

1 Accomplice boryl–metal complexes
The use of suitable transition-metal complexes has several advantages over non-catalyzed boron–boron addition. Chiral complexes enables the formation of new C–B bonds, in a stereoselective manner, using optically active material limited to the relatively small amount of catalyst required.
Other advantages that greatly increase the applicability of boryl–metal complexes in organoboron synthesis [1,2] are the successful activation of less reactive or thermally sensitive boranes and the ability to orientate the functionalized new bonds chemo- and regioselectively.
The decisive role of the boryl–metal complexes is their involvement in a catalytic cycle in which several consecutive steps allow the transformation of unsaturated molecules into organomono- and organodifunctionalized compounds. However, the appropriate selection of the metal and ligands guarantees the success of the global transformation, especially in these cases in which side reactions and metal or borane decomposition can occur. Theoretical studies in this field have also contributed considerably to our understanding of bonding in boryl–metal complexes [3] and have helped to clarify the mechanisms involved in transition-metal-catalyzed asymmetric boron-element additions [4,5].
2 Asymmetric B–B additions
The success of catalytic diboration is based on cleavage of the B–B bond by low-valent metal centers [6] and the kinetic lability of the resulting bis(boryl) complexes [7]. Combining these two factors guarantees firstly the activation of tetraalkoxy- and tetraaryloxydiboranes by oxidative addition (despite their relative high B–B bond energy [8]), and secondly the B–C reductive elimination to afford synthetically suitable chemo- and stereodefined organo-1,2-diborane compounds [9]. The convenience of metal-promoted 1,2-diboration versus the uncatalyzed reaction [10] has motivated researchers to find a suitable catalytic system since Miyaura et al.'s first report [11]. Several studies have focused on metal–phosphine complexes, and others have focused on base-free metal complexes [12]. The diboration of alkynes provides the syn-addition of the B–B bond to afford bis(alkenyl)diborane products through a well-established protocol based on mono-phosphine, platinum-containing catalysts [13]. However, from the mixture of products commonly observed when alkenes and diboranes are reacted in the presence of transition-metal catalysts, it seems to be a more complex transformation.
A highly selective catalyst for the exclusive diboration of alkenes [14], and therefore to avoid the disadvantage of low selectivity, would be much desirable, particularly for the subsequently developing chiral systems to induce asymmetry on the organoboron compounds.
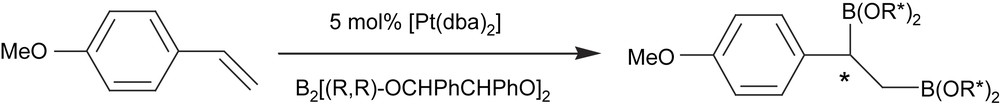
Asymmetric diboration was first achieved using chiral diboranes. Norman et al. [15] studied the addition of enantiomerically pure chiral diboron compounds such as B2[(R,R)-OCHPhCHPhO]2 to vinylarenes in the presence of [Pt(dba)2]. After 3 days of reaction at 4 °C, about 80% of diborated product was obtained with 60% of diastereomeric excess (Scheme 1).
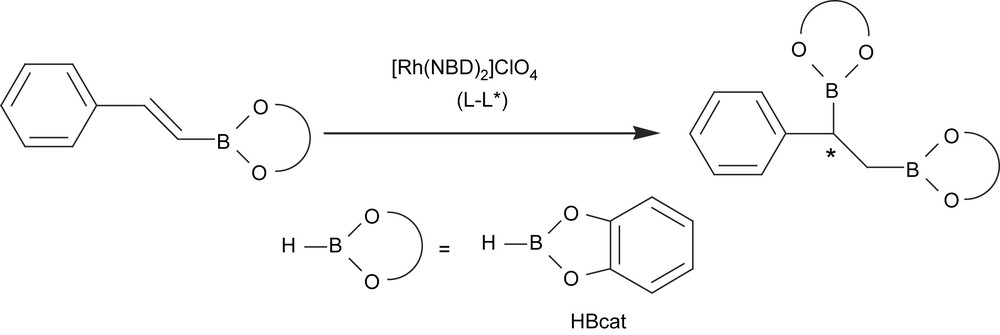
The first example of an asymmetric diboration reaction had the drawback of using stoichiometric amounts of chiral diboron reagent. Another interesting approach at that time was therefore the asymmetric version of the catalyzed regioselective hydroboration of preformed vinylboronate esters [16] (Scheme 2). When the catalytic system involved was [Rh(NBD)2]ClO4/(S,S-chiraphos), enantiomeric excesses of up to 73% were obtained, although the yield on the corresponding diborated/oxidized product was only about 13%. When the chiral diphosphine ligand (−)-diop modified the precursor [Rh(COD)2]BF4, the yield of the diol product could be increased to 87%, though the asymmetric induction was not higher than 11% of enantiomeric excess.
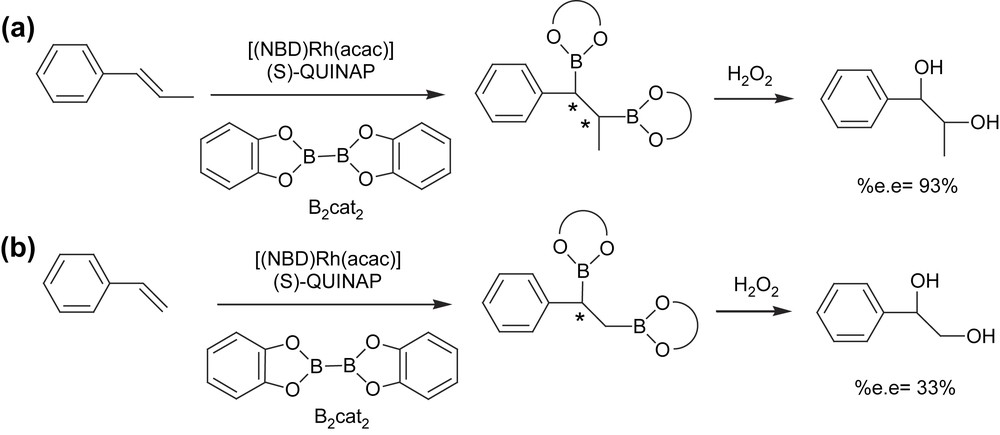
More recently, a new strategy has been developed to induce asymmetry on the diborated products through the asymmetric diboration of alkenes using a chiral phosphine–rhodium complex. Morken et al. [17] transformed trans-alkenes into 1,2-bis(catechol)ester intermediates that were eventually oxidized to the corresponding diols with moderate yield and high enantioselectivity (Scheme 3(a)). The catalytic system used was [(NBD)Rh(acac)]/(S)-QUINAP at room temperature for 24 h. However, the catalytic diboration of cis-alkenes does not appear to be as general as with the trans-substrate geometry. The authors also suggested that monosubstituted and 1,1-disubstituted alkenes will probably require new chiral ancillary ligand structures for effective enantiocontrol (Scheme 3(b)).
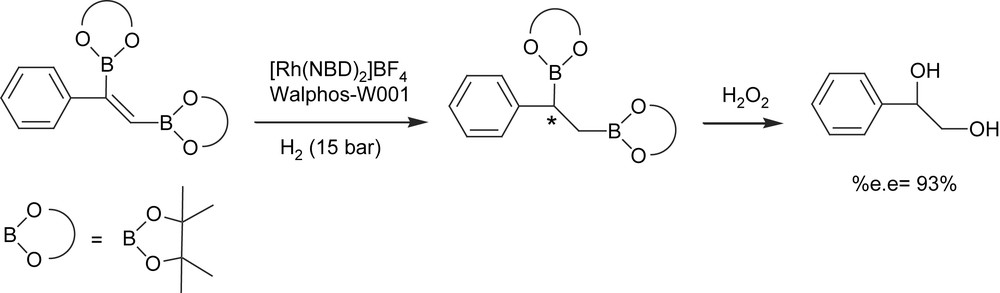
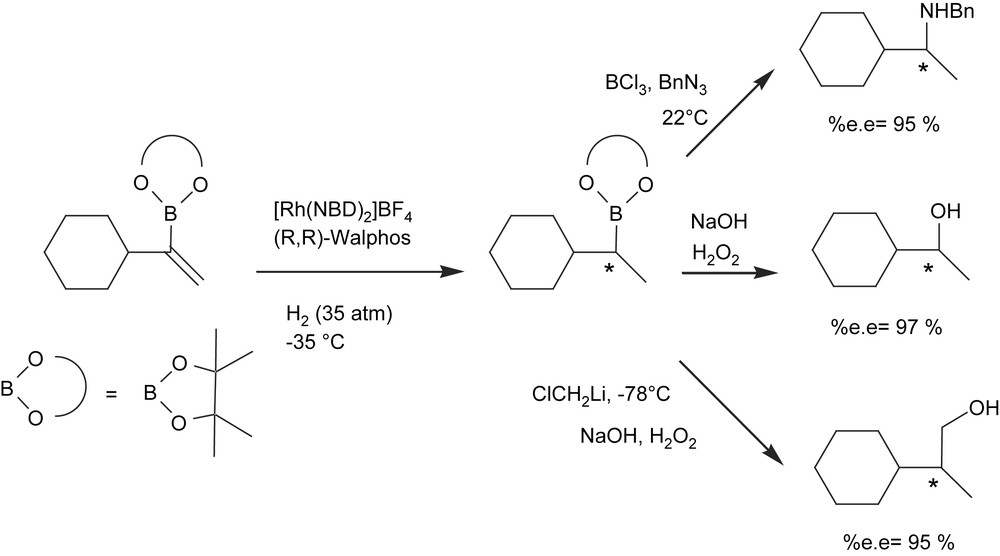
An alternate approach to enantiomerically enriched alkyl 1,2-bis(boronate)ester formation, however, is the catalytic asymmetric hydrogenation of vinyl 1,2-bis(boronate)esters. Morken and Morgan [18] developed a single-pot diboration of alkynes with [Pt(PPh3)2(ethylene)] followed by hydrogenation of the resulting 1,2-bis(boryl)alkenes with Rh(I)/chiral phosphine complex. The oxidative work-up protocol conveniently delivers 1,2-diols with high yield and high enantioselectivity (Scheme 4) with the family of Walphos ligands as the chiral ligands of choice. However, the high level of asymmetric induction is a matter of combined facts: an excess of ligand with regard to catalyst is required (Rh/ligand = 1/2) and toluene seems to be the most convenient solvent.
The effect of the chiral Walphos ligands in asymmetric C–B bond formation on the rhodium-catalyzed hydrogenation of vinylboronic esters has also been described [19] (Scheme 5). In general, high enantiomeric excesses were observed when an excess of ligand with regard to metal was used. Also, very low reaction temperatures (−35 °C) and long reaction times (12 h) were required. These are significant drawbacks compared to the well-established catalytic hydroboration protocols, which perform enantioselectively at room temperature for 1 h with only 0.5 mol% metal/ligand loading [20].
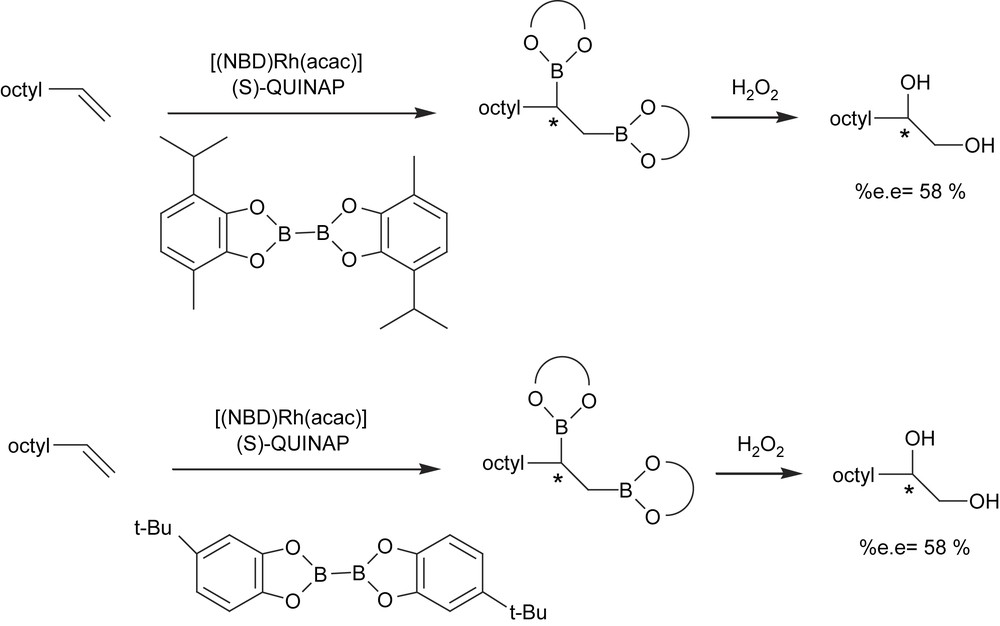
Vinylarenes are still the most challenging class of substrates to catalytically diborate in an asymmetric way. To improve stereoselection with these monosubstituted alkenes, Morken et al. [21] examined alternate diboron reagents using more hindered substituted bis(catecholato)diboron (Scheme 6). However, it seems that the substitution was too far from the metal center to have a significant impact on selectivity.
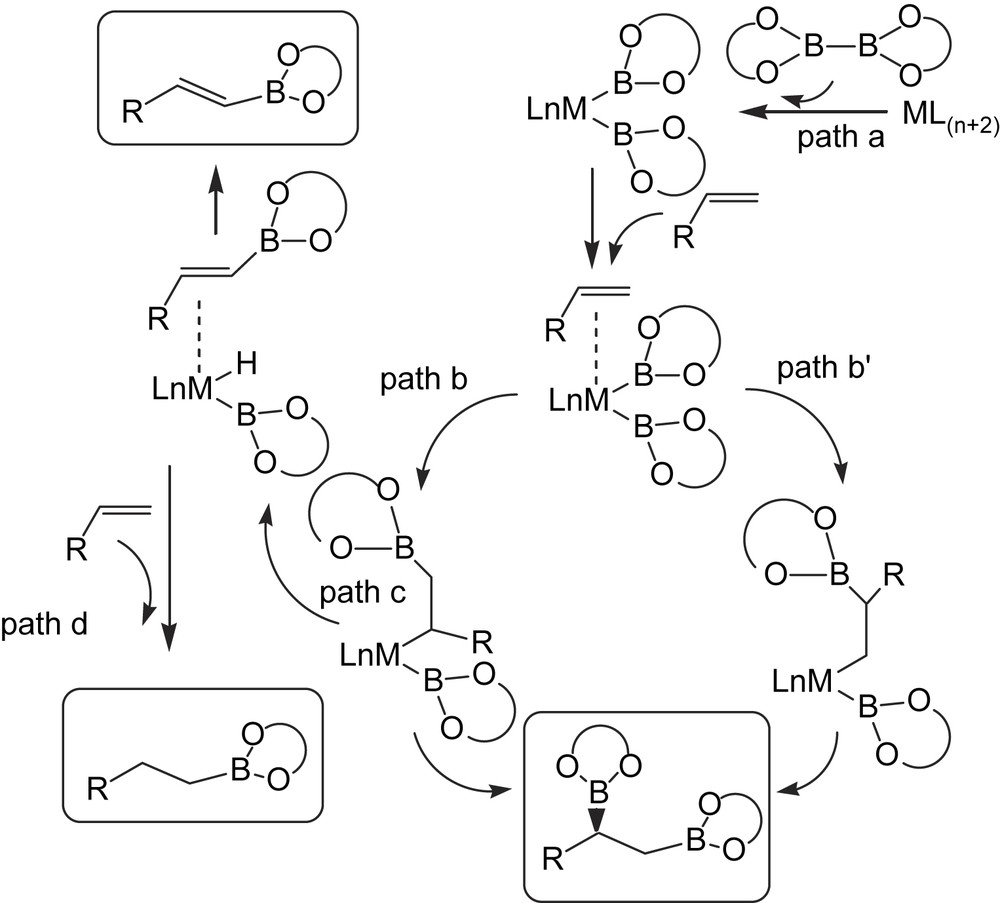
As part of a research program aimed at developing catalytic asymmetric B addition to vinylarenes in order to generate suitable chiral diboranes as intermediates in organic synthesis, we studied the nature of the side reactions and by-products produced during the process [22]. When we reproduced the catalytic asymmetric diboration of styrene with [(NBD)Rh(acac)]/(S)-QUINAP at room temperature for 15 h, we found that while the enantioselectivity was roughly as reported [17], the conversion of the substrate was almost complete but the formation of monoborated by-products became significant. In fact, these findings are in agreement with previous observations by Baker et al. [23] in which, depending on the rhodium(I) catalytic system used, a range of products from mono-, bis- and tris-boronated derivatives can be formed. A plausible explanation from a mechanistic point of view is that the first step is likely to be the oxidative addition of B–B in diborane reagent [6] to the metal, leading to metal–diboryl complex (Scheme 7, path a). However, while the desired 1,2-bis(boronate) ester seems to arise from alkene insertion into one M–B bond followed by B–C reductive elimination involving the second boryl ligand (Scheme 7, paths b–b′), the alkenyl and alkylboronate esters may be produced as a result of a competitive β-H-elimination (Scheme 7, paths c, d).
To analyze how far the relative rates of B–C reductive elimination versus β-hydride elimination are sensitive functions of the chiral ligand, new catalytic 1,2-diborations of styrene were performed with [(NBD)Rh(acac)] modified with (S)-QUINAP, R-BINAP and (S,S)-BDPP. Selectivity on the 1,2-bis(boronate) ester was significantly reduced with the ligands R-BINAP and (S,S)-BDPP (Table 1, 21% and 17%, respectively). However, not only does the size of the bite angle seem to influence the selectivity of the reaction, but also the use of (S)-QUINAP, which like (S,S)-BDPP also forms a six-membered ring with metal, provided higher selectivities towards the 1,2-diborated product (Table 1, 70–76%). QUINAP is less bulky than BINAP and BDPP in the region of the isoquinoline, which replaces one of the diphenylphosphinonaphthalene moieties. Asymmetric induction in the diborated product is also diminished by using both P,P-bidentate ligands. However, the 1,2-diborated product obtained from both the chiral complexes modified with (S)-QUINAP and (R)-BINAP provided the same enantiomeric mixture enriched with the (R)-enantiomer. This contrasts substantially with the trend observed in the hydroboration/oxidation of styrene, where the Rh/(S)-QUINAP catalytic system provided the (S)-1-phenylethanol, and the Rh/(R)-BINAP catalytic system favored the (R)-enantiomer [20]. The chemoselectivity towards the 1,2-diborated product was the most satisfactory (82%) when the electron-releasing aryl substituent was used on the styrene substrate. Noting that during alkene insertion into Rh–boryl complex, chemo- and enantioselection were dependent on alkene electronics, we reasoned that aliphatic 1-alkenes may exhibit different selectivity patterns from those of aromatic olefins. Thus, vinylcyclohexane was mainly converted into the desired 1,2-bis(boronate) ester (78%) with moderate enantiomeric excess (54%). However, the percentage of terminal hydroborated product was twice that of the branched hydroborated product.
Catalytic asymmetric 1,2-diboration/oxidation reaction of vinylarenes with bis(catecholato)diborona
| Entry | Substrate | Catalytic system | Conversion (%)b | %b (%e.e.)c | %b (%e.e.)c | %b |
| 1 | PhCHCH2 | [(NBD)Rh(acac)]/(S)-QUINAP | 90 | 76 (35R) | 24 (34S) | – |
| 2 | PhCHCH2d | [(NBD)Rh(acac)]/(S)-QUINAP | 78 | 70 (37R) | 27 (33S) | 3 |
| 3 | PhCHCH2d | [(NBD)Rh(acac)]/(R)-BINAP | 99 | 21 (21R) | 69 (3S) | 10 |
| 4 | PhCHCH2d | [(NBD)Rh(acac)]/(S,S)-BDPP | 95 | 17 (16R) | 68 (2S) | 14 |
| 5 | PhCHCH2d | [Rh(NBD)2]BF4/(S)-QUINAP | 95 | 55 (35R) | 40 (47S) | 5 |
| 6 | PhCHCH2d | [Rh(COD)2]BF4/(S)-QUINAP | 89 | 66 (36R) | 34 (41S) | – |
| 7 | PhCHCH2d | [Rh(μ-Cl)(NBD)]2/(S)-QUINAP | 92 | 68 (35R) | 25 (40S) | 7 |
| 8 | p-MeO–PhCHCH2 | [(NBD)Rh(acac)]/(S)-QUINAP | 77 | 82 (26R) | 11 (–) | 7 |
| 9 | p-F–PhCHCH2 | [(NBD)Rh(acac)]/(S)-QUINAP | 58 | 58 (20R) | 36 (45S) | 6 |
| 10 | CyCHCH2 | [(NBD)Rh(acac)]/(S)-QUINAP | 100 | 78 (54S) | 7 (ND) | 15 |
a Standard conditions: substrate/bis(catecholato)diboron/Rh complex/chiral ligand = 1/1.1/0.05/0.05; solvent: THF; T: 25 °C; t: 15 h.
b Conversion and selectivity calculated by 1H NMR.
c Enantiomeric excess determined by GC with chiral column, as derivated alcohols for monoalcohols, and derivated ketal for diol.
d Addition of 5 mol% of PPh3.
In an attempt to increase enantioselectivity during this model reaction with vinylarenes as substrates, we performed, for the first time, a double asymmetric induction on the catalytic diboration reaction, using the chiral catalytic system [(NBD)Rh(acac)]/(S)-QUINAP and chiral diborating reagents: bis(diethyl-d-tartrateglycolato)diboron, bis(diisopropyl-d-tartrateglycolato)diboron, and bis((+)-pinanediolato)diboron. As Table 2 shows, when bis(diethyl-d-tartrateglycolato)diboron and bis(diisopropyl-d-tartrateglycolato)diboron were involved in the diboration of styrene, enantiomeric excesses were only 17% and 14%, respectively. Note that in these cases the enantioenriched mixture was with the (S)-enantiomer rather than with the favored (R)-enantiomer formed with bis(catecholato)diboron. The change in the main enantiomer could be influenced by the chiral diborane reagent, as is seen by the reactivity shown when the catalytic system was not modified with (S)-QUINAP. As far as enantioselectivity is concerned, the chiral diborane reagent bis((+)-pinanediolato)diboron showed a similar behavior to that of bis(catecholato)diboron in the 1,2-diboration of p-methoxystyrene and vinylcyclohexane, although again the enantiomeric mixture was enriched with the (S)-enantiomer, and significant percentages of by-products were formed. These results are comparable to those of the platinum-catalyzed diboration of terminal alkenes with other chiral diboranes [15], though isolated yields of the 1,2-diborated product were higher when platinum rather than rhodium was the metal center of the catalytic system.
Catalytic asymmetric 1,2-diboration/oxidation reaction of vinylarenes with [(NBD)Rh(acac)]/(S)-QUINAP and chiral diboranesa
| Entry | Substrate | Diborane reagent | Conversion (%)b | %b (%e.e.)c | %b (%e.e.)c | %b |
| 1 | PhCHCH2 | Bis(neopentylglycolato)diboron | 95 | 39 (–) | 61 (–) | – |
| 2 | PhCHCH2 | Bis(hexyleneglycolato)diboron | 36 | 22 (–) | 78 (5S) | – |
| 3 | PhCHCH2 | Bis(diethyl-d-tartrateglycolato)diboron | 60 | 17 (17S) | 83 (–) | – |
| 4 | PhCHCH2 | Bis(diisopropyl-d-tartrateglycolato)diboron | 100 | 20 (14S) | 80 (–) | – |
| 5d | PhCHCH2 | Bis(diisopropyl-d-tartrateglycolato)diboron | 50 | 12 (23S) | 88 (–) | – |
| 6 | p-MeO–PhCHCH2 | Bis((+)-pinanediolato)diboron | 80 | 36 (14S) | 64 (–) | – |
| 7d | p-MeO–PhCHCH2 | Bis((+)-pinanediolato)diboron | 100 | 25 (15S) | 75 (ND) | – |
| 8 | CyCHCH2 | Bis((+)-pinanediolato)diboron | 100 | 32 (50R) | 3 (–) | 65 |
a Standard conditions: substrate/bis(organo)diboron/Rh complex/chiral ligand = 1/1.1/0.05/0.05; solvent: THF; T: 25 °C; t: 15 h.
b Conversion and selectivity calculated by 1H NMR.
c Enantiomeric excess determined by GC with chiral column, as derivated alcohols for monoalcohols, and derivated ketal for diol.
d Catalytic system based on [(NBD)Rh(acac)]. ND: not determined.
To circumvent the problem of by-product formation in the catalytic diboration reaction, Baker et al. first looked at the right-hand side of the periodic table, where metals with lower d-orbital energies could inhibit alkene–hydride–metal formation through β-H-elimination from alkyl–boryl–metal intermediates, due to the less favored π-backbonding. They chose a gold(I) catalytic system made up of commercially available [Au(PEt3)Cl]/1,2-bis(dicyclohexylphosphino)ethane and exclusive formation of the desired 1,2-bis(boronate)ester was observed [23] from styrene and bis(catecholato)diboron as a model reaction. However, temperatures of 80 °C and long reaction times (48 h) were required. Inspired by this preliminary experiment and motivated to perform enantioselective clean catalytic diboration, we developed a synthetic, structural and catalytic application study into chiral diphosphine–Au(I)–X complexes. These complexes were synthesized from a solution of (dimethylsulfide)–gold(I) chloride in tetrahydrofuran by treatment with the chiral phosphine (R)-BINAP, at room temperature. After removing all volatile components, the complex (R)-BINAP(AuCl)2 was crystallized and isolated (Scheme 8). The chloride derivative could be easily converted into the corresponding iodide complex (R)-BINAP(AuI)2, by metathesis with potassium iodide in tetrahydrofuran [24] (Scheme 8).
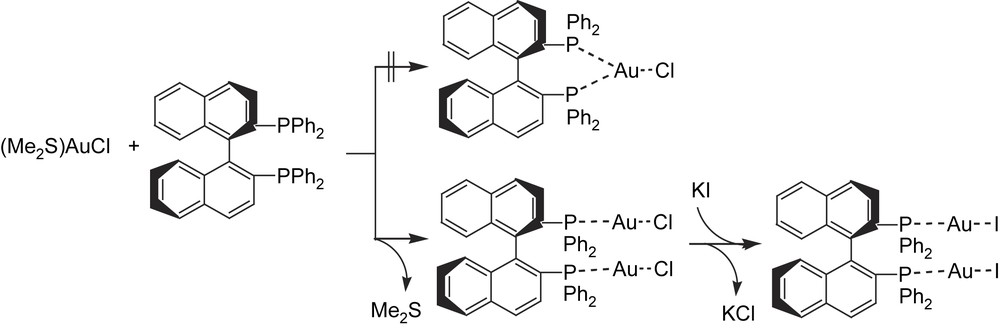
Compounds (R)-BINAP(AuCl)2 and (R)-BINAP(AuI)2 were tested in the diboration of styrene, with bis(catecholato)diboron. For a catalyst loading of 5 mol%, under standard reaction conditions (room temperature in THF), no conversion was observed after 60 h. When the reaction was carried out under reflux, we achieved a very low conversion (7%) into the desired product, though no asymmetric induction was detected. The low activity of the phosphane–gold catalyst precursors could be related to the instability of the complexes under the reaction conditions exposed in a plausible Au(I)/Au(III) mechanism.
In this context, we then turned our attention to an alternative type of ligands, such as N-heterocyclic carbenes (NHCs). NHCs are considered to be better electron donors than phosphines and therefore provide enough electron richness to the metal center to guarantee B–B cleavage of the diborane. Because of the great stability acquired by the metal complexes, N-heterocyclic carbenes have also emerged as a promising family of ligands for the design of efficient homogeneous catalysts [25]. In collaboration with Peris et al., our first attempt to perform the catalytic diboration of alkenes with metal–N-heterocyclic carbene complexes was carried out with the thermally- and air-stable compound [(mentimid)2Ag]AgCl2, (mentimid = 1-methyl-3-(+)-methylmenthoxide imidazolium chloride) [26] (Scheme 9). Among the terminal alkenes we studied from the point of view of catalytic B–B addition, we observed that diboration/oxidation to styrene with [(mentimid)2Ag]AgCl2 provided the single product 1-phenyl-1,2-ethanediol with 76% of conversion (Table 3). The electron-accepting substituted vinylarenes proved to be less reactive than styrene. On the other hand, the more electron-rich vinylcyclohexane was by far the most active substrate in terms of conversion into the corresponding diol. A similar trend has been described in the diboration of para-substituted terminal phenylacetylenes, with Pt(0) catalytic complexes, where electron-withdrawing substituents diminish the reaction rates [13]. Remarkably, the hindered internal alkene indene was cleanly transformed into the desired product, though conversion was low. Note that we performed the same reaction with analogous Ag(I) complexes modified with phosphines (and diphosphines) [27], but they did not allow any conversion of the alkenes towards the desired diborated products, under the same reactions conditions. Even AgNO3 was tested in this reaction to study the possible participation of the ion Ag+ as the catalytic species, but no reactivity was monitored.
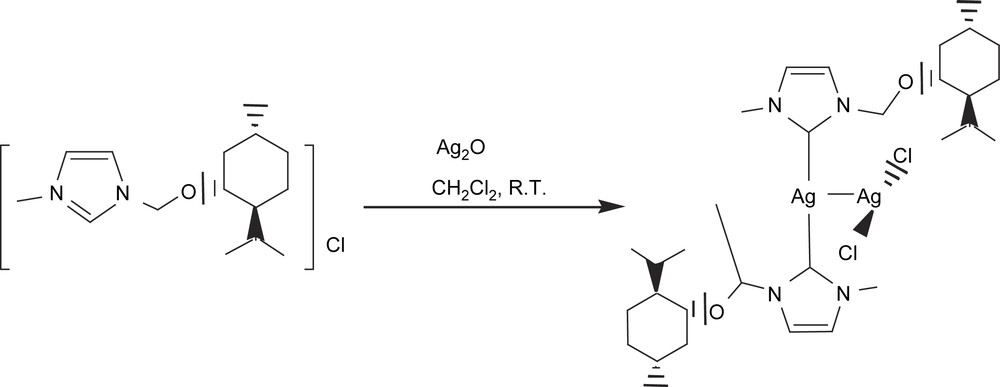
[(mentimid)2Ag]AgCl2 catalyzed diboration/oxidation of alkenes with bis(catecholato)diborona
| Entry | Substrate | Product | Conversion (%)b | Diol (%)c |
| 1 | 76 | 100 | ||
| 2d | 32 | 100 | ||
| 3 | 12 | 100 | ||
| 4 | 14 | 100 | ||
| 5 | 10 | 100 | ||
| 6 | 90 | 100 | ||
| 7 | 20 | 100 |
a Standard conditions: alkene/diborane/Ag complex = 0.5/0.55/0.025; solvent: THF; T: 25 °C, t: 60 h.
b Determined by 1H NMR before oxidation.
c Determined by GC with chiral column FS-Cyclodex B-IP, 50 m × 0.25 mm of the ketal derivatives.
d Reflux at 70 °C, 20 h.
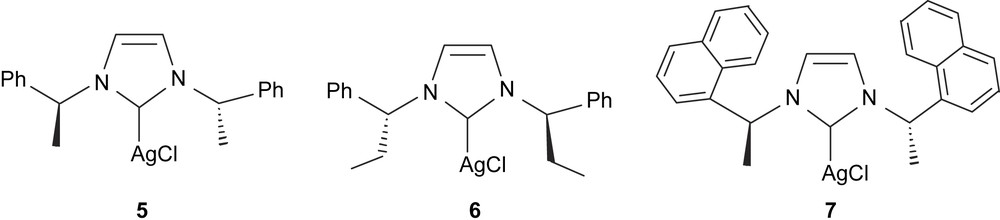
Despite using the chiral imidazolylidene ligand, we observed no asymmetric induction on the reaction products, presumably because the chiral center is far from the metal. Also, the lability of the Ag–Ccarbene bond could favor some equilibrium between [(mentimid)2Ag]AgCl2 and [(mentimid)AgCl] [28], and thus makes it difficult to establish what the real catalytic species is. Alternatively, using chiral diboranes such as bis(diethyl-d-tartrateglycolato)diboron and bis(diisopropyl-d-tartrateglycolato)diboron and bis((+)-pinanediolato)diboron resulted in a totally inhibited diboration reaction, making bis(catecholato)diboron the specific diborating reagent for this reaction with [(mentimid)2Ag]AgCl2. To study whether we could find effective diboration catalysts that could also induce some asymmetry, we decided to test a series of chiral NHC–Ag–Cl complexes (Fig. 1, complexes 5–7) that have previously been described in the literature [29]. These compounds were tested on the catalytic diboration/oxidation of styrene [30] (Table 4, entries 7, 8, 9 and 13) and 6 was tested in the diboration of a series of terminal olefins (entries 10–12). As shown for the catalysts described above, the diborated product is still the main product, though conversion is lower, probably because of the more sterically hindered catalytic system involved. Interestingly, 6 was able to modestly induce some enantioselectivity, with e.e. values between 4% and 9%. We believe this result may be due to the higher sterical crowding of the metal in 6 compared to other chiral catalysts. We are aware that these e.e. values are very low. However, they should be compared to the maximum values (33%) achieved by the sophisticated P,N-QUINAP–Rh(I) complex, and so we truly believe that chiral NHC–M(I)–X complexes may be a useful alternative for the clean asymmetric catalytic diboration of alkenes. We also tested compound 6 in the diboration of vinylcyclohexane at 70 °C, and observed quantitative conversion, but negligible asymmetric induction (entry 10). From the diboration of p-fluorostyrene and p-methoxystyrene, (entries 11 and 12), we established that the more electron-attracting the substituents are, the higher is the conversion to the diborated species.
Catalytic diboration/oxidation reaction of alkenes with M–NHC complexes and bis(catecholato)diborona
| Entry | Catalytic system | Alkene | T (h) | Temperature (°C) | Conversion (%)b | Diol (%) |
| 1 | 2 | Styrene | 60 | 25 | 46 | 100 |
| 2 | 4 | Styrene | 60 | 25 | 94 | 100 |
| 3 | 1 | Styrene | 60 | 25 | 76 | 100 |
| 4 | 3 | Styrene | 60 | 25 | 69 | 100 |
| 5 | 3 | Styrene | 20 | 70 | 86 | 100 |
| 6 | 3 | Vinylcyclohexane | 60 | 25 | 40 | 100 |
| 7 | 5 | Styrene | 60 | 25 | 40 | 100 |
| 8 | 6 | Styrene | 60 | 25 | 13 | 91c,d |
| 9 | 6 | Styrene | 24 | 70 | 42 | 100c,e |
| 10 | 6 | Vinylcyclohexane | 24 | 70 | 100 | 100 |
| 11 | 6 | p-Fluorostyrene | 24 | 70 | 51 | 100 |
| 12 | 6 | p-Methoxystyrene | 24 | 70 | 13 | 100 |
| 13 | 7 | Styrene | 60 | 25 | 10 | 93 |
a Standard conditions: alkene/diborane/M–carbene complex = 0.5/0.55/0.025; solvent: THF.
b Determined by 1H NMR before oxidation.
c Determined by GC with chiral column FS-Cyclodex B-IP, 50 m × 0.25 mm of the ketal derivatives.
d e.e = 9%.
e e.e = 4%.
Finally, we also made an exploratory study of the analogous Au(I)–(NHC) complexes, [NHC–Au(I)–NHC]X (Scheme 10). The Au(I) complexes were obtained in high yield (ca. 90%) by in situ generation of the free carbenes from the corresponding imidazolium salts in THF at −78 °C with t-BuLi, and then addition of Au(PEt3)Cl. Alternatively, 3 and 4 were obtained from the transmetallation of the corresponding carbenes from the silver complexes 1 and 2 to Au(PEt3)Cl, though yields were lower (ca. 50%) than those obtained by direct coordination of the free carbene to Au. Introducing the chlorinated-NHC ligands would also allow us to compare the catalytic implications of the reduction in the electron-donating character of the NHC ligand.
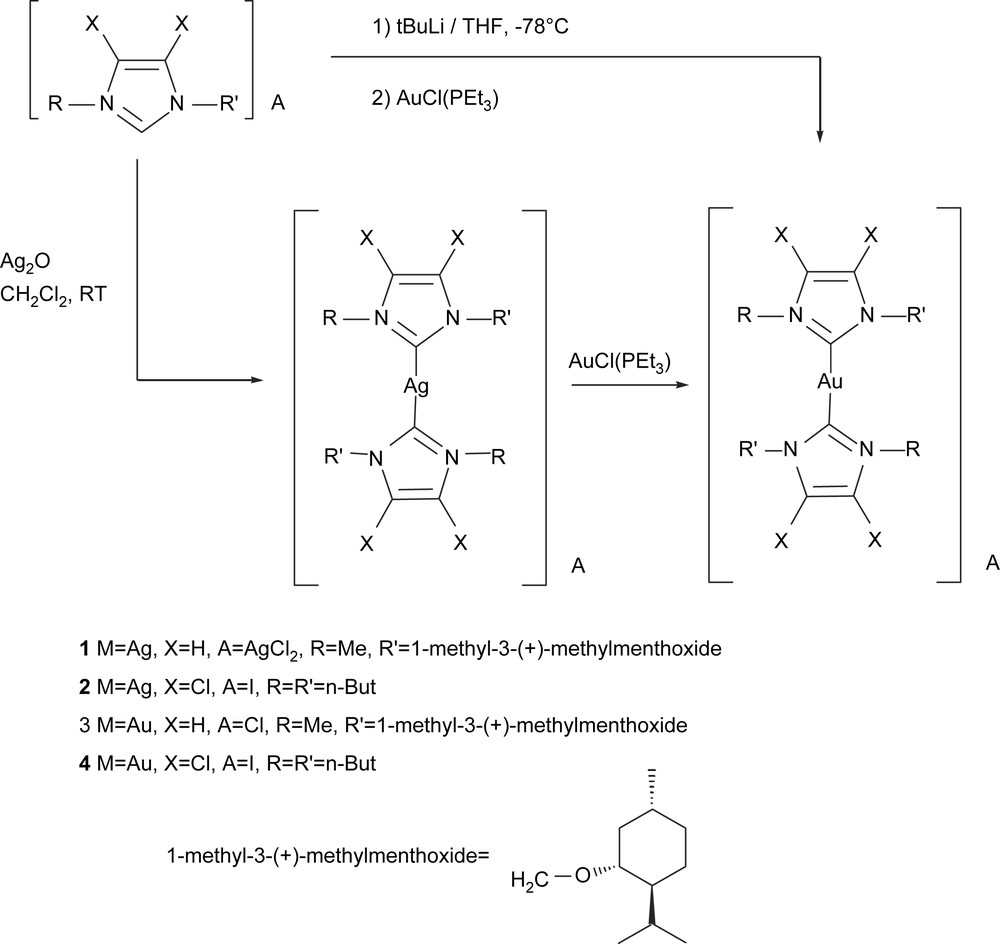
Initially we examined the catalytic activity provided by compounds [NHC–Au(I)–NHC]X. Metal modification with an electron-donating NHC ligand clearly enhanced the catalytic performances, which supports the findings of Baker et al. [23], which suggest that arylphosphine Au complexes are not electron-enriching enough to serve as competent alkene diboration catalysts. We achieved 100% selectivity on the diborated product in the catalyzed diboration of styrene, when 4 converted 94% of the starting material (Table 4). For the chiral complexes 3, total chemoselectivity on the diborated product was observed with moderate to high conversions when the reaction was carried out at 70 °C, though conversions were lower when the catalytic diboration was conducted with more sterically hindered substrates, such as vinylcyclohexane. However, no asymmetric induction was observed on the corresponding diols, after the oxidation procedure.
To develop both active and selective catalysts for this transformation, we, in collaboration with Perez et al., turned our attention to copper, which is the remaining group-11 metal still not known to catalyze the diboration of alkenes. We studied the potential of several complexes containing the (NHC)Cu core as the catalyst for this reaction (Fig. 2) [31] given the success that Cu complexes have displayed in related catalytic transformations [32]. We found that the complexes [(NHC)Cu(NCMe)]BF4 efficiently catalyze the diboration of styrene, when bis(catechol)diboron is used as the boryl source, with a very high degree of conversion. With the appropriate NHC ligand, the reaction proceeded quantitatively towards the diborated derivative PhCH(Bcat)–CH2(Bcat). Note that this new catalytic system based on Cu(I) has two advantages: the metal used is inexpensive and the tunability of the ligand allows the development of the asymmetric version of the catalytic reaction.

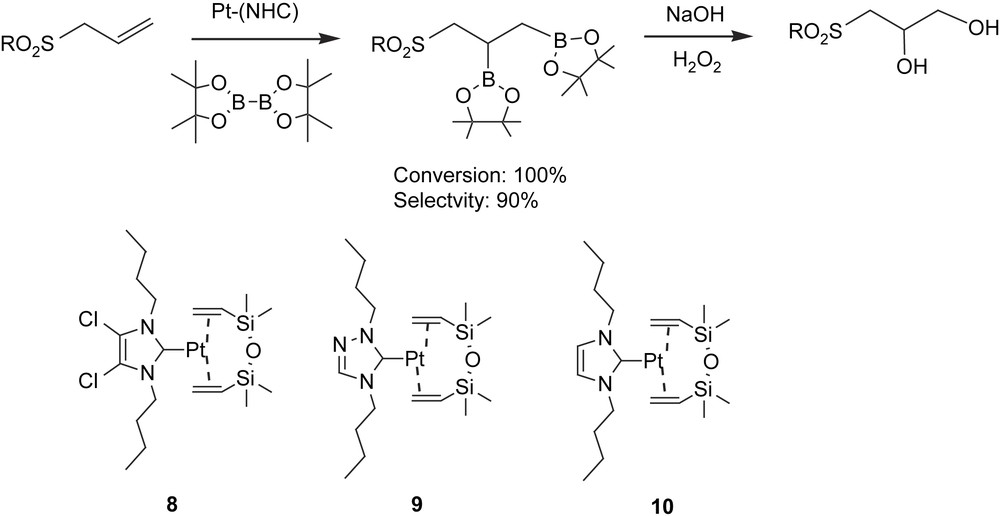
We are currently working on an unprecedented catalytic B–B addition to heteroatom-containing substrates providing total conversion on organosulfur–diboronate esters [33]. Interest in the catalytic diboration of aryl allylic sulfones lies in achieving, after oxidation work-up, an alternative method to obtain 1,2-dihydroxysulfones as valuable intermediates for functionalised cyclic ethers and lactones (Scheme 11) [34]. We observed, for the first time, that Pt–NHC complexes were able to catalyze the 1,2-diboration reaction of alkenes and alkynes. Thus, platinum–carbene-catalyzed diboration improved the B–B addition described with Pt(PPh3)4 [13a] and [Pt(PPh3)2(C2H4)] [13b] and provided an alternative to the more classical base-free platinum [14a,b,d] and mono(phosphine)platinum complexes [13e], though the latter provided slightly faster performances. In fact, heteroatom-containing substrates such as aryl allylic sulfones have been hydroborated in a symmetric [35] and an asymmetric [36] catalytic way to provide, after in situ oxidation, the branched alcohol as a consequence of the directing effects of the sulfone functional group. This electron-attracting terminal group in the allylic system facilitated the simultaneous B–B catalytic addition on the CC bond without any undesired by-product formation due to the isomerisation of the double bond. As Table 5 shows, bis(pinacolato)diboron was added quantitatively under optimized conditions (toluene, 80 °C, 16 h), in the presence of complexes 8–10, and the catalytic system 8 was the most suitable one for this transformation. However, monoalkylboranes were observed in lower percentages as a subsequent β-H elimination competitive pathway. Interestingly, the aryl allylic sulfones were unable to be diborated with metal–phosphine catalytic systems based on rhodium or platinum complexes. An excess of diborane led to total conversion even with electron donor or electron-withdrawing moieties in the substrate. This work opened up a new perspective for future applications of metal-mediated synthesis towards chiral non-racemic 1,2-dihydroxysulfones when asymmetry can be induced through the chiral N-heterocyclic carbenes, as an alternative to the Sharpless asymmetric dihydroxylation method or the hydrolytic resolution of epoxysulfones with Jacobsen's catalyst.
1,2-Diboration of aryl allylic sulfones with Pt(0)–NHCa
| Pt(0)–NHC | Substrate | Aryl allylic sulfone/diborane | Conversion (%)b | Bis(boryl) Alkane sulfone (%)b |
| 8 | PhSO2CH2CHCH2 | 1/1.1 | 90 | 75 |
| 9 | PhSO2CH2CHCH2 | 1/1.1 | 69 | 71 |
| 10 | PhSO2CH2CHCH2 | 1/1.1 | 87 | 85 |
| 8 | PhSO2CH2CHCH2 | 1/2 | 100 | 90 |
| 8 | p(Me)PhSO2CH2CHCH2 | 1/2 | 100 | 91 |
| 8 | p(Cl)PhSO2CH2CHCH2 | 1/2 | 100 | 94 |
a Standard conditions: 5 mol% of Pt(0)–NHC catalytic system, diborane: bis(pinacolato)diboron (B2(cat)2); aryl allylic sulfone/diborane = 1/1.1; solvent: toluene; T: 80 °C; reaction time: 16 h.
b Conversion and selectivity based on 1H NMR.
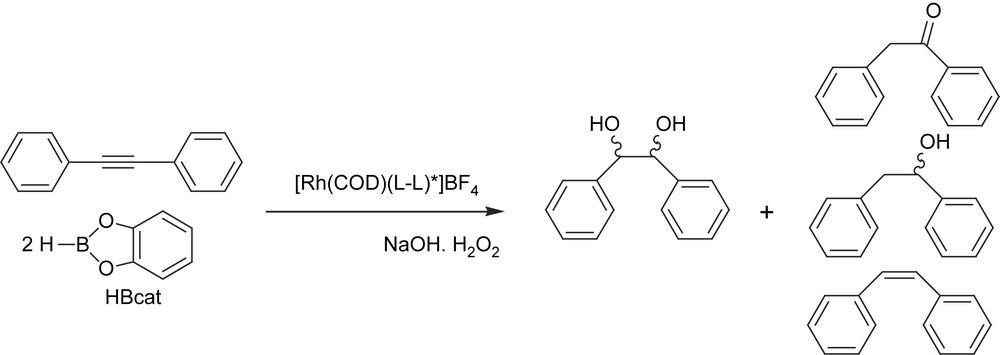
As an alternative to the catalytic diboration synthetic method, we also performed the dihydroboration/oxidation of internal alkynes with catecholborane in the presence of the rhodium catalytic system modified with (S)-QUINAP, in order to explore the viability of the chiral 1,2-diol adduct formation (Scheme 12). In particular, we focused on the synthesis of the hydrobenzoin-type molecule, because the enantiomerically pure hydrobenzoin has proved to be a very useful chiral auxiliary [37] and ligand [38], for stereoselective organic synthesis. These diols, which were previously accessible only through kinetic resolution [39], can now be obtained by the dihydroxylation of olefins [40], the reduction of benzyls [41] or via carbon–carbon bond formation [42]. However, to the best of our knowledge, nobody has yet studied the enantioselective synthesis of hydrobenzoin through the catalytic asymmetric dihydroboration/oxidation of diphenylacetylene with boranes as hydroboration reagents. The chiral complex [Rh(cod)(S,S)-BDPP]BF4 with catecholborane provided about 98% conversion and 68% selectivity on the desired diphenyl-1,2-ethanediol (hydrobenzoin). Other by-products were observed as a result of competitive catalytic reactions such as hydrogenation. However, the diphenyl-1,2-ethanol was characterized mainly as the erythro-compound (96:4), not the desired threo. Unfortunately, even modifying the rhodium complex with other chiral ligands, such as QUINAP and BINAP, did not change this trend towards the formation of the erythro-compound.
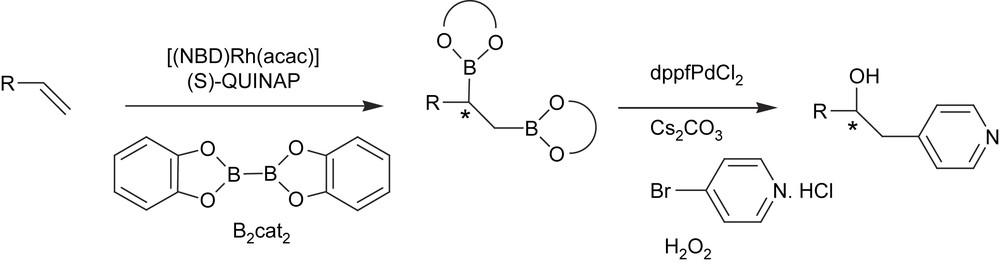
The usefulness of the catalytic asymmetric diboration has been demonstrated in the carbohydroxylation of alkenes by a tandem diboration/Suzuki cross-coupling/oxidation reaction [43]. Chiral non-symmetric 1,2-diboron adducts were therefore generated by [(NBD)Rh(acac)]/(S)-QUINAP and bis(catechol)borane and then reacted in situ with aryl halides. The less hindered C–B bond participated in cross-coupling and the remaining asymmetric C–B bond was then oxidized to provide net asymmetric carbohydroxylation (Scheme 13).
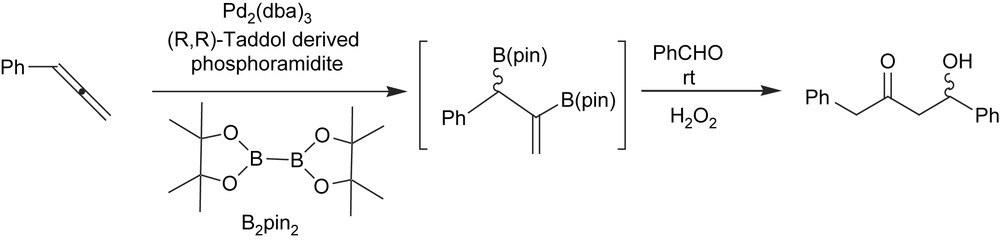
To expand the scope of the catalytic asymmetric diboration reaction, Morken et al. also studied the enantioselective C–B formation from prochiral allenes [44]. The corresponding products are considered highly versatile intermediates in asymmetric synthesis because they contain both allylboronate and vinylboronate functionality. Chiral-rhodium-based catalysts suffered from a lack of activity and selectivity. Bearing in mind the racemic platinum-catalyzed allene diboration described by Miyaura et al. [45], Morken et al. concentrated on developing a catalytic system based on palladium complexes modified with Lewis basic ligands. As far as ligands were concerned, Taddol-derived phosphoramidite afforded moderate yields and high levels of asymmetric induction for a number of prochiral monosubstituted allene substrates (Scheme 14).
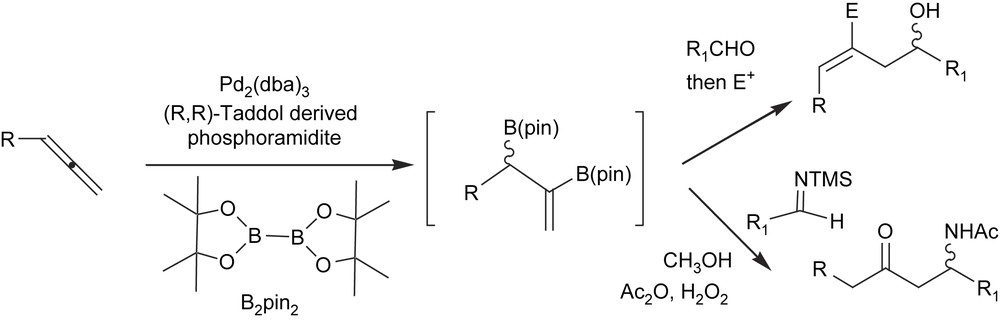
The chiral allylboron formed are effective intermediates for concatenated allylation/functionalisation sequences such as the allylation of carbonyls [46] or aminoallylation [47] (Scheme 15).
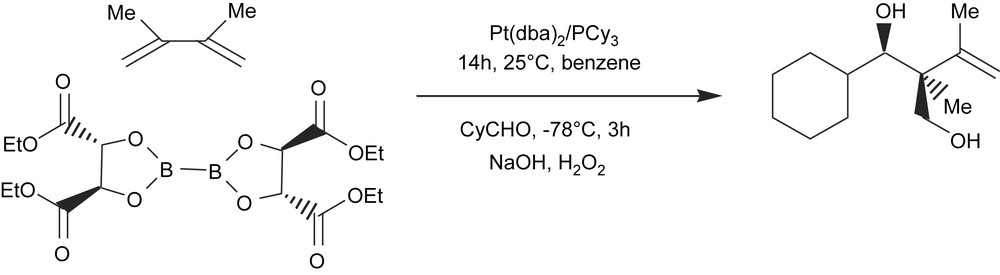
With appropriate substituted diene substrates, the allylmetalation reaction may allow higher product distribution and therefore allow access to quaternary stereocenters. Morken and Morgan [48] have also described a single-pot tandem catalytic diene diboration/carbonyl allylation reaction with chiral diboron reagents. The chirality of the intermediate diboration adduct is transferred through the carbonyl allylation reaction, thus providing access to the stereodefined quaternary carbon center (Scheme 16).
3 Conclusions
Suitable chiral diboranes as intermediates in organic synthesis can be generated to some extent using different strategies: (a) the addition of chiral diboron reagents to alkenes with an achiral catalytic system, (b) the asymmetric version of the catalyzed regioselective hydroboration of preformed vinylboronate esters, (c) the catalytic asymmetric hydrogenation of vinyl 1,2-bis(boronate)esters, (d) the asymmetric diboration of alkenes using chiral metal complexes, (e) double asymmetric induction using a chiral catalytic system and chiral diborating reagents, and (f) the dihydroboration of internal alkynes in the presence of the chiral metal complexes. All of these are valuable synthetic procedures towards the functionalization of organic molecules through B–H or B–B compounds, but the success of the protocol is due to the metal–boryl intermediates involved [3]. Therefore, by correctly choosing the catalytic system and the boryl source, versatile synthetic intermediates with a useful level of stereoisomeric purity can be accessed.
Acknowledgments
We wish to thank the MEC for funding (CTQ2004-04412BQU) and the FPI and the Generalitat de Catalunya for research studentships.