

1 Ynamides: a brief introduction
Ynamides 1, stable surrogates of the more labile ynamines [1], have emerged as remarkably useful building blocks during the last two decades [2]. The nitrogen atom directly connected to the alkyne strongly activates and polarizes the triple bond, whereas the electron-withdrawing group ensures the stability of these building blocks and can also act as a directing group and/or a chiral auxiliary (Fig. 1a). Various classes of electron-deficient ynamines have been developed based on the nature of this electron-withdrawing group, which can be an amide, a carbamate, urea, an imide [3], a sulfonamide, a phosphoramidate [4], a phosphorodiamidate [4] or a hydrazide [5] (Fig. 1b), all corresponding stabilized nitrogen-substituted alkynes falling under the general denomination “ynamides”, which is therefore not restricted anymore to N-alkynyl-amides. Other stable equivalents of ynamines in which the delocalization of the nitrogen lone pair into the alkyne is also restricted because of electronic or geometrical reasons have been reported more recently: they include N-alkynylated N-containing arenes, ynimines [6], yne-sulfoximines [7], yne-sulfonimidamide [8] and N-alkynyl-triazenes [9].
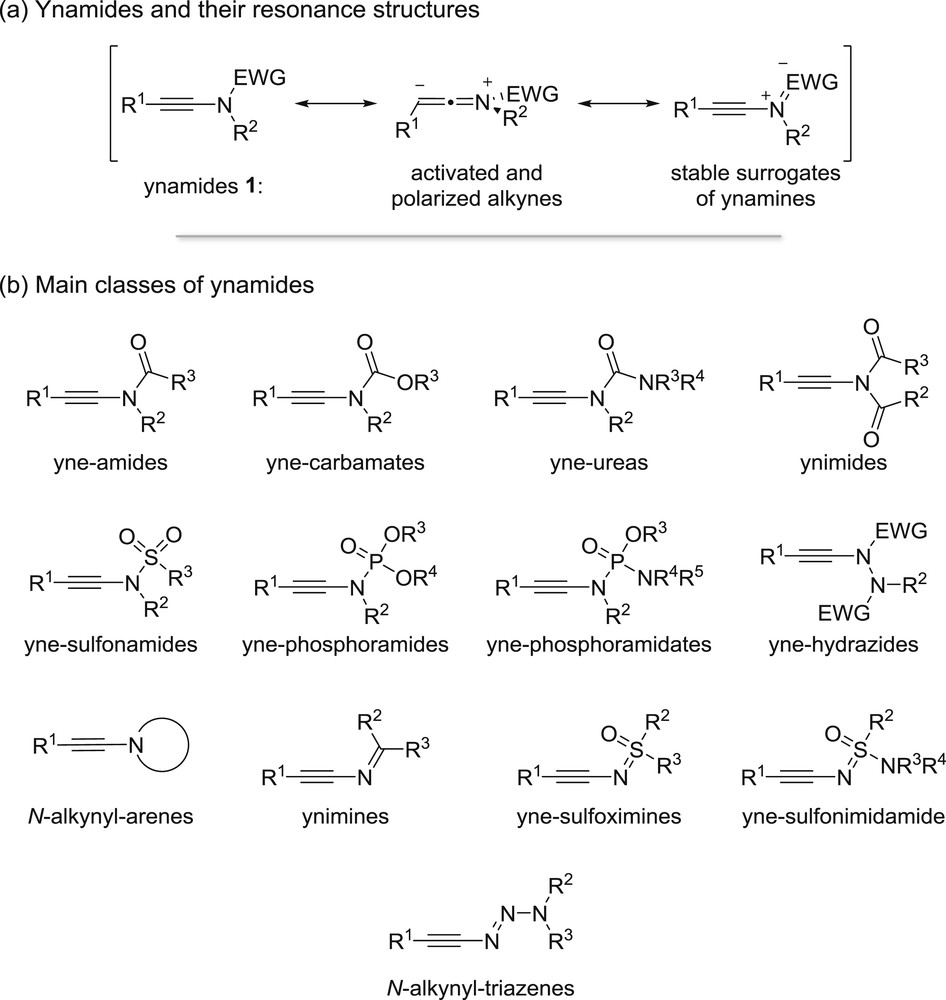
Ynamides: resonance structures and main classes.
The key to the success met by these building blocks was the development of efficient, reliable and broadly applicable methods for their preparation. They can indeed be prepared, on multigram scales, by an array of synthetic methods [10] mostly based on the direct alkynylation of nitrogen nucleophiles with hypervalent alkynyliodonium salts [11], copper acetylides [12], trichloroethylene [13] or copper-catalysed alkynylation with bromoalkynes [14], terminal alkynes [15], gem-dibromoalkenes [16], propiolic acids [17] and potassium alkynyltrifluoroborates [18]. Some of them being now even commercially available, their chemistry was extensively studied, which resulted in the design of an ever-growing number of original methods based on their unique reactivity. Over the years, they have indeed been shown to be remarkable starting materials in a broad range of transformations, which have been recently reviewed [2,19]. They have also been shown to be ideal substrates in metal-mediated [20], pericyclic [21], radical [22], cationic [23] and anionic chemistry, their use in anionic processes being the core of this article.
The enthusiasm generated by the use of ynamides in various anionic processes is actually easily understandable and stands in the nature itself of these reagents, which possess various sites that can be used for the development of anionic reactions. These include the polarized alkyne, which can be used in carbometallation and related processes, the electron-withdrawing group, which can control their regioselectivity and can also be used, depending on its nature, for nucleophilic additions or metallations initiating further transformations, as well as the propargylic position or the acetylenic position in terminal ynamides, which possesses acidic protons (Fig. 2). Although the propargylic metallation of ynamides has not been reported to date, the polarization of the alkyne and the presence/reactivity of the electron-withdrawing group have been exploited for the development of new reactions based on the anionic reactivity of ynamides. These reactions will be overviewed in this review article, which will cover the “anionic” chemistry of ynamides in a broad sense (reactions such as carbopalladations and even some radical processes have been incorporated, mostly for comparison purposes) and they will be classified according to the type of anionic transformation (carbometallation, nucleophilic addition and deprotonation) and its nature (inter- or intra-molecular), starting with processes based on the intermolecular carbometallation of ynamides and related reactions, which will be covered in the following section.
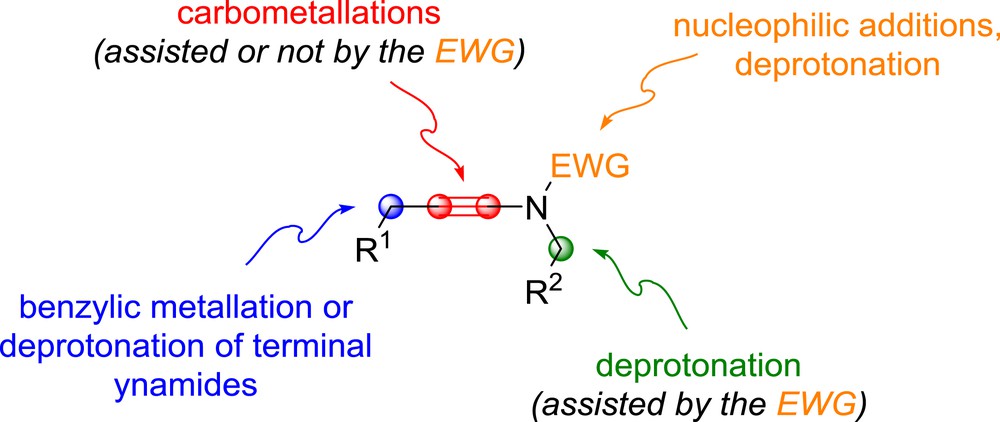
Ynamides: ideal substrates for anionic chemistry.
2 Intermolecular carbometallation of ynamides and related processes: useful entries to polysubstituted enamides and new approaches in asymmetric synthesis
2.1 Intermolecular carbometallation of ynamides and related processes: a straightforward and regioselective entry to highly substituted enamides
The carbometallation reaction of ynamides is probably one of the best examples illustrating that ynamides are more than stable surrogates of the more labile ynamines [24]. Indeed, although the carbocupration of ynamines 2 was shown in the early 1970s to strictly follow the inherent polarization of the triple bond affording a transient β-amino-vinylcopper 3, which was then reacted with a series of electrophiles to provide enamines 4 (Scheme 1a) [25], Marek and colleagues demonstrated some 30 years later that the presence of the electron-withdrawing group in ynamides 1 overcomes the polarization of the alkyne, the carbocupration being now directed by chelation of copper by the electron-withdrawing group to afford an intermediate α-amino-vinylcopper 5, which upon trapping with a series of electrophiles then affords enamides 6 resulting from a syn-carbocupration (Scheme 1b) [26]. As a note, the synthesis of the organocopper/cuprate reagents is not strictly required, because a copper-catalysed carbomagnesiation was also shown to be operative.
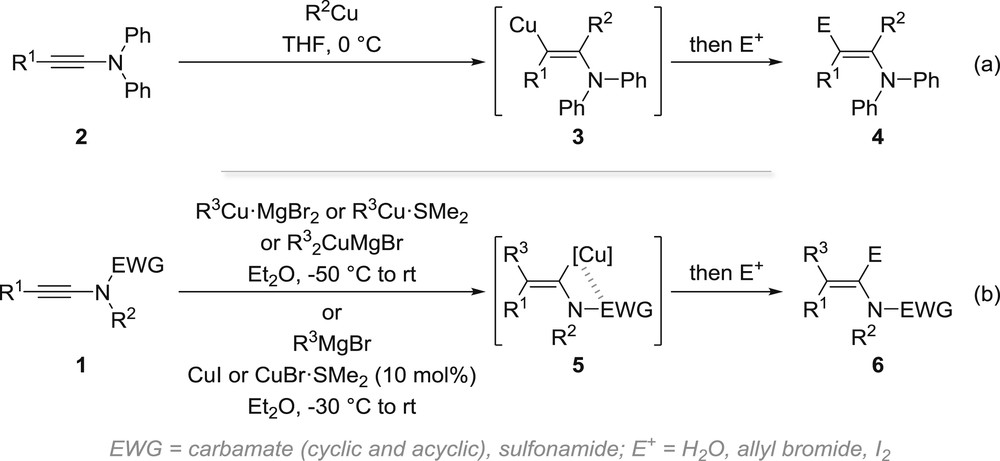
Carbocupration and copper-catalysed carbomagnesiation of ynamines and ynamides: inherent polarization of the alkyne vs chelation with the electron-withdrawing group.
Besides organocopper, organocuprates and Grignard reagents, organozinc halides and diorganozincs can also be used for the carbometallation of ynamides, the main advantage being the high functional group tolerance of these reagents. Organozinc derivatives being unable to perform the carbozincation of ynamides themselves, a catalyst is required in this case, as demonstrated by Lam and colleagues who reported in 2009 the stereoselective synthesis of multisubstituted enamides 6 via a rhodium-catalysed carbozincation of ynamides 1 [27]. In the presence of 5 mol % of Rh(cod)(acac), a series of organozinc halides and diorganozincs could be smoothly added across the triple bond of yne-lactams, yne-oxazolidinones and yne-imidazolidinones with high levels of regio- and stereoselectivity, providing fully substituted enamides 6 after further reaction with an electrophile (Scheme 2a). In addition, intermediate vinylzinc 7 can participate in a Negishi cross-coupling, affording conjugated enamides 8 (Scheme 2b). Although acyclic ynamides such as yne-carbamates or yne-sulfonamides failed to undergo the carbozincation, an interesting observation was made upon investigating the effect of the ligand. Indeed, the use of diethylzinc in the presence of 2.5 mol % of [Rh(cod)Cl]2 and 10 mol % of tris-(2-furyl)phosphine completely altered the reaction course to favour a clean hydrozincation yielding Z-enamides 9 (Scheme 2c) [27b]. The carbozincation could be further extended to the preparation of α,β-dehydroamino acids 10, which could be readily obtained by a copper-catalysed carbozincation with dialkylzinc reagents under an atmosphere of carbon dioxide, the former being chemically compatible with the latter (Scheme 2d) [28]. Compared with other methods available for the preparation of α,β-dehydroamino acids, which often suffer from a lack of stereoselectivity, this formal alkylative carboxylation of ynamides is clearly an excellent alternative.
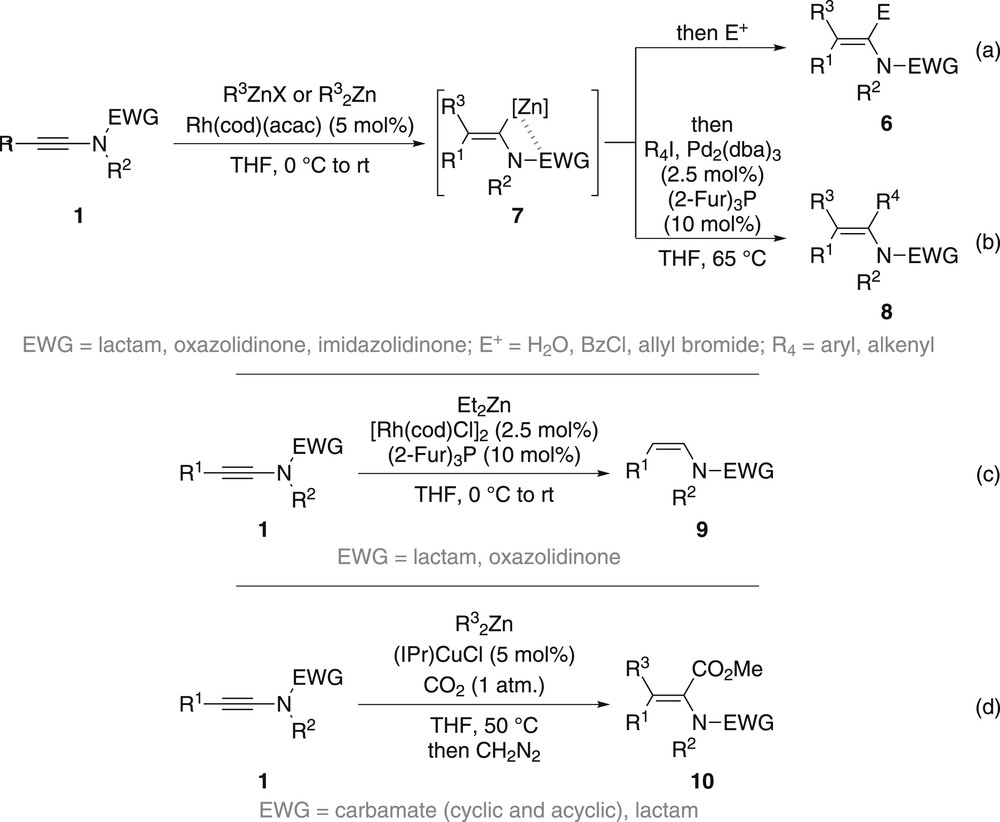
Rhodium- and copper-catalysed carbozincation of ynamides.
The synthetic potential of the regio- and stereocontrolled access to multisubstituted enamides [29] based on the carbometallation of ynamides was further demonstrated by Gourdet and Lam who reported an efficient and innovative total synthesis of (+)-tanikolide 17, an antifungal lactone possessing a chiral tertiary alcohol (Scheme 3) [30]. Capitalizing on the method they developed for the synthesis of multisubstituted enamides, the synthesis began with the preparation of ynamide 13 using the Hsung procedure from bromoalkyne 11 and oxazolidinone 12 and its further rhodium-catalysed carbozincation using functionalized alkylzinc bromide 14. The corresponding disubstituted enamide 15, obtained as a single regioisomer and diastereoisomer resulting from a directed syn-carbozincation, was next subjected to an asymmetric Sharpless dihydroxylation, yielding enantioenriched diol 16 after reduction of the intermediate aminal with sodium borohydride. Further saponification of the ester followed by lactonization provided the desired target molecule 17 with an excellent overall yield, further highlighting the efficiency of the regio- and stereoselective synthesis of enamides—which are not that trivial to prepare—by carbometallation of ynamides.
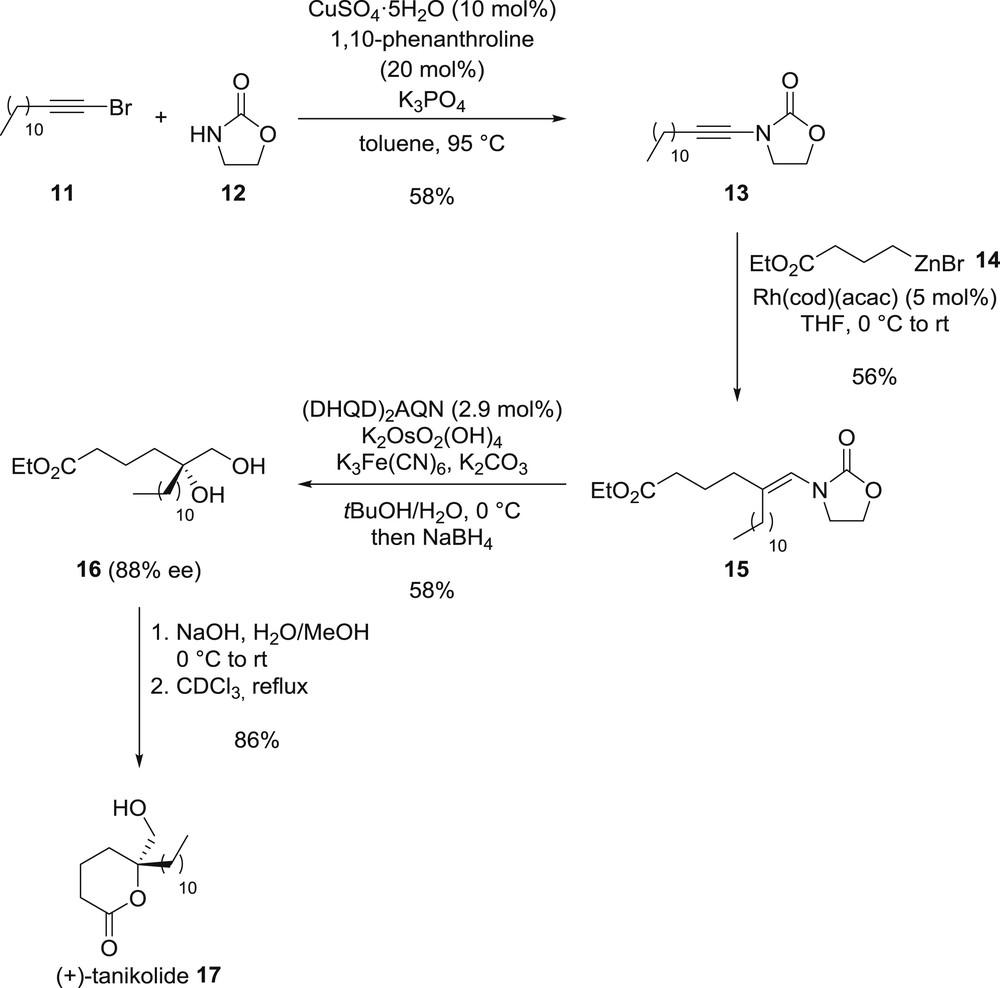
Total synthesis of (+)-tanikolide: highlighting the synthetic efficiency of the carbometallation of ynamides.
Organoboron reagents such as aryl/alkenyl-boronic acids, arylboronates and triarylboroxines were also shown to be suitable reaction partners, in combination with a rhodium catalyst, for the carbometallation of ynamides, which represent an interesting alternative to the use of the corresponding aryl copper, magnesium and zinc reagents because of their greater availability (Scheme 4) [31]. The reaction is performed in a mixture of tetrahydrofuran (THF) and water under microwave irradiation and, as in the previous cases, enables the regio- and stereocontrolled synthesis of β,β′-disubstituted enamides 6 from the corresponding ynamides 1.
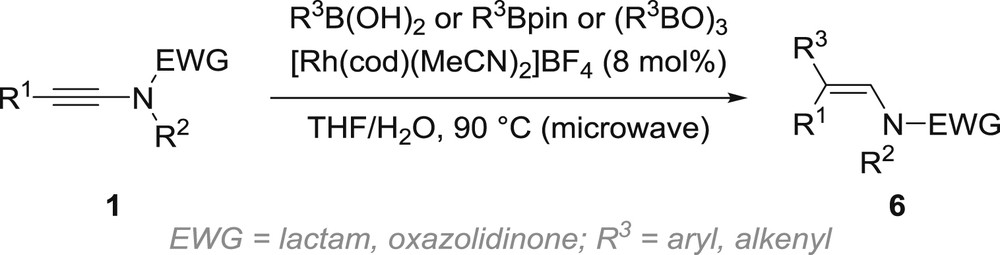
Rhodium-catalysed carbometallation of ynamides with organoboron reagents.
Although they clearly do not fall into the classification of anionic transformations, a series of procedures have been reported for the intermolecular carbopalladation of ynamides: they clearly deserve to be mentioned, at least for comparison purposes. One of the major differences between carbopalladations involving ynamides and the other carbometallations described above is the shift in regioselectivity observed in most cases. Indeed, although carbocuprations and carbozincations proceed to favour the formation of an α-metallated enamide analogous to 18, which benefits from chelation of the metal by the electron-withdrawing group, carbopalladations are less sensitive to directing effects and preferably follow the inherent polarization of ynamides 1 to afford intermediate β-palladated enamides 19 (Scheme 5). The presence of the amino group β to the palladium can in addition facilitate the E–Z isomerization to 21, presumably via a palladium carbene 20, which renders the stereoselectivity of the overall carbopalladation process dependent on the steric properties of the substituent on the starting ynamide.
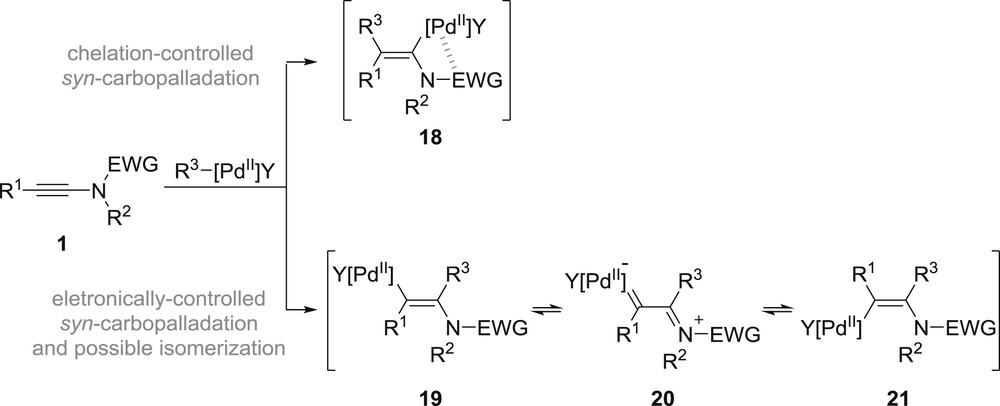
Possible reversal of regio- and stereoselectivity in the carbopalladation of ynamides.
The palladium-catalysed addition of aryl- and alkenyl-boronic acids to ynamides is a remarkable illustration of the reversal of both regio- and stereoselectivities that can be observed with the carbopalladation of ynamides (Scheme 6a). Indeed, the reaction of ynamides 1 with boronic acids in the presence of catalytic amounts of palladium acetate and tris(3-tolyl)phosphine in ethanol at 70 °C gave conjugated enamides 22 resulting from an overall trans addition across the triple bond in which the newly introduced substituent R3 is placed at the α-position, which can be explained by an intermediate vinylpalladium species such as 21 (Scheme 5) [32]. The dramatic influence of the nature of the substituents on the starting ynamide was also noted by the Reddy group who, in continuation of previous studies by the Zhu group [33], examined in detail the selectivity of the palladium-catalysed hydroalkynylation of ynamides [34]. Although the regioselectivity observed in all cases was consistent with an electronically controlled carbopalladation placing the palladium β to the nitrogen, the stereoselectivity observed was found to be strongly influenced by the nature of both the electron-withdrawing groups and the substituent on the alkyne. Indeed, although small electron-withdrawing groups such as oxazolidinones, β-lactam and indoles favoured the formation of the E-ynenamides 23 (via an intermediate such as 19 in Scheme 5), more sterically demanding sulfonamides were found to mostly induce the selective formation of either E-ynenamides 23 when starting from yne-sulfonamides bearing a small R1 substituent such as an alkyl chain or Z-ynenamides 24 (via isomerization of intermediate 19 to 21 to avoid steric clash) when this R1 substituent was replaced by a bulkier aryl group (Scheme 6b). A similar preference was observed starting from a N-Boc-ynamine.
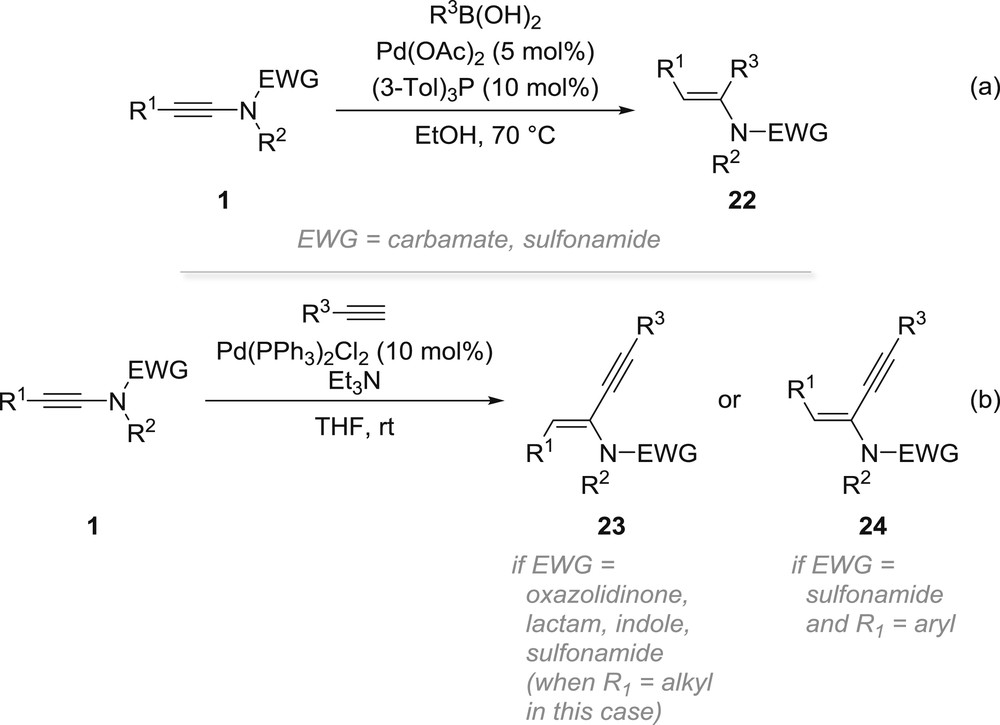
Reactions involving an intermolecular carbopalladation of ynamides.
In addition to their carbometallation, which allows for the selective introduction of a carbon substituent to the starting ynamide, related processes enabling the introduction of heavier group 14 elements such as silicon and tin as well as a boron substituent have been reported. They provide a straightforward access to functionalizable enamides and they will be briefly overviewed in the following section.
2.2 Intermolecular silyl-, stannyl- and boryl-metallation of ynamides: an efficient entry to functionalizable enamides
The first example of a silylcupration of an ynamide was reported in 2008 by the Yorimitsu and Oshima group [35]. Using a large excess of silylcopper reagents in a mixture of THF and hexamethylphosphoramide at −60 °C, phenyl-substituted yne-sulfonamides 25 were found to readily undergo a silylcupration yielding, after hydrolysis or trapping with allylbromide, the corresponding β-silylated enamides 26 obtained with good, although not complete, regio- and stereoselectivities (Scheme 7a). In collaboration with the Riant group [36], we reported a catalytic version of this reaction based on a copper-catalysed silylcupration with Suginome's silylboronates. Upon reaction with these reagents and methanol in THF at room temperature in the presence of copper(I) fluoride tris(triphenylphosphine), a range of ynamides 1 were found to undergo a smooth silylcupration. This reaction, which tolerates various functional groups, affords a straightforward and efficient entry to the corresponding β-silyl-enamides 27 that were formed with high levels of regio- and stereoselectivities (Scheme 7b). This silylmetallation, which was found to be highly E-selective, can also be performed through a silylzincation starting from terminal ynamide 28, which upon reaction with (Me2PhSi)2Zn, followed by transmetallation with CuCN·LiCl and reaction with an electrophile provides the corresponding E-enamides 29 in fair to good yields (Scheme 7c) [37]. An interesting and quite remarkable reversal of selectivity, which was attributed to a switch from a polar to a radical-chain mechanism, was observed when starting from [(Me3Si)3Si]2Zn, which was proposed to be more susceptible to undergo homolytic cleavage of the siliconzinc bond. The Z-enamides 31 could indeed be obtained through a trans-selective radical silylzincation of terminal ynamides 30 using this reagent in combination with diethylzinc as an additive or by using directly (Me3Si)3SiH with an excess of diethylzinc (Scheme 7d) [37,38].
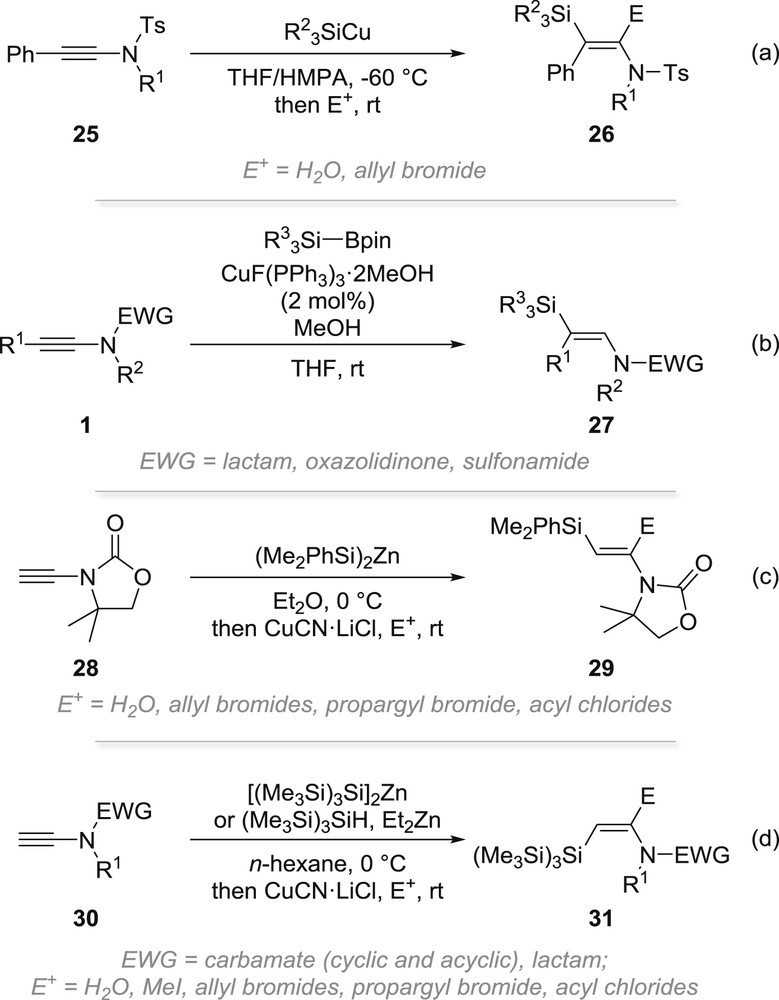
Silylcupration and silylzincation of ynamides.
The only examples of stannylmetallation of ynamides reported to date involve palladium catalysis. As mentioned in the previous section, palladium being less sensitive to chelation, the regioselectivity of the syn-stannylpalladation is controlled by the polarization of the triple bond, which provides E-α-stannyl-enamides 32, although the regioselectivity decreases when starting from terminal and aryl-substituted yne-oxazolidinones or yne-sulfonamides (Scheme 8) [39]. The stannylpalladation is not limited to a formal hydrostannylation because replacing tributyltin hydride by Bu3SnSnBu3, Me3SnSnMe3 or Bu3SnSiMe3 enables clean bis-stannylation or stannylsilylation reactions [39a,b].
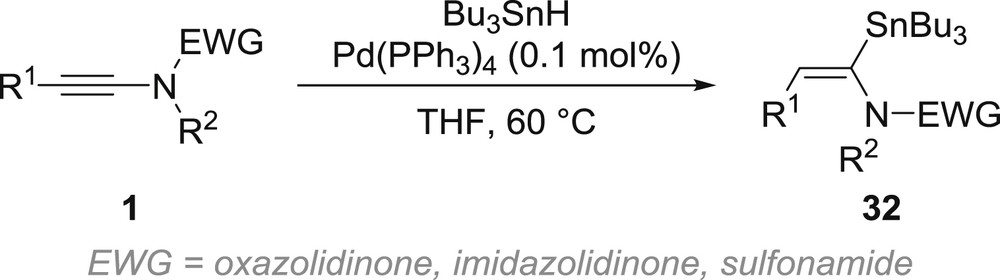
Stannylpalladation of ynamides.
Finally, it ought to be mentioned that the borylmetallation of ynamides has also been investigated using either bis(pinacolato)diboron in combination with a copper catalyst (Scheme 9a) [40] or Suginome's reagent in combination with a palladium catalyst (Scheme 9b) [41]. In the former case, Z-enamides 33 are isolated, their formation being rationalized by a borylcupration yielding an intermediate β-boryl-enamide—whose formation is actually quite surprising because it involves a complete reversal of regioselectivity compared with most cupration reactions involving ynamides—followed by a transmetallation with the copper alkoxide catalyst and final protonolysis. In the latter case, α-silyl-β-boryl-enamides 34 are obtained, the regioselectivity being consistent with the silylborylation proceeding through preferential insertion of the triple bond to the PdB σ bond formed after oxidative addition of Suginome's reagent to palladium(0), which, however, involves a chelation-controlled borylpalladation that is in sharp contrast with the regioselectivity observed for carbopalladations (see Scheme 6).
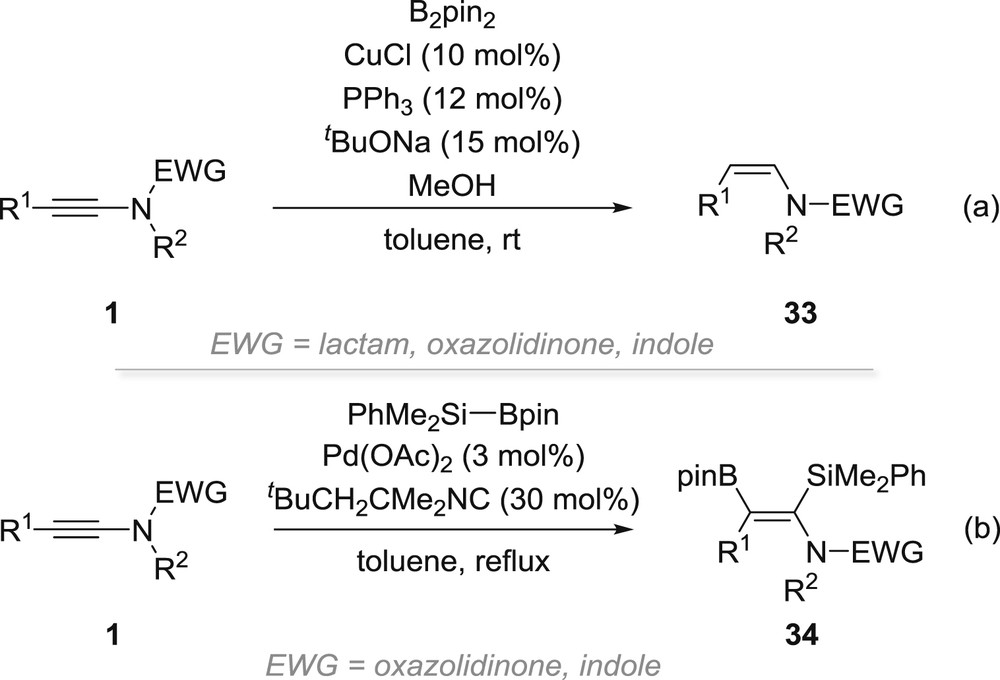
Borylmetallation of ynamides.
As hopefully evidenced by all examples described, the carbo-, silyl-, stannyl- and borylmetallation of ynamides represent remarkably useful entries to highly functionalized and functionalizable enamides, which can be prepared with excellent control, at least in most cases, of the regio- and stereoselectivities. The choice of the metal is crucial for these reactions, which can be controlled either by chelation or by the polarization of the triple bond. These reactions are not limited to the synthesis of enamides because the incorporation of such metallation reactions in cascade processes in which the intermediate metallated enamide further reacts with a substituent judiciously incorporated in the starting ynamide enables the preparation of other building blocks and molecular scaffolds; they will be briefly described in the following section.
2.3 Cascade reactions involving an intermolecular carbometallation of ynamides: efficient access to useful building blocks
The first example of a cascade reaction involving metallation of an ynamide was reported by Yorimitsu and Oshima group in the course of their studies of the copper-catalysed carbomagnesiation of ynamides [35,42]. When starting from N-allyl-ynamides 35 and heating the intermediate vinyl Grignard reagent 36 at 80 °C after addition of 1,2-dimethoxyethane or THF, an aza-Claisen rearrangement occurred, which after elimination of a magnesium sulfinate (which can also proceed before the sigmatropic rearrangement) produced homoallylic nitriles 37 (Scheme 10a).
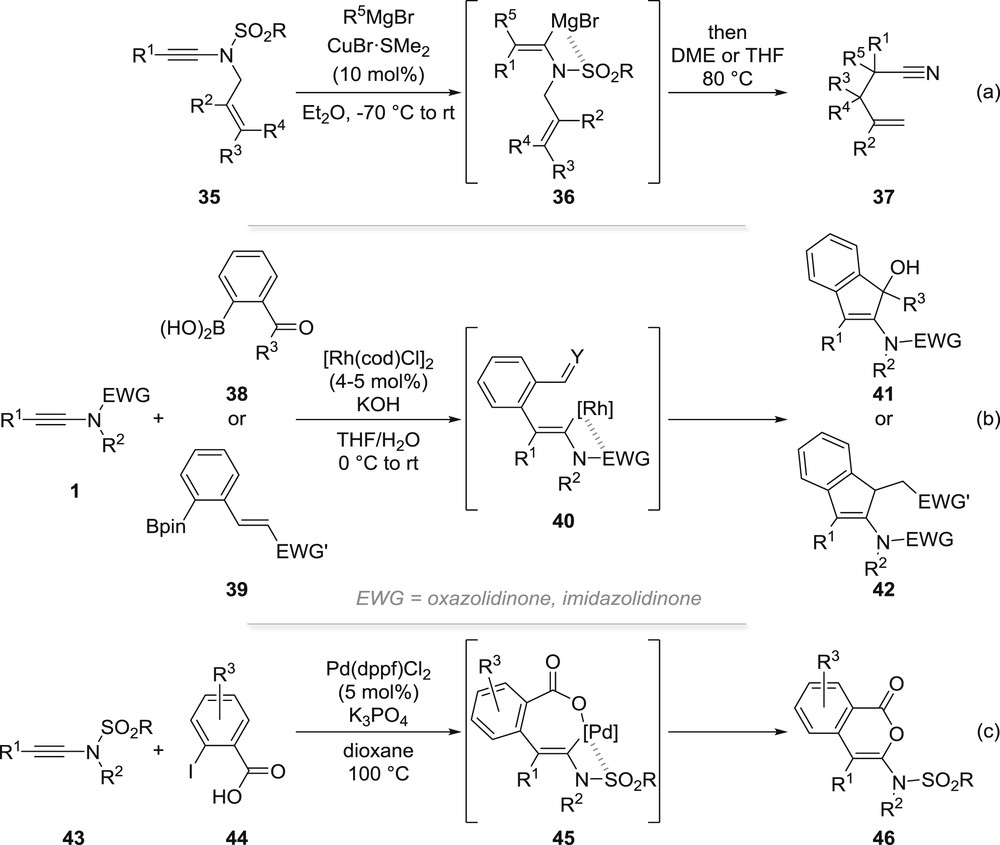
Cascade reactions involving an intermolecular carbometallation of ynamides.
In continuation to their studies on the rhodium-catalysed carbometallation of ynamides with organoboron reagents, the Lam group reported in 2010 an interesting annulation of ynamides starting from arylboron reagents possessing an electrophilic functional group ortho to the boronic acid/boronate such as in 38 or 39 [43]. In this case, the carbometallation yielding 40 is followed by nucleophilic addition to an aldehyde/ketone or a 1,4-addition to an α,β-unsaturated aldehyde, ketone or ester, yielding 2-aminoinden-1-ols 41 or 2-aminoindenes 42 (Scheme 10b).
In a similar vein, the group of Wang and Chang reported another annulation of yne-sulfonamides 43 based on their palladium-catalysed reaction with ortho-iodo-benzoic acids 44. In this case, the carbopalladation was found to be chelation-controlled, yielding intermediate 45, which after further reductive elimination furnishes 3-amino-isocoumarins 46 (Scheme 10c) [44].
Last, but definitely not least, especially elegant and remarkably efficient one-pot sequences involving an intermolecular carbocupration of chiral ynamides were reported in a series of publication by Marek and colleagues. Capitalizing on the high levels of regio- and stereoselectivities observed for the carbocupration of ynamides, including chiral ones such as 47, they had earlier studied (Scheme 1b), they envisioned that the resulting vinylcopper 48 could be further exploited in a series of transformations performed in a one-pot sequence to access aldol products possessing all-carbon quaternary stereocentres, products that cannot be accessed through classical asymmetric aldolization reactions because of the lack of stereocontrol observed for the generation of the enolates that would be required (Scheme 11). In this perspective, Marek and colleagues first envisioned a homologation of 48 with in situ generated ICH2ZnCH2I yielding an intermediate chiral allylzinc. Further reaction of this allylzinc with an aldehyde provided, through a Zimmerman–Traxler transition state, adduct 49, which after a two-step cleavage of the chiral oxazolidinone enables an efficient and highly stereoselective access to the corresponding formal aldol adducts [45]. Although this sequence was found to be remarkably efficient and stereoselective, the main limitation relies on the two-step cleavage of the chiral auxiliary. To address this drawback, they reported 3 years later that the zinc carbenoid used for the homologation of the intermediate vinylcopper 48 could be replaced by an oxenoid generated in situ from equimolar amounts of tert-butylhydroperoxide and n-butyllithium. Although counter-intuitive, the use of this oxenoid turned out to be remarkably well-thought because it was demonstrated to enable a clean and stereoselective generation of fully substituted enolate 50, which was next shown to react smoothly with a series of electrophiles including protic sources, aldehydes and imines to afford, with high levels of diastereoselectivity, chiral imides 51, aldol 52 and Mannich 53 adducts, respectively [46]. In addition, intermediate copper enolate 50 was shown to be an excellent precursor of stereodefined disubstituted ketene aminals 54, which can be generated upon trapping 50 with trimethylsilyl chloride [47]. These ketene aminals, which were shown to be excellent reaction partners for the Mukaiyama aldol reaction (by reaction with an aldehyde in the presence of titanium tetrachloride to give aldol adducts 52), being rather sensitive in the presence of copper, the addition of copper scavengers such as 1,10-phenanthroline before the work up was found to increase the yields.
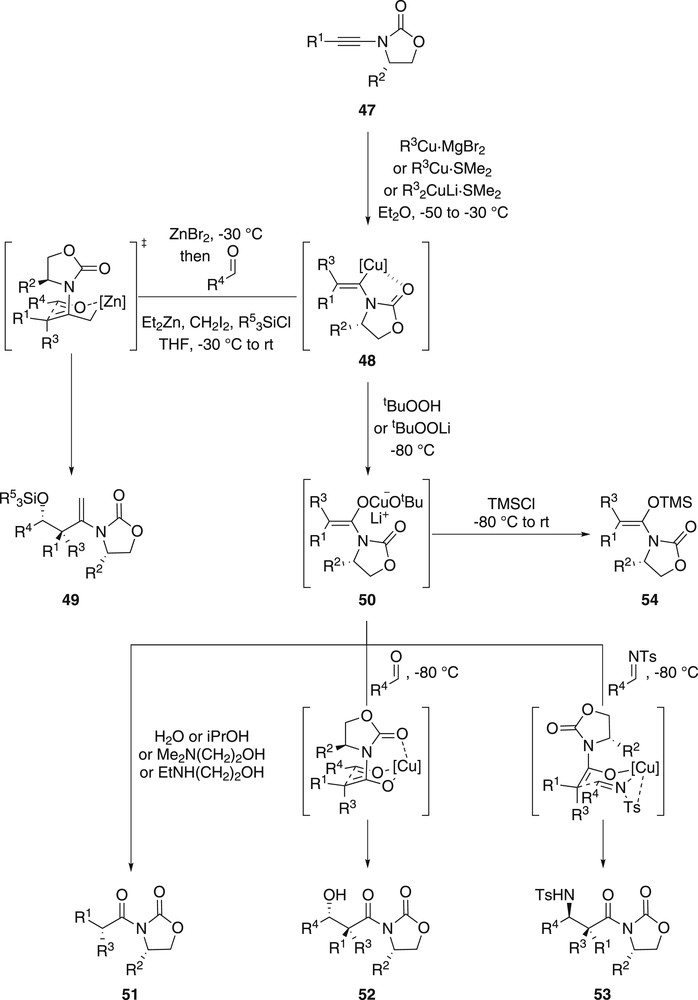
Unique approaches to aldol products possessing all-carbon quaternary stereogenic centres through carbocupration of chiral ynamides.
As overviewed with all examples discussed up to this point in this article, the carbometallation of ynamides has recently evolved from a simple curiosity to a remarkably efficient reaction enabling the preparation of a wide array of building blocks. Indeed, it does not only provide efficient accesses to highly substituted stereodefined enamides but also to an impressive number of building blocks and molecular scaffolds derived from the intermediate metallated enamides. Importantly enough to be mentioned, this simple carbometallation reaction also facilitated the emergence of new synthetic paradigms, notably in asymmetric synthesis as exemplified by the work emphasized above by Marek and colleagues.
The carbometallation of ynamides is not limited to its intermolecular version and has also been recently studied and elegantly exploited in its intramolecular version, which will be overviewed in the next section.
3 Intramolecular carbometallation of ynamides: a remarkably useful reaction for heterocyclic synthesis
In addition to benefiting from the polarization of the ynamide triple bond or directing effect of the electron-withdrawing group, the intramolecular carbometallation of ynamides is of course also favoured by entropy effects. Depending on the relative position of the metal and the alkyne, the nitrogen atom being either in between these two functional groups (such as in 55) or not (such as in 59), the intramolecular reaction can be exploited for the synthesis of nitrogen heterocycles 56–58 or amino-cycles 60–62 (Scheme 12). Both the regio- and stereoselectivity of the intramolecular carbometallation have to be considered and fully controlled for the reaction to be of any synthetic usefulness. Although endo-dig cyclizations yielding 56 or 60 have to proceed through a non-classical anti-carbometallation, their exo-dig counterparts proceed in most cases via syn-carbometallations yielding vinylmetal species such as 57 or 61, although anti-carbometallations have also been reported in some cases. The parameters governing the regio- and stereoselectivity are multiple and include the size of the ring formed [48], the nature of the metal and reagents used (one should keep in mind e.g., that halogen–lithium exchanges can proceed through polar or radical mechanisms), as well as the nature of the electron-withdrawing group, which has been shown to have a dramatic effect.
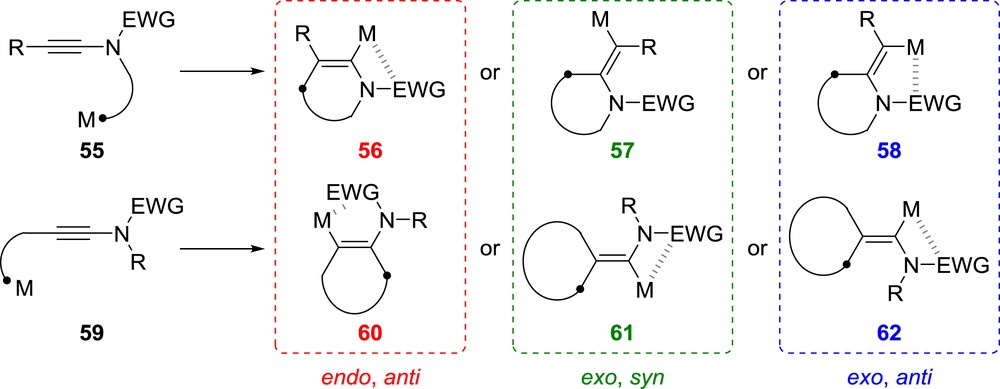
Overall picture of the possibilities for the intramolecular carbometallation of ynamides.
Intrigued and motivated by the possibility of accessing heterocyclic scaffolds for which there were only limited options for their synthesis, we became interested in 2012 in the intramolecular carbometallation of ynamides. In this perspective, we could successfully demonstrate that lithiated N-allyl-ynamides 64, readily generated by deprotonation of the corresponding N-allyl-ynamides 63 with s-butyllithium in the presence of N,N,N',N'-tetramethylethylenediamine, underwent a smooth and extremely rapid metallotropic equilibrium followed by an unprecedented anionic 6-endo-dig cyclization to afford the corresponding 1,4-dihydropyridines 67 in good to excellent yields (Scheme 13a) [49]. Besides providing an efficient access to 1,4-dihydropyridines non-stabilized by the presence of an electron-withdrawing group at C3, which is strictly required in most syntheses of such molecules, we could also demonstrate that by simply replacing the final hydrolysis by a workup under acidic and oxidizing conditions, the corresponding polysubstituted pyridines 68 could be obtained (Scheme 13b). The efficiency of this anionic cyclization could then be further demonstrated in a formal synthesis of the anti-dyskinesia agent sarizotan 69 for which the 3,5-disubstituted pyridine core could be readily prepared by intramolecular carbolithiation of the corresponding suitably functionalized ynamide [50].
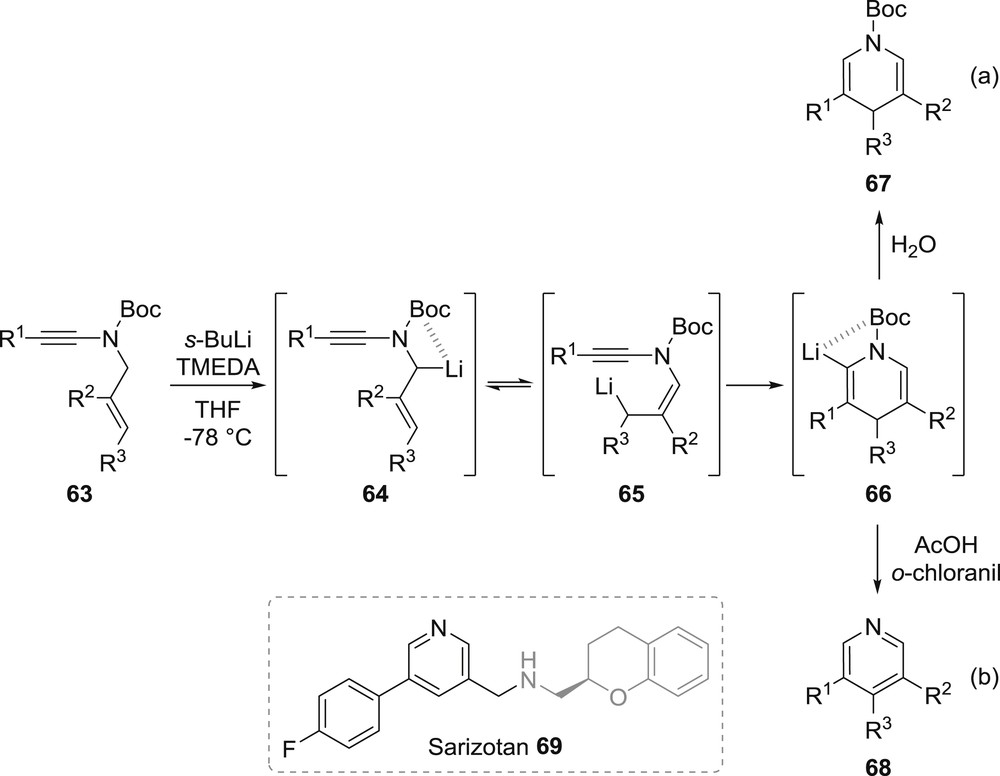
Synthesis of 1,4-dihydropyridines and pyridines by intramolecular carbolithiation of ynamides.
The success of this intramolecular carbolithiation led us to investigate related systems, and we reported later on other nitrogen heterocycles, such as indoles, which could also be obtained using a similar strategy. Indeed, we could demonstrate that polyfunctionalized indoles 72 could be readily obtained by carbocupration starting from ynamides such as 70, the related carbolithiation being inoperative in this case (Scheme 14a) [51]. A similar cyclization using either a carbocupration or a copper-catalysed carbomagnesiation after a bromine–magnesium exchange (73 → 75, Scheme 14b) was reported by Knochel and colleagues who also extended it to the synthesis of azaindoles 78 and pyrrolopyrimidines 83 starting from the corresponding N-(pyridin-3-yl)- and N-(pyrimidin-4-yl)-ynamides 76 and 81, respectively (Scheme 14c,e) [52]. Capitalizing on the acidity of the proton at C4 in 76 and on the possibility of a directed metallation, they in addition reported a divergent carbocupration based on a direct deprotonation yielding 80 (after transmetallation/carbocupration) instead of the halogen–lithium exchange (Scheme 14d) [52a]. In all cases, a single regioisomer resulting from a 5-endo-dig cyclization was observed.
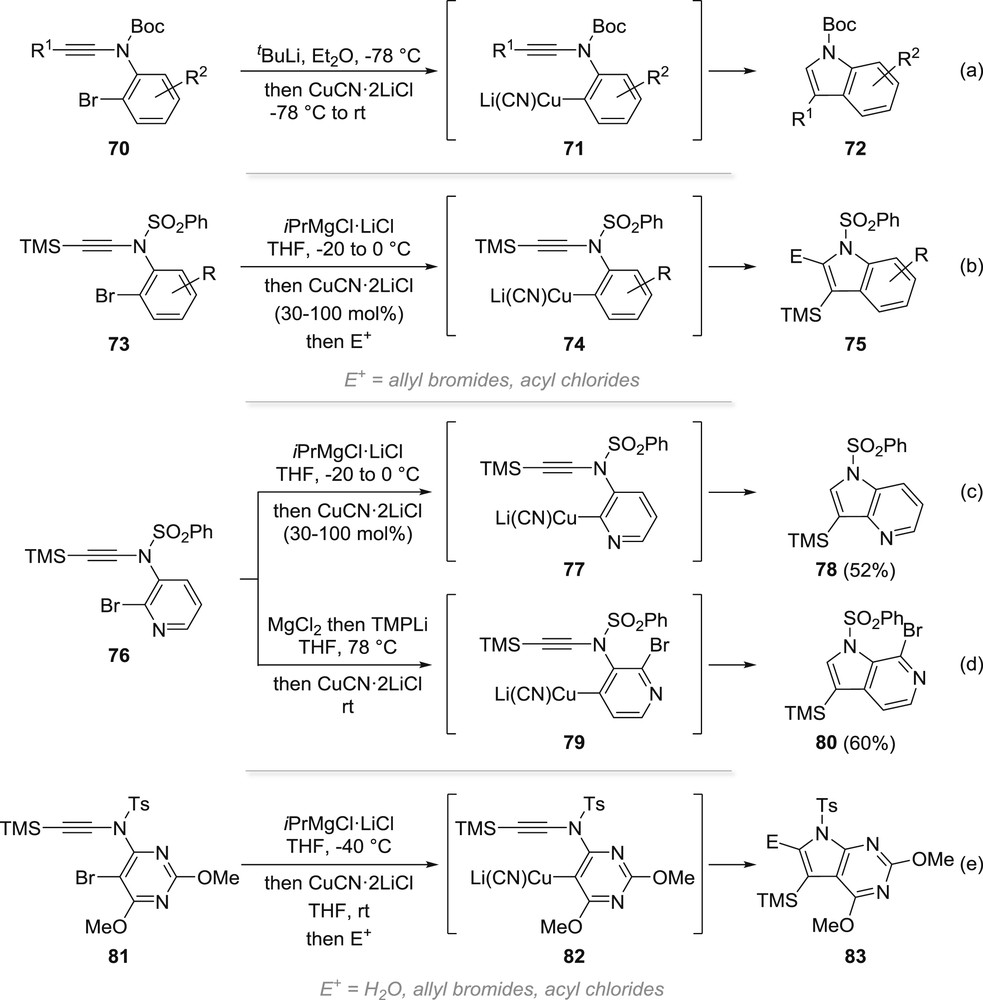
Synthesis of indoles and related heterocycles by intramolecular carbocupration of ynamides.
The intramolecular carbopalladation of suitably functionalized ynamides was also demonstrated to be quite efficient for the synthesis of a range of heterocycles, ranging from simple azacycles to complex polycyclic systems. In all cases, regardless of the size of the heterocycle formed, the carbopalladation seems to follow the inherent polarization of the triple bond and the electron-withdrawing group on the starting ynamides does not seem to have any directing effect on these cyclizations, which is in accordance with results previously discussed, in the intermolecular series, in Section 2. The first example of an intramolecular carbopalladation of ynamides was actually reported in 2004 by Meyer, Cossy and colleagues who reported an efficient access to methylene-isoindolinones 85 based on a carbopalladation of ynamides 84 in the presence of an arylboronic acid, which traps the vinylpalladium(II) species formed after the oxidative addition/carbopalladation sequence (Scheme 15a) [53]. The efficiency of this cyclization, in which the boronic acid can be replaced by ammonium formate to replace the aryl group by a hydrogen atom in the final cyclized product, was later on used as a key step in a remarkably efficient and short total synthesis of lennoxamine 86 [54]. Capitalizing on the success of this intramolecular carbopalladation, Anderson and colleagues reported later a similar cyclization starting from bromoenynamides 87. As in the previous case, the intermediate vinylpalladium 88 resulting from the oxidative addition/carbopalladation could either be reduced with ethanol, yielding simple cyclized product 89 (Scheme 15b) or used to transmetallate a vinyltin or vinylboron derivative present in the reaction mixture to furnish, after 6π-electrocyclization, polycyclic azacycles 90 (Scheme 15c) [55]. In the presence of an additional triple bond suitably placed on the starting ynamide such as in 91, a remarkable cascade polycyclization involving consecutive carbopalladations was found to occur, yielding tricycles 92 in good to excellent yields (Scheme 15d) [56].
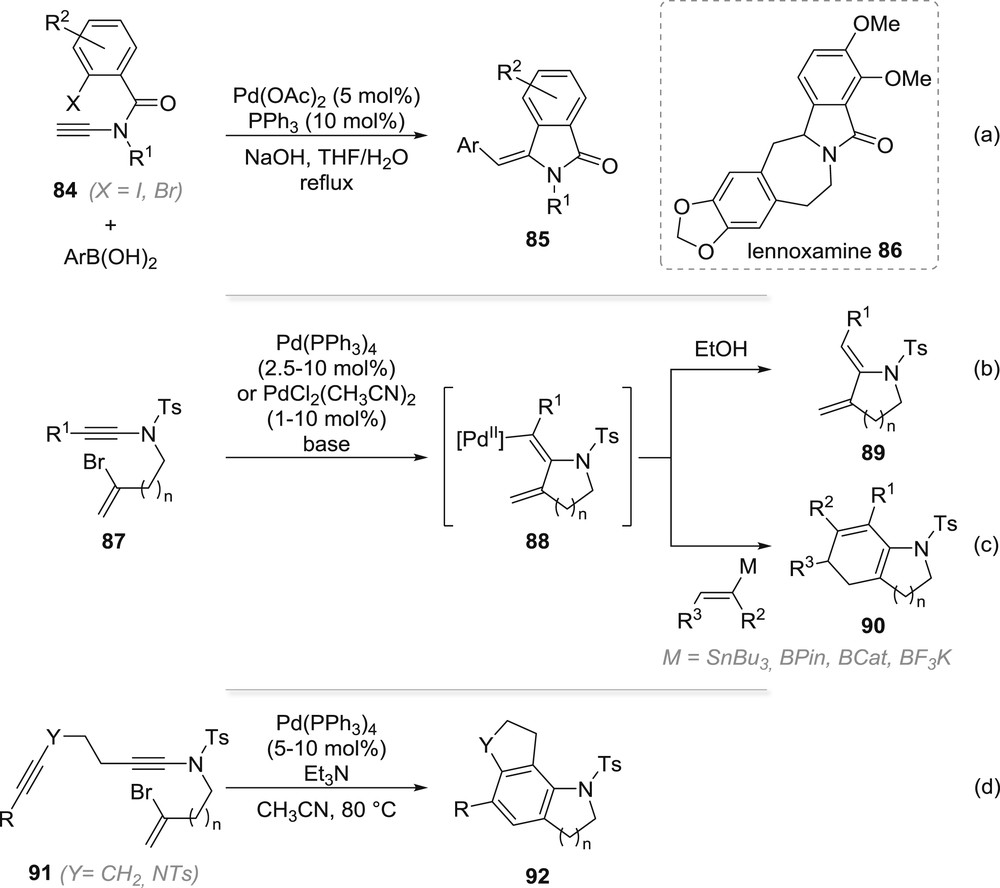
Intramolecular carbopalladation of ynamides from isoindolinones to polycylic nitrogen heterocycles.
Although in all examples mentioned above, the nitrogen atom is located between the triple bond and the site where the metallation occurs, which provides efficient entries to a broad variety of nitrogen heterocycles, a different connectivity enables the synthesis of nitrogen-substituted cyclic systems. This strategy was explored by Durandetti, Maddaluno and colleagues who investigated the intramolecular carbolithiation of a series of ynamides 93: although a chelation-driven 5-exo-dig anti-carbolithiation yielding aminomethylene-dihydrobenzofuran derivatives 94 was observed in most cases, a complete switch in regioselectivity in favour of a 6-endo-dig cyclization to 95 was observed starting from an yne-phosphoramide, irrespective of the organolithium species used to trigger the iodine–lithium exchange (Scheme 16) [57]. If Density Functional Theory calculations revealed that the unusual anti-carbolithiation could be explained by the affinity of the electron-withdrawing group in the starting ynamides for the lithium cation, the peculiar behaviour of the yne-phosphoramide could not be rationalized.
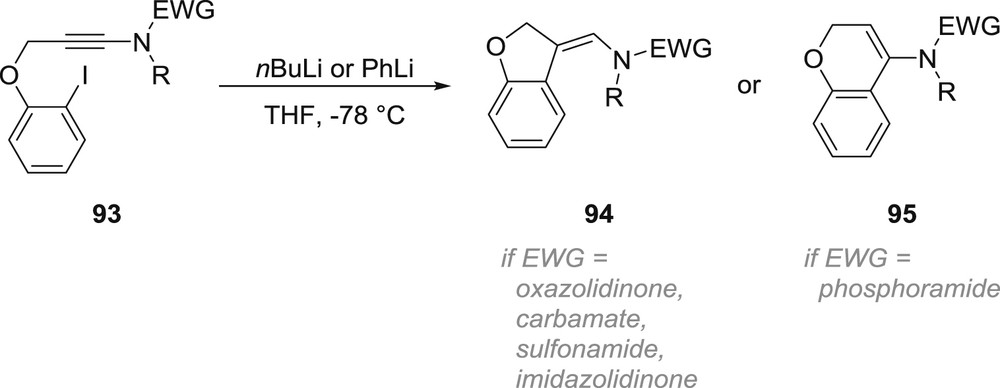
Dramatic effect of the electron-withdrawing group in the intramolecular carbolithiation of ynamides.
As evidenced by all results described in this section, the intramolecular carbometallation of ynamides provides effective accesses to numerous heterocyclic systems. Efficient reactions could be designed based on such a step, provided that the parameters controlling both the reactivity and the selectivity are carefully understood. The nature of the metal and the electron-withdrawing group on the starting ynamides clearly have a strong influence on the outcome of intramolecular carbometallations, and there is now clearly enough experimental data—although still not fully understood—for the design and development of other processes based on the unique reactivity of ynamides towards carbometallations.
If such reactions clearly constitute the corner stone of the anionic chemistry of ynamides, other reactions based on the unique reactivity of ynamides and related building blocks under anionic conditions have been reported. They include reactions triggered by deprotonation or nucleophilic addition to the electron-withdrawing group, which will be overviewed in the following section.
4 Deprotonation/nucleophilic addition to the electron-withdrawing group of ynamides: efficient generation of otherwise hardly accessible reactive intermediates
Reactions involving the anionic chemistry of the electron-withdrawing group of ynamides are actually rather scarce, which is fairly logical because this group usually serves as a protecting group responsible for the stability of ynamides. Playing around with this group might therefore result in a total loss of stability and it might therefore seem safer not to fool around with strong bases or nucleophiles that might react with this electron-withdrawing group.
For a while, the only example of a reaction involving metallation of the electron-withdrawing group was indeed limited to the metallation at the 2-position of N-alkynyl-imidazoles 96 followed by trapping of the resulting anion, in which the stability brought to the nitrogen-substituted alkyne by delocalization into the imidazole ring is intact, with various electrophiles (Scheme 17) [58]. Everything therefore happens as in the absence of the nitrogen-substituted alkyne, which does not react under the reaction conditions and is not affected by the metallation of the imidazole.
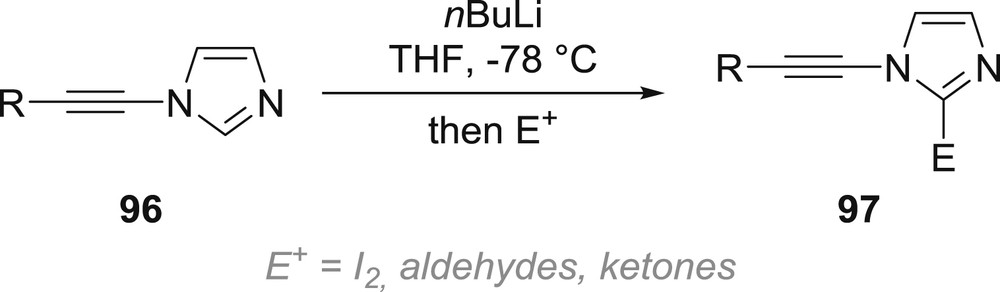
Metallation of N-alkynyl-imidazoles at C2.
The synthetic usefulness of a reaction sequence involving an anionic reaction at the electron-withdrawing group of ynamides was actually reported in 2014 only when we observed that ynimines such as 98 and 110 were excellent precursors of otherwise hardly accessible metallated ketenimines 100 and 112 (Scheme 18) [59]. We could indeed demonstrate that the addition of an organolithium reagent to ynimine 98 regioselectively took place on the imine moiety and that the resulting metallated ketenimine 100 could then be trapped by an alkyl halide, generating a transient unstable ketenimine 101, which after homolytic fragmentation and recombination provided highly substituted alkanenitriles 105 in modest to good yields (Scheme 18a). Introduction of a trimethylsilyl group on the starting ynimine resulted in a higher stability of the lithiated ketenimine, which could also be trapped by an alkyl halide, the resulting trimethylsilyl-ketenimine 106 being now perfectly stable (Scheme 18b). Alternatively, trapping the 2-lithio-2-silyl-ketenimine 100 with an aldehyde triggered a Peterson-type elimination generating an intermediate cumulene 108, which upon hydrolysis gave α,β-conjugated amide 109 (Scheme 18c).
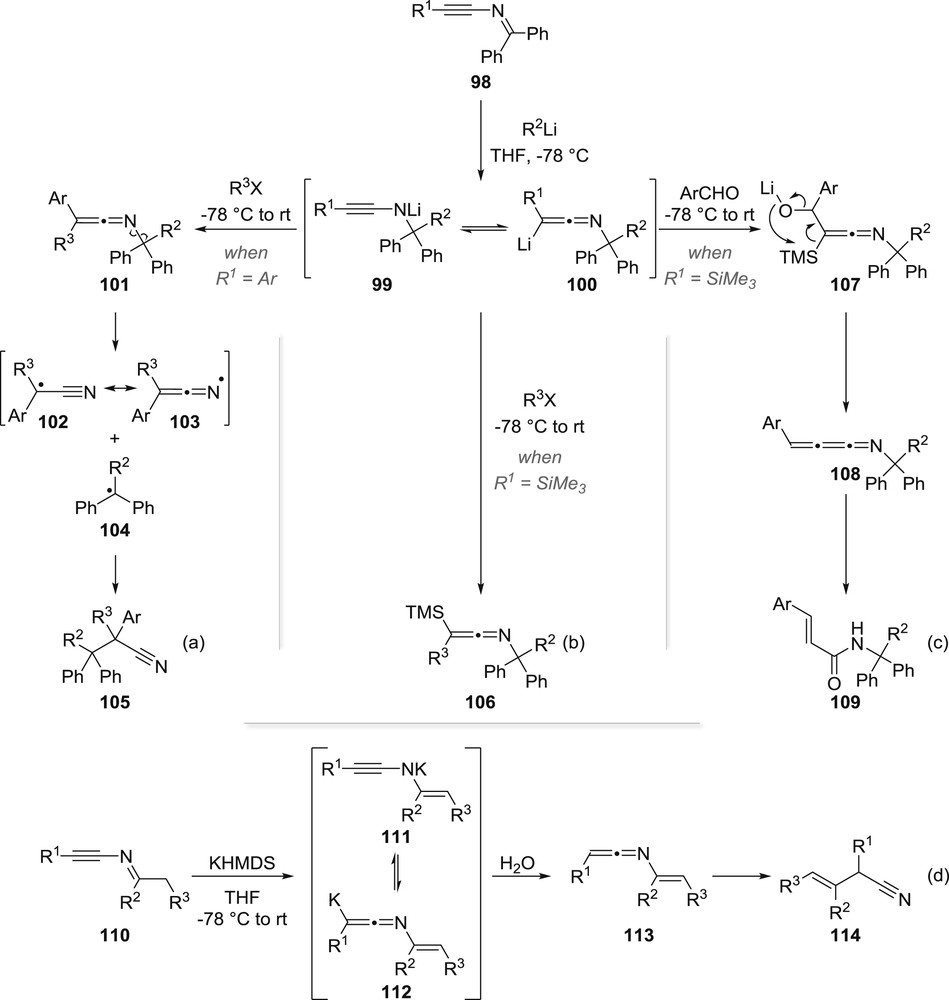
Ynimines: useful precursors of metallated ketenimines.
Alternatively, we could also show that metallated ketenimines 112 could be generated by deprotonation of the imine in ynimines such as 110 with potassium bis(trimethylsilyl)amide rather than by nucleophilic addition of an organolithium. Further hydrolysis then afforded a transient ene-ketenimine 113 whose radical rearrangement gave allylic nitriles 114 (Scheme 18d).
It finally ought to be mentioned that related metallated ketenimines have been postulated as intermediates in the reaction between indole and ynamides [60]. In this case, sodium tert-butoxide has been postulated to, surprisingly, deprotect the starting ynamide 1, generating an intermediate sodium ketenimine 115, which would be protonated by indole 116. Further addition of the deprotonated indole 117 to the ketenimine 118 and final protonation would account for the formation of acylated indole 119 (Scheme 19).
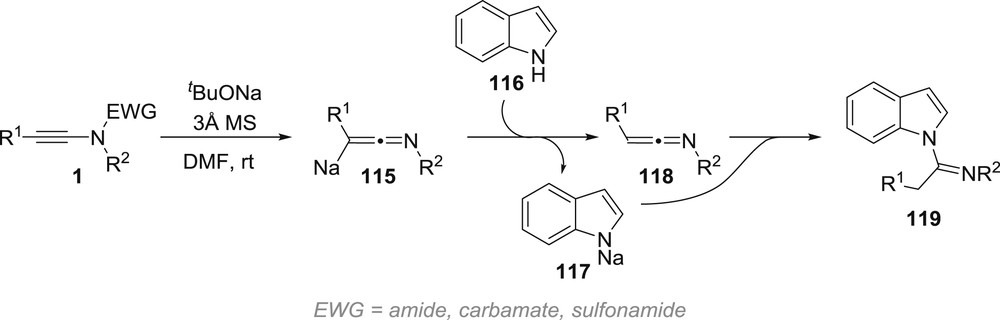
Generation of sodium ketenimines by deprotection of ynamides.
Finally, we could recently demonstrate that organolithium reagents could react in a highly chemoselective manner with yne-oxazolidinones 120 to afford the ring-opened acyclic yne-amides 121, useful building blocks for chemical synthesis whose synthesis is still rather challenging and often low yielding, without competitive carbolithiation or propargylic lithiation (Scheme 20) [61].
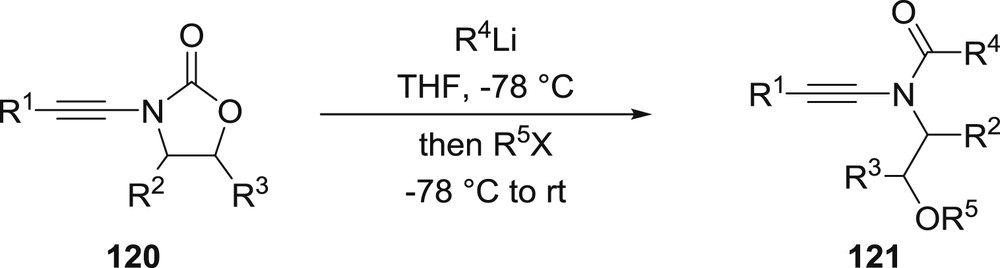
Ring-opening of yne-oxazolidinones to acyclic yne-amides.
Although the anionic transformations involving nucleophilic addition or deprotonation of the electron-withdrawing group of ynamides are still quite underdeveloped to date, there is no doubt that this area of the anionic chemistry of ynamides will be the focus of further investigations because it clearly has a strong synthetic potential, both for the generation of anionic intermediates that are still challenging to prepare or to explore new pathways in carbanionic chemistry.
Before ending this review article, reactions involving metallated terminal ynamides will be covered in the final section.
5 Reactions involving metallated terminal ynamides: a straightforward entry to functionalized ynamides
The last area of the anionic chemistry of ynamides is indeed based on the trapping of metallated terminal ynamides with a series of electrophiles, which enables the synthesis of complex ynamides from simpler ones (Scheme 21). If metallated ynamides 124 can of course be prepared by direct deprotonation of terminal ynamides 30 [62], although the deprotonation that can be performed with a series of strong bases is usually not as simple as it seems [62a], they can also be prepared in situ from different precursors. Indeed, β,β-halo- [62a,63] and α,β-dichloro- [13] enamides 122 and 123 were found to be excellent precursors of metallated ynamides 124, which are generated depending on the nature of the halogen through deprotonation/elimination/lithium–halogen exchange (for chlorinated enamides) or lithium–halogen exchange/Fritsch–Buttenberg–Wiechell rearrangement/deprotonation (for brominated enamides) sequences. The metallated ynamides 124 generated in situ can then be trapped by a broad range of electrophiles including alkyl halides, chlorosilanes, epoxides, aldehydes, ketones, enones, anhydrides, chloroformates, CO2, isocyanates, imines, sulfinylimines, disulfides or chlorophosphates, although the list might not be exhaustive [13,62,63]. Further transmetallation with zinc bromide also enables the synthesis of aryl-substituted ynamides 126 via a Negishi coupling [64]. As a note, it should also be mentioned that copper acetylides derived from terminal ynamides, usually generated under catalytic conditions, can also be used for the Glaser–Hay dimerization of terminal ynamides [64b,65], their Cadiot–Chodkiewicz cross-coupling with bromoalkynes [66], or for their addition to activated electrophiles such as acyl chlorides and N-acyl-pyridinium salts [67] and that Carreira's enantioselective addition of terminal alkynes to aldehydes was recently extended to the use of terminal ynamides [68].
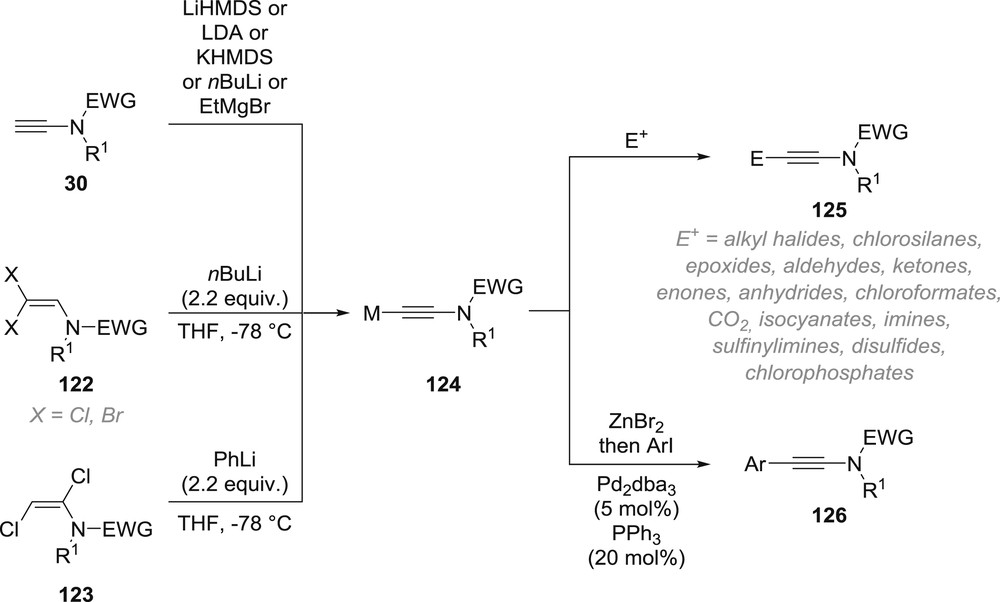
Generation and functionalization of metallated terminal ynamides.
6 Conclusions and outlook
As evidenced by all examples overviewed in this article, the anionic chemistry of ynamides has proven over the years to be extremely fruitful and productive. The development of efficient and robust methods for their preparation have facilitated the design of an ever-growing number of original methods based on their unique reactivity under anionic conditions. All together, these studies enabled not only the design and development of remarkably efficient and original reactions but also brought general answers to long-standing problems in organic chemistry and the emergence of new synthetic paradigms. Considering the number of reactive sites that can be used to trigger anionic transformations in ynamides, they can clearly be considered as unique building blocks in anionic chemistry provided that their reactivity can be finely understood, tuned and controlled. There is now clearly enough data, most of them being summarized in this article, to predict the anionic reactivity of ynamides, even if further developments will certainly be, at least in part, based on new, unpredictable and unforeseen reactivities. Let us wait and see.
Acknowledgements
The authors thank the Université libre de Bruxelles, the Fédération Wallonie–Bruxelles (ARC Consolidator 2014–2019) and the ANR (project DYNAMITE ANR-2010-BLAN-704). C.Z. acknowledges the Chinese Scholarship Council (CSC) for a graduate fellowship.