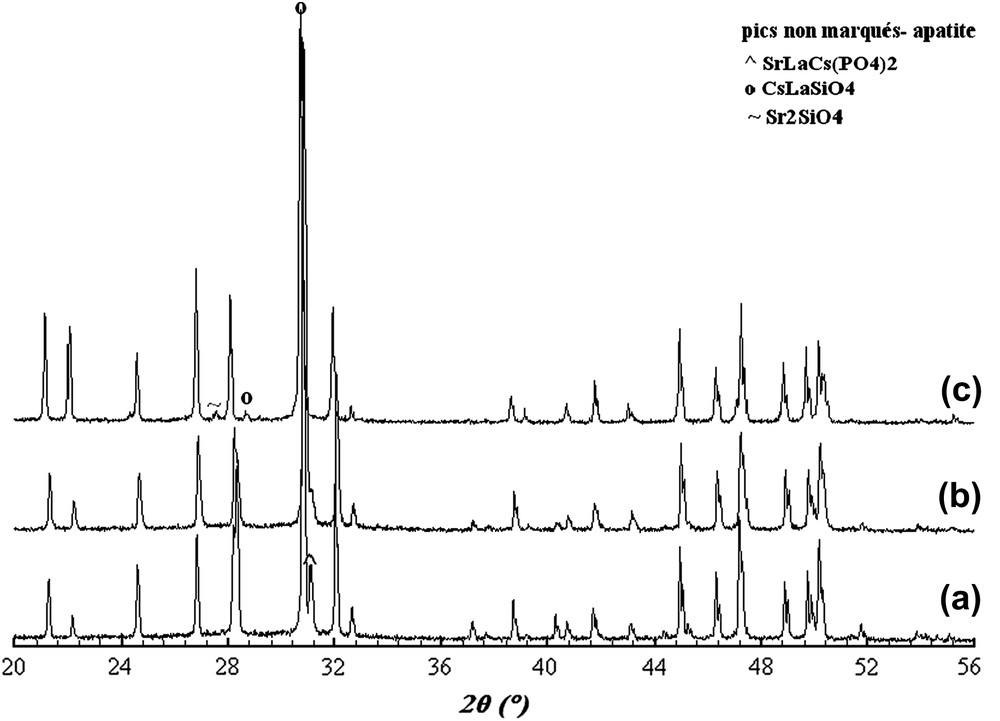
Diagrammes DRX des britholites dopées au césium, de compositions Sr8LaCs(PO4)6F2 (a), Sr7La2Cs(PO4)5(SiO4)F2 (b) et Sr2La7Cs(SiO4)6F2 (c).
Les spectres d'absorption infrarouge des britholites lacunaires présentés sur la Fig. 3 mettent en évidence la présence des bandes d'absorption des groupements PO4 et SiO4. Leur attribution a été réalisée par comparaison aux spectres des phases Sr10(PO4)6F2, Sr8La2(PO4)4(SiO4)2F2 et Sr4La6(SiO4)2F2 [30]. Les spectres d'absorption infrarouge des échantillons dopés au césium (Fig. 3) sont similaires à ceux des britholites lacunaires. Cependant, on note le déplacement des bandes d'absorption des groupements PO4 et SiO4 du côté des faibles nombres d'ondes (Tableau 2). Ce déplacement serait dû à l'expansion de la maille [31]. Ce résultat corrobore ceux obtenus par diffraction des rayons X et confirme l'incorporation du césium dans la maille apatitique. Les bandes supplémentaires apparaissant sur certains spectres vers 3450, 1640 et 1380 cm−1 seraient relatives aux groupements OH de surface, dus à la réhydratation de la poudre après synthèse [32].

Spectres IR des britholites lacunaires et dopées au césium de compositions Sr8La(PO4)6F (a), Sr8LaCs(PO4)6F2 (b), Sr7La2(PO4)5(SiO4)F (c), Sr7La2Cs(PO4)5(SiO4)F2 (d), Sr2La7(SiO4)6F (e) et Sr2La7Cs(SiO4)6F2 (f).
Positions (en cm−1) des bandes d'absorption IR et Raman des britholites lacunaires et dopées au césium
| Spectroscopie infrarouge | Spectroscopie Raman | |||||||
| υ1 (cm−1) | υ2 (cm−1) | υ3 (cm−1) | υ4 (cm−1) | υ1 (cm−1) | υ2 (cm−1) | υ3 (cm−1) | υ4 (cm−1) | |
| PO43− | ||||||||
| Sr8La(PO4)6F | 944 | 450 | 1082 | 589 | 952 | 442 | 1073 | 593 |
| 1047 | 583 | |||||||
| – | 421 | 1028 | 574 | |||||
| 1038 | 564 | – | – | 1001 | ||||
| Sr7La2(PO4)5(SiO4)F | 918 | 509 | – | 594 | 951 | – | 1073 | – |
| – | – | 1037 | 564 | – | 417 | – | – | |
| – | – | – | 574 | |||||
| – | – | 1001 | ||||||
| Sr2La7(SiO4)6F | – | – | – | – | – | – | – | – |
| Sr8LaCs(PO4)6F2 | 942 | 446 | 1080 | 586 | 950 | 440 | 1070 | 591 |
| – | 419 | 1045 | 581 | |||||
| – | ||||||||
| 1035 | 560 | – | 1026 | 571 | ||||
| 1001 | ||||||||
| Sr7La2Cs(PO4)5(SiO4)F2 | 912 | 506 | 1076 | 589 | 950 | 442 | 1071 | 592 |
| 1035 | 560 | – | 415 | 1047 | 579 | |||
| – | 400 | 999 | 571 | |||||
| Sr2La7Cs(SiO4)6F2 | – | – | – | – | – | – | – | – |
| SiO44− | ||||||||
| Sr8La(PO4)6F | – | – | – | – | – | – | – | – |
| Sr7La2(PO4)5(SiO4)F | – | 436 | – | 564 | 845 | – | – | – |
| 870 | 404 | 946 | – | – | – | – | – | |
| Sr2La7(SiO4)6F | 919 | 455 | – | 542 | 851 | 395 | 919 | 526 |
| 848 | 407 | 964 | 492 | 513 | ||||
| 872 | ||||||||
| Sr8LaCs(PO4)6F2 | – | – | – | – | – | – | – | – |
| Sr7La2Cs(PO4)5(SiO4)F2 | – | 432 | – | 560 | 845 | – | – | – |
| 865 | 402 | 942 | 544 | – | – | – | – | |
| Sr2La7Cs(SiO4)6F2 | 916 | 450 | 1034 | 540 | 850 | 392 | 943 | 524 |
| 846 | 401 | 956 | 489 | – | – | 916 | 512 |
Les spectres de diffusion Raman des mélanges intermédiaires traités et non traités au césium ont été acquis à la température ambiante dans le domaine 300–1200 cm−1 (Fig. 4). L'attribution des bandes d'absorption des groupements PO4 et SiO4 a été effectuée par comparaison aux spectres de phases apatitiques de compositions similaires [33–35]. Le Tableau 2 récapitule l'ensemble des fréquences observées pour les différentes compositions étudiées. Les groupements PO4 sont caractérisés par les raies situées respectivement à 950 cm−1 (υ1), 540–620 cm−1 (υ4) et 380–460 cm−1 (υ2). Les raies correspondant au mode υ3 et apparaissant à des fréquences supérieures à 950 cm−1 sont de très faible intensité. Ces raies, qui correspondent aux quatre modes de vibration du groupement PO4 apatitique (Fig. 4a et b) sont absentes du spectre du composé totalement silicaté (Fig. 4c). Pour le composé phosphosilicaté, la raie détectée vers 850 cm−1 est attribuable au mode de vibration de valence symétrique υ1 du groupement SiO4. Les autres modes relatifs à ce groupement ne sont observés que dans la phase purement silicatée. Ils donnent lieu aux raies à 920 cm−1 (υ3), 512–525 cm−1 (υ4) et 395 cm−1 (υ2) (Fig. 4c). Après insertion du césium, les positions des bandes de diffusion ont subi un déplacement dans le même sens que celui observé sur les spectres d'absorption infrarouge (Fig. 4d–f). Sur certains spectres (Fig. 4e et d), une bande supplémentaire vers 690 cm−1 a été observée. Comme on l'a indiqué plus haut, elle serait due aux groupements OH de surface issus de la réhydratation de la poudre après synthèse [32].
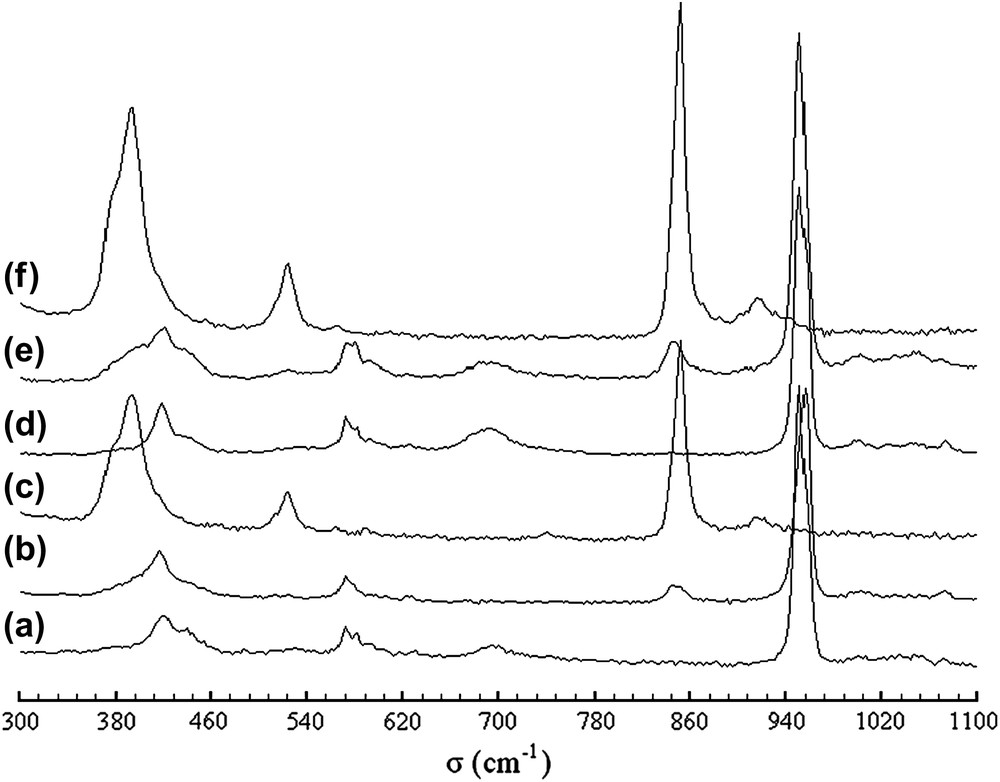
Spectres Raman des britholites lacunaires et dopées au césium de compositions Sr8La(PO4)6 (a), Sr8LaCs(PO4)6F2 (b), Sr7La2(PO4)5(SiO4)F (c), Sr7La2Cs(PO4)5(SiO4)F2 (d), Sr2La(SiO4)6F (e) et Sr2La7Cs(SiO4)6F2 (f).
L'observation au microscope électronique à balayage en électrons secondaires des mélanges intermédiaires indique la présence de grains de morphologies et de tailles différentes, donnant à penser que les échantillons sont constitués de plusieurs phases (Fig. 5a–c). De même, cette figure met en évidence une évolution de la morphologie des échantillons après incorporation du césium (Fig. 5d–f), confirmant l'apparition des nouvelles phases, révélées par l'analyse DRX. La microanalyse X à dispersion en énergie a été effectuée sur les différents grains constituant chaque échantillon. Les résultats obtenus sont regroupés dans le Tableau 3. Chaque valeur correspond à la moyenne de six mesures. Les pourcentages atomiques initialement introduits dans les mélanges sont également inclus. On constate que seuls les grains de l'échantillon Sr7La2Cs présentent la même composition, alors que les deux autres échantillons sont constitués de grains de compositions différentes. Aussi, on note que le pourcentage molaire du césium n'est proche du pourcentage escompté que pour l'échantillon Sr7La2Cs. Les pourcentages atomiques des différents éléments conduisent aux formulations consignées dans le Tableau 4. Elles correspondent aux formules chimiques des phases identifiées par la diffraction des rayons X. Une confirmation de ces résultats a été recherchée par analyse par fluorescence X. Le Tableau 4 montre que, pour l'échantillon Sr7La2Cs, la composition déterminée est très voisine de celle obtenue par microanalyse X. Senamaud a attribué la faible teneur en césium dans le cas des pôles phosphaté et silicaté à sa volatilisation au cours de la calcination [36]. Aussi, il n'est pas exclu, comme il a été avancé par Compayo [24], que du césium aurait servi à la formation d'autres phases secondaires, riches en cet élément et qui n'ont pas été identifiées par diffraction des rayons X.

Micrographies des britholites lacunaires et dopées au césium prises au MEB en mode électrons secondaires : Sr8La(PO4)6F (a), Sr8LaCs(PO4)6F2 (b), Sr7La2(PO4)5(SiO4)F (c), Sr7La2Cs(PO4)5(SiO4)F2 (d), Sr2La7(SiO4)6F (e) et Sr2La7Cs(SiO4)6F2 (f).
Pourcentages atomiques expérimentaux déterminés par microanalyse à dispersion en énergie (EDX) et ceux introduits dans les mélanges initiaux
| Sr8LaCs | Sr7La2Cs | Sr2La7Cs | |||||||
| % Atom introduits | % Atom Phase 1 | % Atom Phase 2 | % Atom introduits | % Atom Phase | % Atom introduits | % Atom Phase 3 | % Atom Phase 4 | % Atom Phase 5 | |
| Sr | 19,4 | 19,4 ± 0,2 | 2,1 ± 0,2 | 16,6 | 17,6 ± 0,1 | 4,7 | 5,6 ± 0,2 | – | 26,9 ± 0,2 |
| La | 2,4 | 2,5 ± 0,1 | 0,8 ± 0,4 | 4,8 | 4,3 ± 0,2 | 16,6 | 16,5 ± 0,2 | 17,0 ± 0,2 | 1,1 ± 0,4 |
| Cs | 2,4 | 1,4 ± 0,3 | 1,1 ± 0,4 | 2,5 | 2,2 ± 0,3 | 2,4 | 1,3 ± 0,3 | 13,0 ± 0,1 | – |
| P | 14,2 | 14,3 ± 0,4 | 1,9 ± 0,3 | 11,9 | 11,4 ± 0,4 | – | – | – | – |
| Si | – | – | – | 2,4 | 2,8 ± 0,3 | 14,3 | 14,3 ± 0,2 | 14,0 ± 0,3 | 14,4 ± 0,2 |
| O | 57,1 | 57,5 ± 0,5 | 7,6 ± 0,5 | 57,1 | 57,1 ± 0,1 | 57,1 | 57,4 ± 0,1 | 56,0 ± 0,2 | 57,5 ± 0,6 |
| F | 4,7 | 4,7 ± 0,6 | – | 4,7 | 4,5 ± 0,2 | 4,7 | 4,7 ± 0,4 | – | – |
Formulation des différentes compositions à partir des résultats obtenus par microanalyse à dispersion en énergie (EDX) et fluorescence X et rendement d'incorporation
| Compositions | Analyse par EDX | Analyse par fluorescence X | Rendement d'incorporation |
| Sr8LaCs | Sr8,1La1,05Cs0,6(PO4)6F1,95 | Sr8,2La0,90Cs0,8(PO4)6F1, 9 | 0,80 |
| Sr2,1La0,8Cs1,1(PO4)1,9 | |||
| Sr7La2Cs | Sr7,4La1,8Cs0,91(PO4)4,8(SiO4)1,2F1,91 | Sr7,1La1,8Cs0,97(PO4)5,1(SiO4)0,9F1,67 | 0,97 |
| Sr2La7CS | Sr2,35La6,9Cs0,57(SiO4)6F1,97 | Sr2,61La6,4Cs0,63(SiO4)5,99F1,75 | 0,63 |
| Cs1,18La0,9(SiO4)0,97 | |||
| Sr2,04La0,08(SiO4)1,08 |
Le mélange Sr7La2, CsCO3 et NH4F, conduisant à la phase Sr7La2Cs, a été soumis à une analyse thermogravimétrique. La courbe obtenue présentée sur la Fig. 6 montre qu'au cours de son chauffage, le mélange a subi plusieurs pertes de masse. La première, de 1%, qui a eu lieu dans le domaine 25–283 °C, est attribuée au départ de l'eau moléculaire adsorbée. La deuxième, comprise entre 283 et 345 °C, correspond à la volatilisation de l'ammoniac formé après décomposition du fluorure d'ammonium (NH4F). La perte de masse théorique correspondante est 1,02%. Au-delà de cette température, on note la décomposition du carbonate de césium, qui se produit en deux étapes, respectivement à 390 et 650 °C [36]. Chaque étape est suivie par le départ de l'eau de déshydratation selon l'équation (2), respectivement à 450 et 745 °C. Ces différents départs correspondent à une perte de masse totale de 2%. Elle est du même ordre de grandeur que celle attendue (1,86%). La dernière perte de masse (0,2%), intervenant entre 820 et 960 °C, est relative à la volatilisation d'une partie du césium, soit 3% environ de la masse introduite. Cette faible valeur, qui est compatible avec le résultat obtenu par fluorescence X, peut être expliquée par l'atmosphère autostabilisante utilisée. Elle pourrait être aussi due à des effets structuraux. En effet, le strontium, de taille relativement importante, permet à la structure de s'accommoder aux contraintes engendrées par l'insertion de cet élément.
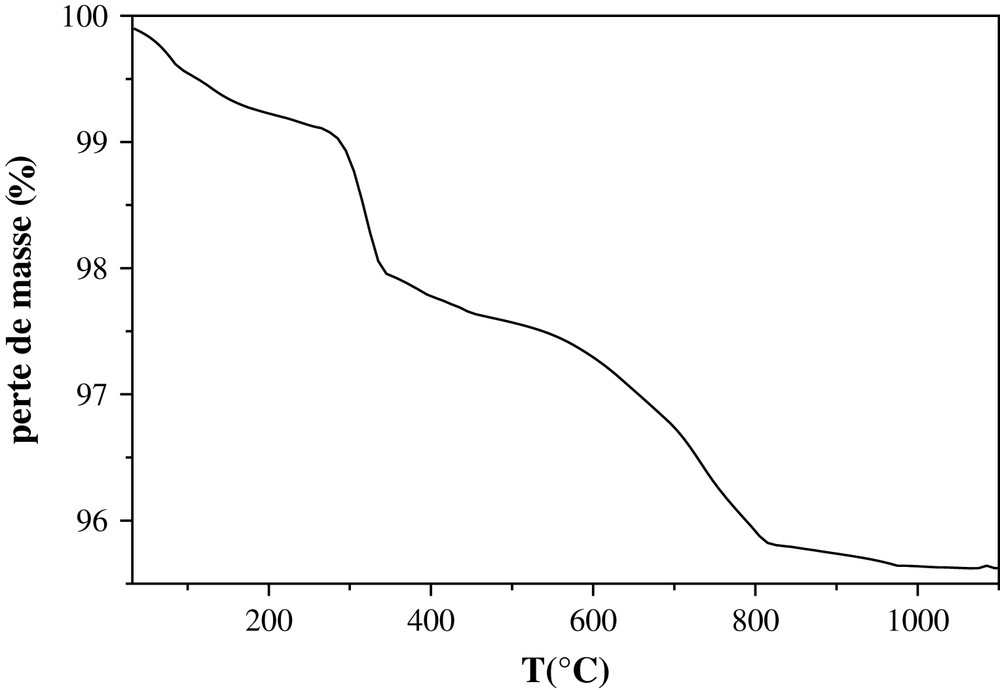
Analyse thermogravimétrique du mélange britholite lacunaire (Sr7La2(PO4)5(SiO4)F), carbonate de césium et fluorure d'ammonium.
4 Conclusion
Des britholites lacunaires ont été préparées par réaction à l'état solide à 1400 °C. Après avoir été mélangées avec du carbonate de césium et du fluorure d'ammonium, elles ont subi un traitement thermique à 1100 °C pendant 4 h. La caractérisation des produits obtenus par microanalyse X à dispersion en énergie et fluorescence X, DRX, spectroscopies d'absorption infrarouge et Raman et microscopie électronique à balayage montre que le césium a été incorporé dans la structure apatitique. Par ailleurs, les résultats obtenus conduisent aux conclusions suivantes :
- (1) la quantité de césium insérée n'est pas uniforme d'une phase à une autre. Elle n'est proche de celle introduite initialement dans les mélanges que pour la composition Sr7La2Cs(PO4)5(SiO4)F2. Aussi, parmi les trois compositions étudiées, seule cette dernière a été obtenue à l'état pur. Un tel résultat suppose que cette phase pourrait convenir au conditionnement du césium. Toutefois, une étude sur sa durabilité demeure nécessaire ;
- (2) le faible taux de césium inséré dans les compositions Sr8LaCs(PO4)6F2 et Sr2La7Cs(SiO4)6F2 serait dû à sa volatilisation et/ou à la formation d'autres phases secondaires, non identifiées par DRX. En se référant aux travaux de L. Compayo, qui a étudié les britholites substituées au néodyme, ces phases secondaires, riches en césium, mais solubles, seraient rédhibitoires pour le conditionnement de cet élément ;
- (3) la comparaison de nos résultats avec ceux obtenus par Senamaud sur des Ca(Nd)-britholites de mêmes compositions montre que le taux de césium inséré dans ces dernières est plus faible. Ceci serait dû à sa volatilisation au cours de la calcination et/ou à des contraintes stériques engendrées par la difficulté de la structure apatite à accommoder les distorsions induites par les tailles différentes des ions Cs+ et Ca2+, limitant ainsi l'incorporation du césium dans la matrice apatitique.