

1 Introduction
Various types of metal complexes, such as Y(III) [1], Zn(II) [2], Al(III) [3], B(III) [4], Ni(II) [5], Cu(II) [6–8] with chelating anilido-imine (Fig. 1, I) ligands have received extensive attention in recent years due to their applications in coordination chemistry and catalysis. The anilido-imine compounds have similar frameworks and combine the steric and electronic features of the β-diketiminate (Fig. 1, II) and salicylaldiminato (Fig. 1, III) ligand frameworks extensively researched in bioinorganic and transition metal chemistry [9,10]. The general method for the synthesis of anilido-imine compounds involved the condensation of the ortho-fluorobenzaldehyde with 1 equiv of amine to form a Schiff base and the subsequently nucleophilic substitution of the Schiff base by aromatic amide lithium (Scheme 1). Previously, we have reported the luminescent properties and coordination chemistry of Zn(II) complexes supported by anilido-imine and salicylaldiminato ligands [2c,11]. As part of our continuing study, we designed a new multidentate anilido-imine compound (Fig. 1, 2) containing o-OMe-anilinyl, which has two N and two O donor atoms and could be used to synthesize polynuclear metal complexes with Zn(II) ions. Thus we tried to synthesize ortho-C6H4F(CHNC6H4OMe-2) with ortho-flurobenzaldehyde and 2-methoxyaniline as the starting materials according to the literature [1]. However, a new compound 1 with formula ortho-C6H4F[CH(NHC6H4OMe-2)2] was always obtained.

Molecular structure of anilido-imine (I), β-diketiminate (II), salicylaldiminato (III) and object compound (2).
Synthetic routes for anilido-imine compounds.
As we know, the reaction of 1,2-diamine with substituted aldehydes produces the corresponding imidazolidine, which are the intermediates for the synthesis of substituted dihydroimidazole [12]. To the best of our knowledge, there are few reports on the reaction of amine with substituted aldehydes to form the phenylmethanediamine. Furthermore, compound 1 was readily converted into the anilido-imine compound by treatment with n-BuLi. The new anilido-imine compound could be used as a potentially multidentate ligand for preparing polynuclear metal complexes. Herein, we wish to report the synthesis and characterization of a new multidentate anilido-imine compound containing o-OMe-anilinyl and the corresponding zinc complex, as well as their specific structural features.
2 Experimental
2.1 General comments
All organometallic reactions were performed using standard Schlenk techniques under a high-purity argon atmosphere or glovebox techniques. n-Hexane, THF, and toluene were dried by refluxing over sodium and benzophenone and distilled under argon prior to use. n-BuLi was purchased from Aldrich and used as received. 1H and 13C NMR spectra were measured using a Varian Mercury-300 or Bruker Avance 500 NMR spectrometer. The elemental analyses were performed on an Elementar Vario EL cube analyzer. IR spectra were recorded on an IRAffinity-1 spectrometer using KBr pellets. All melting points were determined by an X-5 micro-melting point apparatus and are uncorrected.
2.2 Synthesis of ortho-C6H4F[CH(NHC6H4OMe-2)2] (1)
A mixture of ortho-flurobenzaldehyde (5.00 mL, 47.5 mmol) and 2-methoxyaniline (10.70 mL, 95.0 mmol) in n-hexane (50 mL) was stirred at room temperature overnight. A lot of white solid is formed. The mixture was filtered and washed with n-hexane (4 mL × 3) under reduced pressure. The white solid product was dried in vacuo. Yield: 15.90 g, 95.0%. mp 76–78 °C. Anal. calcd for C21H21FN2O2 (352.4): C 71.57, H 6.01, N 7.95. Found: C 71.58, H 5.91, N 7.95%. 1H NMR (300 MHz, DMSO-d6, 298 K): δ = 3.74 (s, 3H, OCH3), 3.80 (s, 2 × 3H, OCH3), 4.67 (br, 2H, ArNH), 6.48–6.54 (m, 1H), 6.60–6.69 (m, 2H), 6.77 (dd, 1H, J = 1.2 Hz, 9.0 Hz), 6.97 (dt, 1H, J = 1.2 Hz, 9 Hz), 7.08 (dd, 2H, J = 1.5 Hz, 8.1 Hz), 7.19–7.25 (m, 1H), 7.32–7.39 (m, 2H), 7.57–7.64 (m, 1H), 8.08 (dt, 1H, J = 1.8 Hz, 9 Hz), 8.71 (s, 1H) ppm. 13C NMR (75 MHz, DMSO-d6, 298 K): δ = 55.1 (OCH3), 55.5 (OCH3), 110.5, 112.1, 113.8, 116.1, 120.5, 120.8, 120.9, 124.8, 124.9, 127.0, 127.7, 127.8, 133.4, 133.6, 137.6, 141.0, 146.3, 151.8, 153.8 ppm. IR (KBr, cm−1): υ 3425 (N–H), 3364 (N–H), 3065, 3042, 3016, 2962, 2936, 2902, 2834, 1844, 1802, 1598, 1507, 1487, 1458, 1419, 1361, 1339, 1320, 1251, 1243, 1224, 1176, 1151, 1136, 1123, 1104, 1088, 1061, 1051, 1019, 949, 897, 855, 832, 810, 779, 763, 732, 647, 591, 521, 461.
2.3 Synthesis of ortho-C6H4(2-OMeC6H4)(CHNC6H4OMe-2) (2)
A solution of n-BuLi (8.9 mL, 1.60 mol/L, 14.2 mmol) in n-hexane was added to a solution of ortho-C6H4F[CH(NHC6H4OMe-2)2] (5.00 g, 14.2 mmol) in THF (40 mL) at –78 °C. The mixture was allowed to warm to room temperature and stirred for four days. The reaction was quenched with H2O (20 mL). The water phase was extracted with ethyl ether (20 mL × 2). The combined organic phase was dried over anhydrous MgSO4 and evaporated to dryness to give the crude product as a brown–red oil, which was further purified by column chromatography on silica gel with ethyl acetate/petroleum ether (1:2 in volume) as the eluent to give the pure product as yellowish crystals (4.10 g, 87.0%). mp 78–80 °C. Anal. calcd. for C21H20N2O2 (332.4): C 75.88, H 6.06, N 8.43. Found: C 75.84, H 6.04, N 7.97%. 1H NMR (500 MHz, CDCl3, 298 K): δ = 3.90 (s, 2 × 3H, OCH3), 6.83 (t, 1H, J = 7.0 Hz), 7.03 (m, 5H), 7.14 (dd, 1H, J = 1.5, 8.0 Hz), 7.21 (dt, 1H, J = 1.5, 7.0 Hz), 7.29 (m, 1H) 7.38 (d, 1H, J = 12.5 Hz), 7.45 (d, 1H, J = 7.5 Hz), 7.57 (d, 1H, J = 3.5 Hz), 8.69 (s, 1H, CH = NAr), 11.25 (s, 1H, NH) ppm. 13C NMR (125 MHz, CDCl3, 298 K): δ = 55.8 (OCH3), 56.1 (OCH3), 113.4, 117.0, 119.6, 120.6, 120.9, 121.1, 121.2, 123.2, 126.4, 130.8, 131.6, 134.8, 140.9, 146.0, 151.9, 152.5, 163.4 (CHN) ppm. IR (KBr, cm−1): υ 3462, 3006, 2956, 1844, 1811, 1771, 1620, 1589, 1571, 1524, 1495, 1456, 1383, 1338, 1249, 1173, 1159, 1114, 1047, 1028, 972, 913, 839, 752, 743.
2.4 Synthesis of trinuclear zinc complex 3
A solution of ortho-C6H4(2-MeO-C6H4NH)(CHNC6H4OMe-2) (0.25 g, 0.8 mmol) in toluene (10 mL) was slowly added to a solution of ZnEt2 (1.20 mmol) in toluene (10 mL) at room temperature under stirring. The mixture was stirred at room temperature for 1 h and at 80 °C for an additional 4 h. The solvent was removed in vacuo, and the obtained orange–red residue was recrystallized from n-hexane/toluene (v/v = 5:1, 5 mL) to give an orange–red solid (0.17 g, 45%). 1H NMR (300 MHz, C6D6, 298 K): δ = 0.54 (q, J = 7.5 Hz, 2H, ZnCH2CH3), 0.84 (t, J = 7.5 Hz, 3H, CHCH2CH3), 1.17 (t, J = 7.5 Hz, 3H, ZnCH2CH3), 1.74–1.86 (m, 2H, CHCH2CH3), 3.30 (s, 6H, OCH3), 3.40 (s, 6H, OCH3), 4.72 (d, J = 6.0 Hz, 2H, CHCH2CH3), 5.86 (d, J = 6.0 Hz, 1 H, Ph-H), 6.10 (d, J = 6.0 Hz, 1H, Ph-H), 6.26 (d, J = 6.0 Hz, 1H, Ph-H), 6.38–6.44 (m, 2H, Ph-H), 6.51 (t, J = 6.0 Hz, 1H, Ph-H), 6.60–6.68 (m, 4H, Ph-H), 6.71–6.75 (m, 2H, Ph-H), 6.82–6.87 (m, 4H, Ph-H), 6.90–6.95 (m, 2H, Ph-H), 7.06–7.11 (m, 2H, Ph-H), 7.29–7.38 (m, 2H, Ph-H), 7.45–7.55 (m, 2H, Ph-H) ppm. IR (KBr, cm−1): υ 2934 (w), 2842 (w), 2360 (w), 1594 (m), 1490 (s), 1450 (m), 1231(s), 1171(m). 1115 (m), 1015 (m), 890 (w), 732 (s), 502 (w), 441 (w).
2.5 X-ray structure determinations of 1–3
The single-crystal X-ray diffraction data for 1–3 were collected on a Rigaku R-AXIS RAPID IP diffractometer equipped with graphite-monochromated Mo Kα radiation (λ = 0.71073 Å), operating at 293 ± 2 K. The structures were solved by direct method [13] and refined by full-matrix least squares based on F2 using the SHELXTL 5.1 software package [14]. All non-hydrogen atoms were refined anisotropically. Unless otherwise noted, hydrogen atoms were included in idealized position and were allowed to ride.
3 Results and discussion
3.1 Synthesis of compounds 1 and 2
A lot of known Schiff compounds ortho-C6H4F(CH = NAr′) (Ar′ = 2,6-iPr2C6H3, 2,6-Me2C6H3, 2,6-Et2C6H3, 4-MeC6H4, Ph) [1,2c,3,5,7,11] have been readily synthesized by condensation reaction of ortho-fluorobenzaldehyde with 1 equiv of the relevant amine in n-hexane in the presence of anhydrous MgSO4. However, a new compound, ortho-C6H4F[CH(NHC6H4OMe-2)2] 1, was formed by the reaction of ortho-fluorobenzaldehyde with 1 equiv of 2-methoxyaniline in similar condition, with trace amounts of ortho-C6H4F(CHNC6H4OMe-2) (Pre1) obtained as a by-product (Scheme 2). The new compound ortho-C6H4F[CH(NHC6H4OMe-2)2] 1 was readily obtained when the two liquid raw materials were mixed together in n-hexane or free of solvent in any ratio. The highest yield was obtained when the mole ratio of the ortho-flurobenzaldehyde to 2-methoxyaniline is 1:2.

Synthetic routes for compounds 1 and 2.
Compound 1 is insoluble in water, slightly soluble in n-hexane, while soluble in hot n-hexane, toluene, and THF. Fortunately, white crystals suitable for X-ray crystal structure determination were obtained in n-hexane at room temperature. The detailed crystal structure information will be shown in the crystal description section. Compound 2 was readily synthesized by the reaction of compound 1 with 1 equiv of n-BuLi in THF (Scheme 2) and purified by chromatography on silica gel with ethyl acetate/petroleum ether as the eluent to give pure products as yellowish crystals. Compound 2 is soluble in common solvents, such as n-hexane, methylene chloride, chloroform, ethyl acetate, toluene, and THF.
Both compounds 1 and 2 were characterized by 1H and 13C NMR spectroscopy, IR along with elemental analysis, and satisfactorily analytic results were obtained. Compound 1 was found to decompose gradually in chloroform due to its sensitivity to acids or acidic solvents. The 1H NMR and 13C NMR spectra of compound 1 in a deuterated DMSO solution were obtained. The 1H NMR spectrum of 1 exhibits a broad resonance at δ = 4.67 ppm for the NH proton. The methylene CH proton of compound 1 exhibits a resonance at 8.71 ppm and the corresponding methylene CH carbon exhibits a resonance at 153.8 ppm. The 1H NMR spectrum of 2 exhibits a resonance at δ = 8.69 ppm for the imino CH proton, while the corresponding 13C NMR resonance is at δ = 163.4 ppm. Compared with the 1H NMR spectrum of 1, the NH proton resonance at 4.67 ppm disappeared and a characteristic NH proton resonance for anilido-imine compound 2 at 11.25 ppm appeared, which were comparable to other reported compounds of this type 10.53–11.64 ppm for ortho-C6H4{NH(C6H3Ar)}(CH = NAr′) [2c,3a,3b,15].
The IR data is consistent with the presented structures. The middle strong band at ca. 1123 cm−1 associated with the C–F stretching vibration is present in the IR spectrum of compound 1. The characteristic strong band at ca. 1620 cm−1 associated with the imine CN stretching vibration is present in the IR spectrum of compound 2.
3.2 Synthesis of trinuclear zinc complex 3
The reaction of compound 2 with 1.5 equiv of ZnEt2 at 80 °C caused the elimination of ethylane and alkylation of the imino group of the ligand, and further yields the trinuclear zinc complex 3 (Scheme 3). 1H NMR analysis of complex 3 revealed a characteristic set of peaks for the dianionic tetradentate ligand and the coordinated ethyl group. The N–H proton signal of the free ligands disappeared, and the new Zn–CH2CH3 proton signals appeared at a higher field (0.54–0.84 ppm), which is indicative of the formation of a Zn–N bond in the new complex. The absence of the signal for the imino proton suggested the alkylation of the imino group of the ligand, which was also confirmed by the formation of a CH(CH2CH3)N group exhibiting discrete multiple resonances, assigned to the methylene protons. The OMe protons showed a high field shift at 3.30 and 3.40 ppm compared to those at 3.90 ppm for the free ligand, suggesting that the OMe moiety coordinated to the zinc ion in a η1-fashion. The IR spectrum of complex 3 showed a strong band at 1231 cm−1 attributed to the C–N stretching vibration and the disappearance of the CN stretching vibration bonds at 1620 cm−1, which also indicated the alkylation of the imino group of the ligand. Moreover, the Zn–N stretching vibration is observed at 441 and 502 cm−1.

Synthetic route for complex 3.
3.3 The probable mechanisms for the formation of anilido-imine 2
Two probable mechanisms involving the transformation of the compound 1 to compound 2 in the presence of n-BuLi have been proposed, as shown in Scheme 4. When treated the compound 1 with 1 equiv of n-BuLi, one of the secondary amines was deprotonated. Then, the nucleophilic nitrogen anion could attack the C–F bond in the central aromatic ring (Scheme 4 route a) or the C–N bond in the same carbon atom (Scheme 4 route b). According to the route a, the C–F bond cleavage occurs and a new C–N bond forms, resulting in a new four-membered nitrogen-containing heterocycle. Then, the other secondary amine was further deprotonated by 1 equiv of n-BuLi. The formed nitrogen anion could attack the C–N bond in the four-membered ring to cause a ring-opening reaction of the four-membered nitrogen-containing heterocycle, resulting in the formation of a CN bond and an anilido anion. After hydrolysis with 1 equiv of H2O, ortho-C6H4(2-OMeC6H4)(CHNC6H4OMe-2) is produced. According to route b, the Schiff base ortho-C6H4F(CHNC6H4OMe-2) and lithium 2-methoxy-phenylamine are produced in the nucleophilic reaction. The C–F bond in ortho-C6H4F(CHNC6H4OMe-2) should be further substituted by lithium 2-methoxyphenylamine in a nucleophile way to produce ortho-C6H4(2-OMeC6H4)(CHNC6H4OMe-2). In route a, 2 equiv of n-BuLi are needed, while 1 equiv of n-BuLi is needed in route b. Compound 1 was treated with 1 equiv of n-BuLi and 2 equiv of n-BuLi, respectively. The results indicated that only 1 equiv of n-BuLi was needed. So, route b is more probable, as indicated by the total amount of n-BuLi during the reaction (Scheme 4).

The probable mechanism for the formation of compound 2.
3.4 Crystal structures of 1–3
The molecular structures of 1–3 were determined by X-ray crystallographic analysis. Crystals of compound 1 suitable for X-ray crystal structure determination were grown from n-hexane at room temperature. Crystals of compound 2 suitable for X-ray crystal structure determination were grown from ethyl acetate/petroleum ether at room temperature. Crystals of trinuclear zinc complex 3 suitable for X-ray crystal structure determination were grown from toluene/n-hexane at room temperature. The ORTEP drawings of molecular structures of 1–3 are shown in Figs. 2–4, respectively. The crystallographic and refinement data for 1–3 are summarized in Table 1. Hydrogen bond geometries for 1 and 2 are given in Table 2. Selected bond lengths and angles for 3 are given in Table 3.

Molecular structure of compound 1 (the other molecule has been omitted for clarity). The thermal ellipsoids are drawn at 30% probability levels.

Molecular structure of compound 2 (the thermal ellipsoids are drawn at 30% probability levels).

Molecular structure of the trinuclear zinc complex 3 (all the hydrogen atoms have been omitted for clarity. The thermal ellipsoids are drawn at 30% probability levels).
Crystal data and structural refinements details for 1–3.
| 1 | 2 | 3 | |
| Formula | C21H21FN2O2 | C21H20N2O2 | C50H58N4O4Zn3 |
| Fw | 352.4 | 332.39 | 975.11 |
| Temperature/K | 293(2) | 293(2) | 293(2) |
| Crystal system | Triclinic | Monoclinic | Monoclinic |
| Space group | P | P2(1) | C2/c |
| a/Å | 12.268(3) | 11.736(2) | 17.954(4) |
| b/Å | 12.391(3) | 7.3385(15) | 13.364(3) |
| c/Å | 13.236(3) | 11.975(2) | 19.999(4) |
| α/˚ | 75.84(3) | 90 | 90 |
| β/˚ | 67.39(3) | 119.23(3) | 108.54(3) |
| γ/˚ | 76.59(3) | 90 | 90 |
| Volume (Å3) | 1779.5(6) | 900.1(3) | 4549.3(16) |
| Z | 4a | 2 | 4 |
| Dcalcd (Mg.m−3) | 1.315 | 1.226 | 1.424 |
| F(000) | 744 | 352 | 2032 |
| θ range for data collection | 3.23–27.48 | 3.39–27.48 | 3.05–27.47 |
| Limiting indices | –15 ≤ h ≤ 15, –16 ≤ k ≤ 16, –17 ≤ l ≤ 17 | –14 ≤ h ≤ 15, –9 ≤ k ≤ 9, –15 ≤ l ≤ 15 | –21 ≤ h ≤ 23, –17 ≤ k ≤ 16, –25 ≤ l ≤ 25 |
| Data/restraints/parameters | 7949/0/482 | 3940/1/232 | 5160/18/281 |
| Goodness-of-fit on F2 | 1.046 | 1.040 | 1.041 |
| Final R indices [I > 2σ(I)] | R1b = 0.1258, wR2c = 0.3184 | R1b = 0.0401, wR2c = 0.0819 | R1b = 0.0841, wR2c = 0.2031 |
| R indices (all data) | R1b = 0.2130, wR2c = 0.3737 | R1b = 0.0634, wR2c = 0.0888 | R1b = 0.1606, wR2c = 0.2421 |
| Largest diff. peak and hole/e·A−3 | 0.672/–0.339 | 0.101/–0.107 | 1.398/–0.520 |
a There are two crystallographically independent molecules in the asymmetric unit.
b R1 = ∑||Fo|–|Fc||/∑|Fo|.
c wR2 = [∑[w (Fo2–Fc2)2]/∑[w (Fo2)2]]1/2.
Hydrogen bond geometries for 1 and 2.
| Structure | D–H···A | d(D–H)(Å) | d(H···A)(Å) | d(D···A)(Å) | < (DHA) (deg) |
| 1a | N(2)–H(102)···O(1)#1 | 0.86 | 2.60 | 3.382(6) | 151.2 |
| N(1)–H(101)···O(1) | 0.83(6) | 2.16(6) | 2.620(5) | 115(5) | |
| N(2)–H(102)···O(2) | 0.86 | 2.36 | 2.631(6) | 98.9 | |
| N(3)–H(103)···O(4) | 0.93(6) | 2.13(6) | 2.645(6) | 114(5) | |
| N(3)–H(103)···O(3)#2 | 0.93(6) | 2.43(7) | 3.281(6) | 152(5) | |
| N(4)–H(104)···O(3) | 0.86 | 2.24 | 2.611(5) | 106.3 | |
| C(3)–H(3)···N(1) | 0.93 | 2.55 | 2.867(7) | 100.3 | |
| C(24)–H(24)···N(4) | 0.93 | 2.52 | 2.857(7) | 101.4 | |
| C(3)–H(3)···O(2)#1 | 0.93 | 2.70 | 3.438(7) | 136.5 | |
| C(42)–H(42B)···F(1)#3 | 0.96 | 2.72 | 3.270(7) | 116.8 | |
| C(7)–H(7)···F(1) | 0.98 | 2.50 | 2.817(5) | 98.3 | |
| C(14)–H(14B)···F(2)#4 | 0.96 | 2.81 | 3.394(7) | 120.4 | |
| C(28)–H(28)···F(2) | 0.98 | 2.53 | 2.811(6) | 96.4 | |
| C(24)–H(24)···O(4)#2 | 0.93 | 2.64 | 3.412(6) | 140.9 | |
| 2b | C(14)–H(14C)···O(2)#1 | 0.96 | 2.61 | 3.500(3) | 154.2 |
| N(1)–H(1)···N(2) | 0.85(2) | 2.018(18) | 2.698(2) | 136.9(17) |
a #1 – x, –y, –z + 1; #2 – x + 1, –y + 1, –z + 2; #3 x, y + 1, z + 1; #4 x – 1, y, z.
b #1 – x, y – 1/2, –z + 1.
Selected bond lengths [Å] and angles [°] for 3.
| Complex 3a | |||
| Zn(1)–N(1) | 2.009(5) | N(1)–Zn(1)–Zn(2)#1 | 130.85(15) |
| Zn(1)–N(2) | 2.040(5) | N(2)#1–Zn(1)–Zn(2)#1 | 49.83(16) |
| Zn(2)–N(1) | 2.118(5) | N(2)–Zn(1)–Zn(2)#1 | 129.75(16) |
| Zn(2)–N(2) | 2.130(6) | C(24)–Zn(2)–N(1) | 134.1(4) |
| Zn(2)–O(1) | 2.387(5) | C(24)–Zn(2)–N(2) | 132.7(4) |
| Zn(2)–O(2) | 2.441(5) | N(1)–Zn(2)–N(2) | 93.2(2) |
| Zn(2)–C(24) | 1.980(8) | C(24)–Zn(2)–O(1) | 103.0(3) |
| C(1)–N(2) | 1.459(9) | N(1)–Zn(2)–O(1) | 71.80(19) |
| C(7)–N(1)#1 | 1.486(9) | N(2)–Zn(2)–O(1) | 90.68(19) |
| Zn(1)–Zn(2)#1 | 2.7684(10) | C(24)–Zn(2)–O(2) | 101.7(3) |
| N(1)#1–Zn(1)–N(1) | 129.8(3) | N(1)–Zn(2)–O(2) | 92.33(18) |
| N(1)#1–Zn(1)–N(2)#1 | 99.4(2) | N(2)–Zn(2)–O(2) | 70.8(2) |
| N(1)–Zn(1)–N(2)#1 | 102.2(2) | O(1)–Zn(2)–O(2) | 155.21(17) |
| N(1)#–Zn(1)–N(2) | 102.2(2) | C(7)#1–N(1)–Zn(1) | 108.8(4) |
| N(1)–Zn(1)–N(2) | 99.4(2) | C(7)#1–N(1)–Zn(2) | 113.5(4) |
| N(2)#1–Zn(1)–N(2) | 127.6(3) | Zn(1)–N(1)–Zn(2) | 84.2(2) |
| N(1)#1–Zn(1)–Zn(2)#1 | 49.56(15) | Zn(1)–N(2)–Zn(2) | 83.2(2) |
| C(1)–N(2)–Zn(1) | 111.2(4) | C(22)–N(1)–Zn(1) | 117.1(4) |
| C(10)–N(2)–Zn(1) | 117.6(4) | C(22)–N(1)–Zn(2) | 114.2(4) |
| C(1)–N(2)–Zn(2) | 111.5(4) | C(10)–N(2)–Zn(2) | 116.1(4) |
a Symmetry transformations used to generate equivalent atoms: #1 – x, y, –z + 1/2.
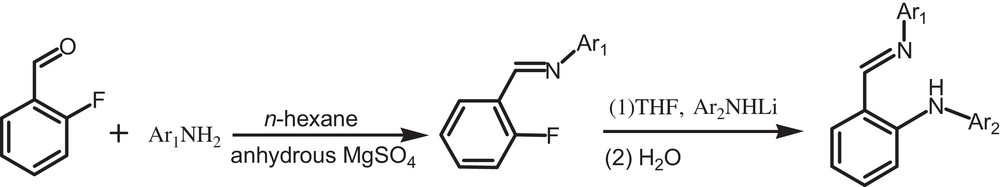
X-ray analysis reveals that the unit cell of 1 contains two independent molecules, one (molecule A) of which is shown in Fig. 2. The dihedral angles among phenyl rings are 94.4°, 102.7° and 71.2° (102.8°, 99.4° and 88.2° in molecule B), respectively. In molecule A, there exist an intramolecular C–H···F interaction with an S(5) motif, a 16C–H···N interaction with an S(5) motif and a N–H···O interaction with an S(5) motif. Two adjacent molecules A form a dimer A through intermolecular N–H···O and C–H···O interactions. There are similar interactions in molecules B. Dimer A and dimer B were further linked through the intermolecular C–H···F interactions to form a 3-D network (Fig. 5).
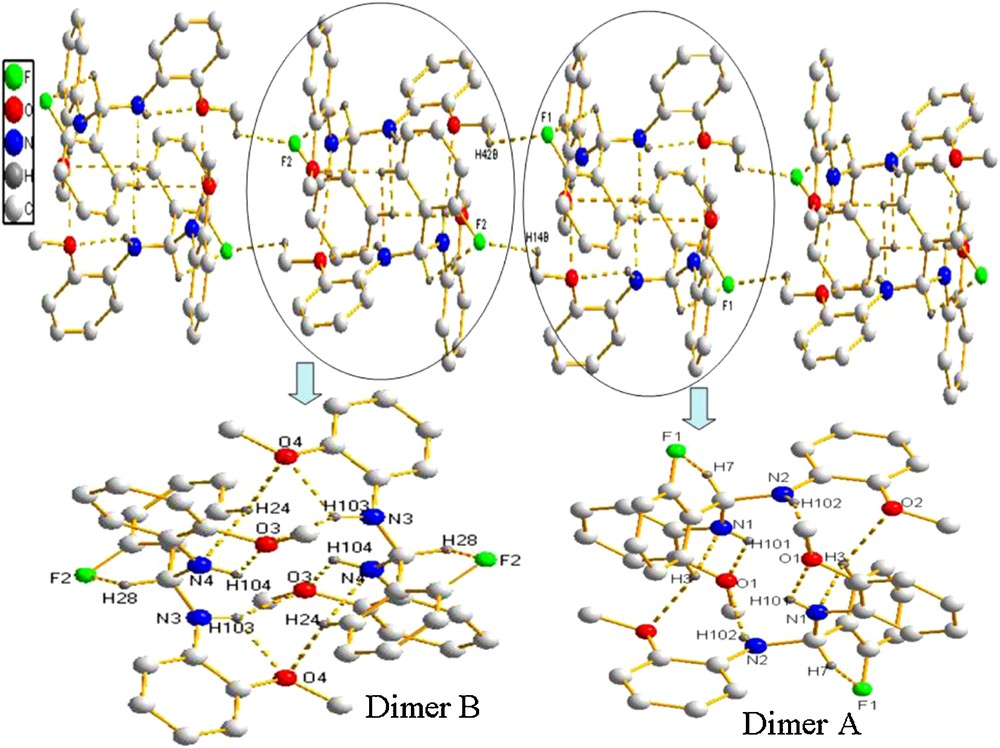
(Color online.) Packing of compound 1 (hydrogen bonds are indicated by dashed lines; the hydrogen atoms not involved in hydrogen bonds are omitted for clarity).
In molecule 2, the bond lengths and angles are within normal range. The CN bond length [1.272(2) Å] is comparable to those found in similar anilido-imine compounds, such as ortho-C6H4{7-NH(2,4-Me2)C9H4N}(CHNC6H3Me2-2,6) [1.271(2) Å] [2c], ortho-C6H4{7-NH(2,4-Me2)C9H4N}(CHNC6H3Et2-2,6) [1.267 (4), 1.275 (4) Å] [2c], and ortho-C6H4(4-OMeC6H4)(CHNC6H3Et2-2,6) [1.2730 (15) Å] [15]. The dihedral angles between the central benzene ring (C1/C2/C3/C4/C5/C6) and the two MeO-substituted benzene rings of the anilido-imine compound are 57.8° (C8/C9/C10/C11/C12/C13), and 132.8° (C15/C16/C17/C18/C19/C20), respectively. The dihedral angle between the two MeO-substituted benzene rings (ring C8/C9/C10/C11/C12/C13 and ring C15/C16/C17/C18/C19/C20) is 109.5°. An intramolecular N–H···N hydrogen bond forms a six-membered ring, generating an S(6) motif [16]. In the packing of the crystal, the adjacent molecules were linked through intermolecular C–H···O interactions [3.500(3) Å, 154.2°,–x, y – 1/2, –z + 1] arising from the interactions between the oxygen atom of the methoxyl in the phenyl ring bonding to the anilido nitrogen atom and the hydrogen atom of the methoxyl in the phenyl ring bonding to the imino nitrogen atom to form a 1-D S-shaped chain (Fig. 6). There are no interactions between each adjacent chain.
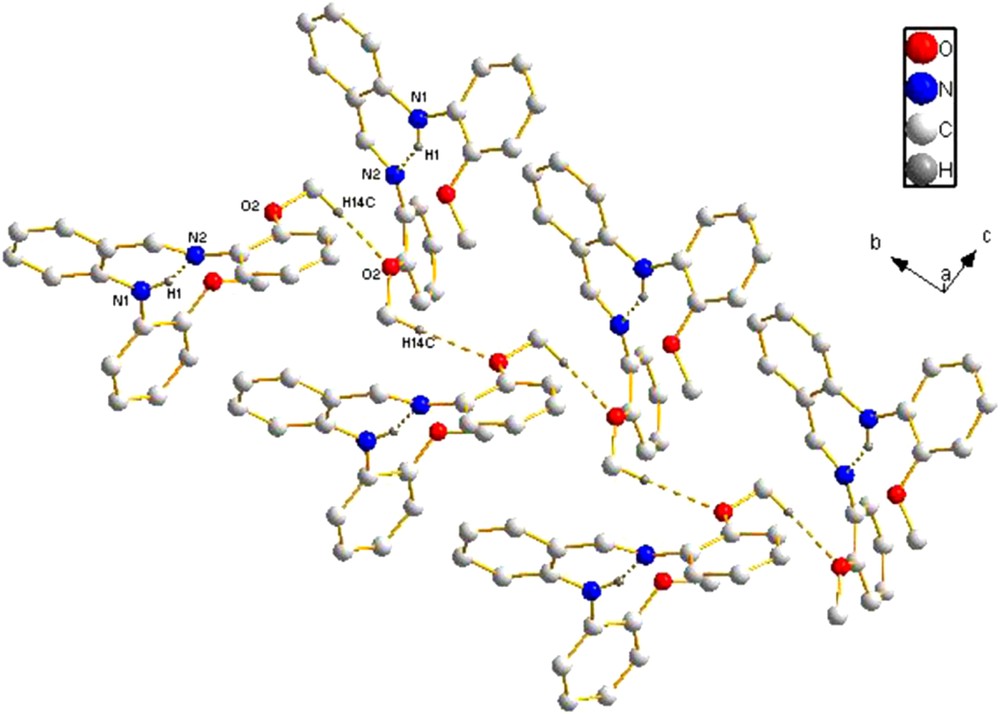
(Color online.) Packing of compound 2 (hydrogen bonds are indicated by dashed lines; the hydrogen atoms not involved in hydrogen bonds are omitted for clarity).
Trinuclear zinc complex 3 crystallizes in the monoclinic space group C2/c. The molecule has a C2 symmetry axis. As shown in Fig. 4, the Zn1 cation locates on a two-fold axis and adopts a distorted tetrahedral geometry with the metal centre chelated by two bidentate bisanilido ligands. The N1–Zn1–N2 bite angle is 99.4(2)°. The Zn2 and Zn2#1 (#1 – x, y, –z + 1/2) ions are five-coordinated and display the slightly distorted trigonal–bipyramidal geometry with the equatorial plane defined by the N1, N2, and C24 atoms, and the O1 and O2 atoms in the axial positions. The mean deviation of the Zn2 ion from the equatorial plane is only 0.0025 Å. The angles in the equatorial plane range from 93.2(2)° to 134.1(4)°, and the axial Zn2–O bonds subtend an angle of 155.21(17)°. The Zn2–N1 (2.118(5) Å) and Zn2–N2 (2.130(6) Å) bond lengths are slightly longer than the corresponding Zn1–N bond lengths (Zn1–N1: 2.009(5) Å; Zn1–N2: 2.040(5) Å). The Zn2–O1 and Zn2–O2 distances are 2.387(5) and 2.441(5) Å, respectively, indicating the weak coordination interaction between the zinc atom and the oxygen atoms in the methoxyl groups. The Zn–C bond length of 1.980(8) Å is in the normal range of the corresponding reported values (1.85–2.01 Å for the distances of Zn alkyl bonds) [17]. The Zn–Zn separation distance is 2.7684(10) Å, indicative of strong metal–metal interaction. The dihedral angle between the ring Zn1/Zn2/N1/N2 and the ring Zn1/Zn2A/N1A/N2A is 69.2°. There also exist π–π interactions between the two methoxyl substituted phenyl rings with the separation distances 3.0542 Å (distance between the ring C17/C18/C19/C20/C21/C22 and the ring C17A/C18A/C19A/C20A/C21A/C22A) and 3.6409 Å (distance between the ring C10/C11/C12/C13/C14/C15 and the ring C10A/C11A/C12A/C13A/C14A/C15A), respectively.
4 Conclusions
In conclusion, a new adduct ortho-C6H4F[CH(NHC6H4OMe-2)2] has been synthesized and characterized. Furthermore, an efficient and new method has been found for the synthesis of anilido-imine compound ortho-C6H4(2-OMeC6H4)(CHNC6H4OMe-2). The packing of 1 was stabilized by C–H···O, C–H···N, C–H···F and N–H···O hydrogen bonds. The packing of 2 was stabilized by C–H···O and N–H···N hydrogen bonds. The novel trinuclear zinc complex 3 was obtained from the reaction of ZnEt2 with ortho-C6H4(2-OMeC6H4)(CHNC6H4OMe-2) by alkane elimination along with the alkylation of the imino group of the ligand. The trinuclear zinc complex 3 represents a rare example of zinc alkyl derivative stabilized by two bianionic ONNO tetradentate ligands in four to five coordination mode.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant Nos. 21004026 and 21074043). We are also grateful for support by the Frontiers of Science and Interdisciplinary Innovation Project of Jilin University (Nos. 450060445023 and 450060445027). We thank Prof. Yang Guangdi for assistance with the crystal resolution.