

1 Introduction
Enaminoketones (or β-ketoamines) and related compounds possessing the structural unit N–CC–Z (Z = COR, CO2R, CN, etc.) are versatile synthetic intermediates that combine the ambident nucleophilicity of enamines with the ambident electrophilicity of enones [1].
Some specific procedures reported for the synthesis of β-enaminocarbonyl derivatives include the reaction of amines and 1,3-diketones over clay K10 or silica under microwave irradiation [2], reaction of β-aminoketones in the presence of triethylamine prompted by Pd(II) [3], and reactions of α-metalated imines with esters [4]. The generally employed method for the preparation entails the reaction between ammonia or a primary or secondary amines with 1,3-dicarbonyl compound in benzene solution with azeotropic removal of the water formed [5–9].
Recent years have witnessed an impressive revival of interest in bidentate, β-diketiminate ligands. Much of the recent works has focused on more sterically hindered derivatives that feature bulky substituents at the nitrogen [10,11].
This ligand family is a subset of Schiff base ligands [12] and has been known for many years with initial interest driven by their influence on the electronic structure of coordination compounds [13,14]. In their deprotonated form, these uninegative, four-electron donors can be viewed as hemiporphyrinate ligands with lower coordination numbers than cyclopentadienyl and tris(pyrazolyl)borate ligands. Diketiminate ligands share some similarities with amidinate ligands as both are monoanions in their deprotonated forms [15,16]. In addition to having a larger bite angle, β-diketiminate ligands have six electrons in the π-manifold and can be viewed as heteroatom analogues of pentadienyl ligands. The steric and electronic properties of β-diketiminate can be readily altered through an appropriate choice of amine and β-diketone used in their synthesis [17].
Recently, we have been interested in the synthesis of cationic methallyl complexes supported by the β-diimine ligand, the conjugate acid of the β-diiminate [18,19]. We have found that the coordination chemistry of this ligand was different in comparison with otherwise identical α-diimine [20].
However, whilst there are now many examples of this ligand in the deprotonated monoanionic form as chelate, there are no examples to date of the unsymmetrical and free protonated parent ligand itself. We report here a simple procedure for the synthesis of β-ketoamine starting from β-diketone and primary amines.
2 Results and discussion
Direct acid-catalysed condensation is a one-pot synthesis, employed to extend the range of β-iminoamine ligands available to coordination chemists with cases of variable bulk, electron donicity, rigidity and functionality.
Two ways of synthesis were used to prepare these compounds. The first one applied for the symmetrical β-diketone (pent-2,4-dione), requires two steps which translate the difference in reactivity of the two carbonyls. The intermediary β-ketoamine is initially obtained with high yields by heating β-diketone in the presence of the corresponding primary amine. All β-ketoamines were obtained in the solid form and are stable to the air and moisture. Their purification can be carried out by a simple crystallization from n-hexane.
In the second stage, the carbonyl group reacts with the ammonium salt of the aromatic amine in protic solvents to lead the β-iminoamine hydrochloride. The heating of a mixture of β-iminoamine hydrochloride and sodium hydroxide gives the corresponding β-iminoamine.
The low reactivity of the second carbonyl function with the ammonium salt of the aromatic amine has explained the low yield obtained [13]. Indeed it was noticed that the yield and the reaction speed during this stage were sensitive to the electronic effects and the steric encumberment of the aromatic amine used (Scheme 1(A)).
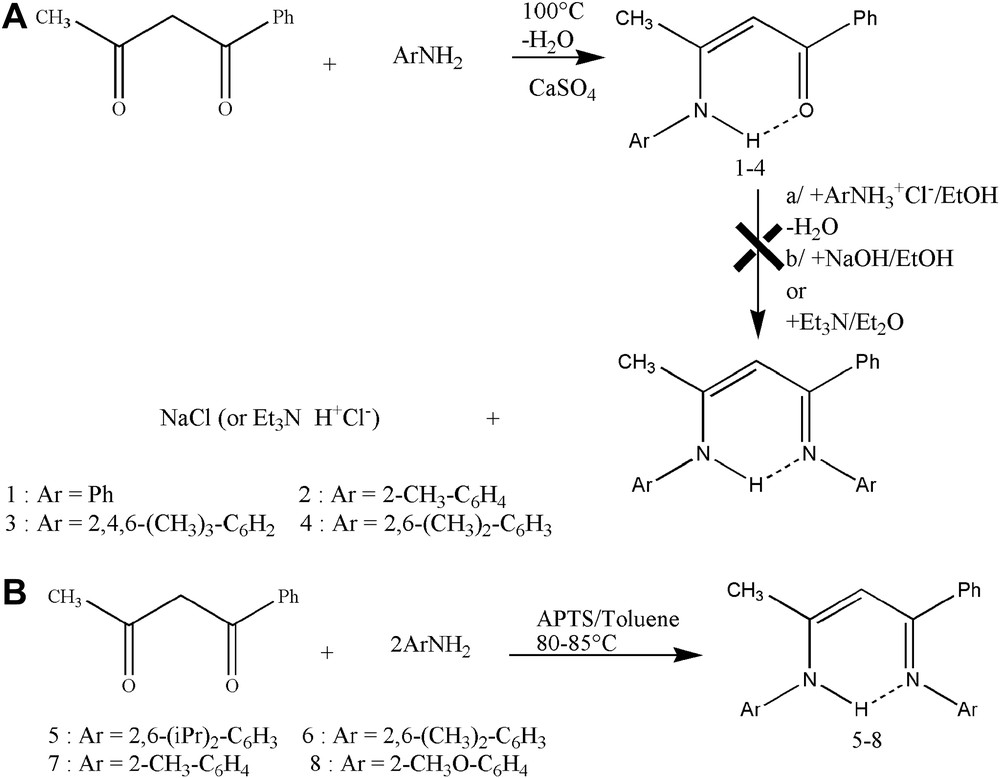
Synthesis of β-ketoamines and unsymmetrical β-iminoamines.
With an aim of varying the steric, electronic properties and the rigidity of the β-iminoamines, we used substituted diketones (1-methyl-3-phenylpent-2,4-dione and 1,3-diphenylpent-2,4-dione). The first synthesis gave access only to the β-ketoamines in the case of the 1-methyl-3-phenylpent-2,4-dione (Scheme 1(A)), whereas 1,3-diphenylpent-2,4-dione did not react. The second synthesis was applied for the ortho-substituted aniline with benzoylacetone as well as pent-2,4-dione. It consists of a condensation reaction of a β-diketone with 2 equiv of aromatic primary amines in the presence of an excess of paratoluene sulfonic acid (Scheme 1(B)).
Indeed heating in the toluene of a mixture of amine, β-diketone and 1.3 equiv of paratoluene sulfonic acid led in only one stage and in high yield to the corresponding β-iminoamine. The water is carried out by a simple azeotropic distillation using a Dean–Stark apparatus. The 1,3-diphenylpent-2,4-dione remains inactive.
All β-iminoamines used in this work were obtained in the solid form as yellow powders. They are slightly sensible to the air and moisture. These compounds were characterized by 1H, 13C NMR and IR spectroscopies.
All 1H NMR spectra of the ketoamines present a broad signal, which corresponds to the protons of the amine function (N–H). The chemical shift of this proton appears between 12.37 and 12.94 ppm. It is generally very sensitive to concentration, solvent nature and temperature. The protons of the methyl carried by the amino group appear between 1.68 and 2.43 ppm. The vinylic protons appear in the form of singlet between 5.82 and 5.94 ppm.
The NMR data of ligands are consistent with the hydrogen-bridged “β-iminoamine” structure shown in Scheme 1, although complexes incorporating neutral ligands of this type show that they are coordinated to nickel and palladium as a β-diimine tautomer [18,19].
The 1H NMR spectrum of the β-iminoamine ligands presents a singlet around 1.81 ppm, which corresponds to protons of the CH3 group. The vinylic protons of β-iminoamines are blinded and appear around 5.16 ppm, contrary to those for β-ketoamines, which appear towards 5.90 ppm. The presence of tautomeric β-iminoamine is confirmed by a broad signal corresponding to the amino proton (N–H).
The 13C NMR spectra of β-ketoamines present a signal characteristic of the sp2 carbon of the ketone function between 188.60 and 188.82 ppm, whereas the sp3 carbon of the amine function appears around 165 ppm. Vinylic carbons appear between 92.39 and 94.36 ppm.
Contrary to β-iminoamines having a symmetrical structure, unsymmetrical compound enabled us to locate carbons carrying the functions amine and imino. Indeed the first carbon (sp3) resonates between 154 and 159 ppm, whereas the second gives one signal around 165 ppm (unsymmetrical molecule). Vinylic carbon atoms of different β-iminoamines appear between 96.10 and 102.10 ppm.
The IR spectra of β-ketoamines 1–4 present ν(CO) stretching frequencies around 1650 cm−1, which correspond to the ketone. The wide broad ν(N–H) stretching frequencies around 3400 cm−1 are more significant and confirm well the obtention of β-ketoamine and not of β-ketoimine. The ν(CN) stretching frequencies of the β-iminoamine 5–8 are different from those of the β-ketoamine. They appear between 1610 and 1622 cm−1. The presence of ν(N–H) stretching frequencies confirm that they are indeed β-iminoamine tautomers. The substituents on the nitrogen atoms influence the frequencies CN and N–H. The ν(N–H) stretching frequencies are located around 3600 cm−1.
The compound 5 crystallizes in P21/n space group with a monoclinic system; the observed bond lengths are consistent with the delocalization of the imino and alkenes double bonds across the N–C–C–C–N backbone (Table 1 and Figs. 1 and 2)
Selected bond lengths [Å] and angles [°] for 5
| Bond lengths (Å) | Angles (°) | ||
| C(1)–C(2) | 1.513 (5) | N(1)–C(2)–C(3) | 120.4 (5) |
| C(2)–N(1) | 1.300 (5) | N(1)–C(2)–C(1) | 122.9 (5) |
| C(2)–C(3) | 1.334 (6) | C(3)–C(2)–C(1) | 116.7 (5) |
| C(3)–C(4) | 1.355 (6) | C(2)–C(3)–C(4) | 126.4 (5) |
| C(4)–N(2) | 1.339 (5) | N(2)–C(4)–C(3) | 122.3 (5) |
| C(4)–C(5) | 1.498 (6) | N(2)–C(4)–C(5) | 119.4 (5) |
| N(1)–H(2n) | 1.874 (2) | C(3)–C(4)–C(5) | 118.3 (5) |
| N(2)–H(2n) | 0.984 (2) | C(3)–C(4)–C(5)–C(10) | −56.9 (5) |
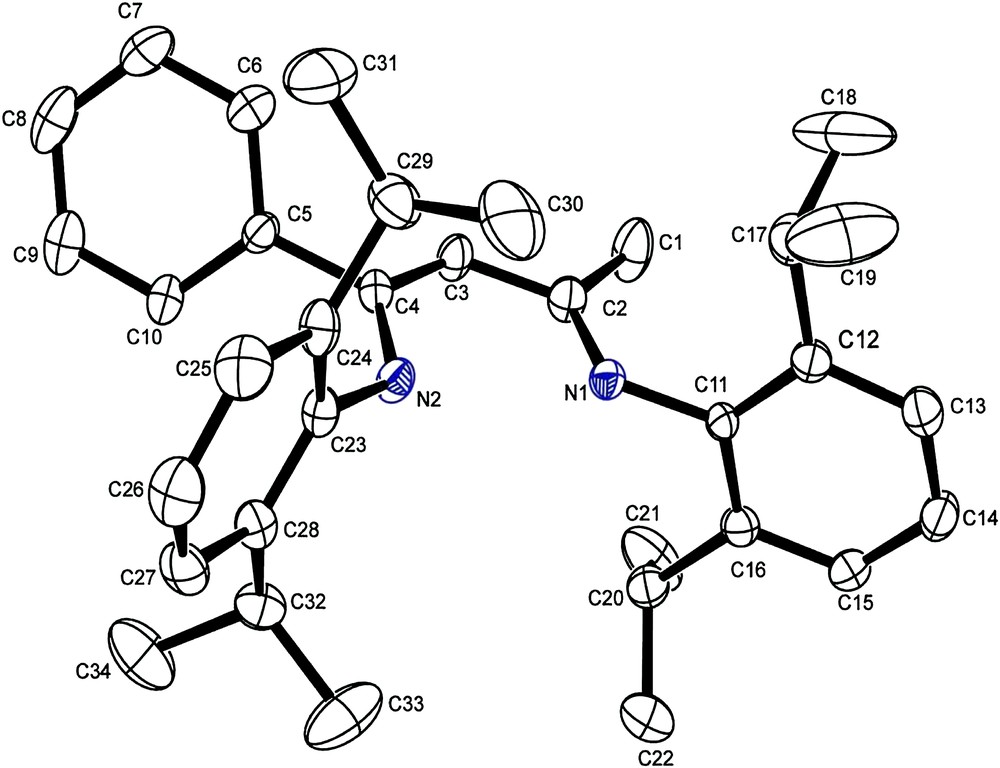
Perspective ORTEP diagram of β-iminoamine 5. Thermal ellipsoids are at 50% probability. Hydrogen atoms are omitted for clarity.

View of the chelate ring in 5. The aryl rings are omitted for clarity.
The N–C and C–C bond lengths of the N–C–C–C–N backbone are in the range associated with aromatic N–C and C–C bonds [21]. Variation between the N–C [N(1)–C(2) and N(2)–C(4)] and C–C [C(2)–C(3) and C(3)–C(4)] bonds is statistically insignificant.
Compound 5 thus crystallizes as a mixture of the two possible imine–enamine tautomer; inspection of the difference map around C(3) gives no evidence to suggest the presence of the β-iminoamine tautomer. As a consequence, the amine proton is disordered in 66:34% (50:50 for 2,6-(iPr)2-C6H3-NC(Me)CHC(Me)NH-2,6-(iPr)2-C6H3) [22] occupancy over both N(1) and N(2) with bond lengths of 0.90 (2) and 0.984 (2) Å, respectively.
The two backbone methyl and phenyl groups (C(1) and C(5)) adopt a syn orientation; this is surprising as an anti conformation would lead to a less congested conformer. It is likely that small steric, electronic and entropic factors, or crystal-packing forces, are responsible for this. The two Aryl rings adopt a synperiplanar arrangement in order to minimize steric crowding (Fig. 1).
The phenyl group carried by carbon C(4) is distorted out of the N(2)–C(4)–C(3)–C(2)–N(1) backbone plane by about 62°, as indicated by the torsion angle of C(3)–C(4)–C(5)–C(6) of 118.3°. This can be allotted to a steric effect caused by the isopropyl group carried by the aromatic rings related to the atom of nitrogen N(2) (Fig. 2).
3 Conclusion
In summary, we have developed a straightforward synthesis of new unsymmetrical β-iminoamine ligands in high yields. These compounds are the precursors of the β-diimine ligands. Their coordination to the palladium is currently under study. It gives new complexes supported by the unsymmetrical β-diimine ligands.
4 Experimental
4.1 General
All manipulations were carried out under an atmosphere of dry argon. Toluene was distilled from sodium benzophenone. Aniline, 2-methylaniline, 2,6-dimethylaniline, 2-methoxyaniline, 2,4,6-trimethylaniline and 2,6-diisopropylaniline were distilled from potassium hydroxide prior to use. Melting points were determined on a Büchi SMP-20 capillary apparatus. NMR spectra were recorded on a Brucker AC-300 spectrometer. H and C chemical shifts are given in ppm and referenced to the residual solvent resonance relative to TMS. Infrared spectra were recorded on a Brucker Vector 22.
4.2 General procedure for the preparation of β-ketoamines and β-iminoamines
The β-ketoamines were prepared by adding 50 mol% excess of the appropriate primary amine directly to the β-diketone (benzoylacetone). The mixture was heated at 100 °C for several hours in the presence of calcium sulfate. The products were purified by vacuum distillation or crystallization.
For the synthesis of the unsymmetrical β-iminoamine precursor, a mixture of amine (2 equiv), benzoylacetone (1 equiv) and paratoluene sulfonic acid (1.3 equiv) was refluxed in toluene for 24 h under an atmosphere of dry argon. Water was eliminated by an azeotropic distillation using a Dean–Stark apparatus. The toluene was then decanted off, and the solid residue was treated with diethyl ether, water and Na2CO3·H2O. After stirring for 30 min, the ether layer was separated, dried under MgSO4, and the solvent was removed in vacuum. The yellow solid was crystallized from hexane.
4.3 3-N-(Phenylamino)-1-phenylbut-2-en-1-one (1)
Following the general procedure, from aniline (55 mmol, 5 mL) and benzoylacetone (55 mmol, 8.89 g) were obtained 12.47 g of 1 as a yellow solid after crystallization from hexane. Yield = 83%. M.p. (°C): 111–112. IR (KBr): νCO = 1665 cm−1, νN–H = 3360 cm−1. 1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 2.41 (s, 3H, (NH)C–CH3), 5.91 (s, 1H, CHCO(Ph)), 7.17–7.95 (m, 10H, Harom), 12.84 (s, 1H, H–N). 13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 20.52 (CH3), 94.36 (CHCO(Ph)), 124.81–140.07 (Carom), 162.32 (C–N), 188.71 (CO).
4.4 3-N-(2-Methylphenylamino)-1-phenylbut-2-en-1-one (2)
Following the general procedure, from 2-methylaniline (46 mmol, 5 mL) and benzoylacetone (46 mol, 7.45 g) were obtained 8.67 g of 2 as a yellow solid after crystallization from hexane. Yield = 75%. M.p. (°C): 115–117. IR (KBr): νCO = 1645 cm−1, νN–H = 3406 cm−1. 1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 2.03 (s, 3H, 2-CH3–C6H4), 2.43 (s, 3H, (NH)C–CH3), 5.93 (s, 1H, CHCO(Ph)), 6.69–7.96 (m, 9H, Harom), 12.94 (s, 1H, H–N). 13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 18.22 (2-CH3–C6H4), 20.27 (CH3), 93.76 (CHCO(Ph)), 126.34–140.16 (Carom), 163.23 (C–N), 188.70 (CO).
4.5 3-N-(2,4,6-Trimethylphenylamino)-1-phenylbut-2-en-1-one (3)
Following the general procedure, from 2,4,6-trimethylaniline (35 mmol; 5 mL) and benzoylacetone (35 mmol; 5.67 g) were obtained 7.6 g of 3 as a yellow solid. Yield = 78%. M.p. (°C): 113–115. IR (KBr): νCO = 1658 cm−1, νN–H = 3395 cm−1. 1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 1.68 (s, 3H, (NH)C–CH3), 2.10 (s, 6H, 2,6-(CH3)2–C6H2), 2.20 (s, 3H, 4-CH3–C6H2), 5.82 (s, 1H, CHCO(Ph)), 6.59–7.87 (m, 7H, Harom), 12.37 (s, 1H, N–H). 13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 18.61 (4-CH3–C6H2), 19.86 (2,6-(CH3)2–C6H2), 21.36 (CH3), 92.66 (CHCO(Ph)), 122.22–160.26 (Carom), 165.48 (C–N), 188.82 (CO).
4.6 3-N-(2,6-Dimethylphenylamino)-1-phenylbut-2-en-1-one (4)
Following the general procedure, from 2,6-dimethylaniline (40 mmol; 5 mL) and benzoylacetone (40 mmol; 6.55 g) were obtained 8.38 g of 4 as a yellow solid after crystallization from hexane. Yield = 79%. M.p. (°C): 109–110. IR (KBr): νCO = 1660 cm−1, νN–H = 3371 cm−1. 1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 1.79 (s, 3H, (NH)C–CH3), 2.26 (s, 6H, 2,6-(CH3)2–C6H3), 5.94 (s, 1H, CHCO(Ph)), 6.94–7.97 (m, 8H, Harom), 12.55 (s, 1H, H–N). 13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 18.36 (2,6-(CH3)2–C6H3), 19.53 (CH3), 92.39 (CHCO(Ph)), 127.06–140.10 (Carom), 164.83 (C–N), 188.60 (CO).
4.7 3-N-(2,6-Diisopropylphenylamino)-1-phenyl-1N-(2,6-diisopropylphenylimino)but-2-ene (5)
Following the general procedure, from 2,6-diisopropylaniline (3.8 mL; 20 mmol), benzoylacetone (1.62 g; 10 mmol) and paratoluene sulfonic acid (13 mmol; 1.3 equiv) were obtained 3 g of 5 as a yellow solid. Yield = 63%. M.p. (°C): 130–132. IR (ATR): νCN = 1615 cm−1, νN–H = 3675 cm−1. 1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 0.93 (d, 3H, CH3–iPr, J = 6.9 Hz), 1.07 (d, 3H, CH3–iPr, J = 6.6 Hz), 1.13 (d, 3H, CH3–iPr, J = 6.6 Hz), 1.21 (d, 3H, CH3–iPr, J = 6.9 Hz), 1.81 (s, 3H, H4), 3.09 (sept, 2H, CH–iPr), 3.26 (sept, 2H, CH–iPr), 5.13 (s, 1H, H2), 6.96–7.23 (m, 11H, Harom), 12.35 (s, 1H, N–H). 13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 22.09 (CH3–CN), 22.55 (CH3–iPr), 23.71 (CH3–iPr), 24.45 (CH3–iPr), 25.56 (CH3–iPr), 28.50 (CH–iPr), 28.77 (CH–iPr), 96.10 (CC(CH3)), 123.32–144.08 (Carom), 159.29 (C–N), 165.42 (CN).
4.8 3-N-(2,6-Dimethylphenylamino)-1-phenyl-1N-(2,6-dimethylphenylimino)but-2-ene (6)
From 2,6-dimethylaniline (35 mmol; 5 mL), benzoylacetone (2.84 g; 17.5 mmol) and paratoluene sulfonic acid (23 mmol; 1.3 equiv) were obtained 4.6 g of 6 as a yellow solid. Yield = 71%. M.p. (°C): 128–129. IR (ATR): νCN = 1610 cm−1, νN–H = 3674 cm−1. 1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 1.79 (s, 3H, CH3), 2.17 (s, 12H, 2,6-(CH3)2–C6H3), 5.12 (s, 1H, CHC(C6H5)), 6.88–7.26 (m, 11H, Harom), 12.4 (s, 1H, N–H). 13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 18.79 (2,6-(CH3)2–C6H3), 19.31 (2,6-(CH3)2–C6H3), 21.47 (CH3–CN), 96.23 (CC(CH3)), 123.81–146.43 (Carom), 159.97 (C–N), 165.42 (CN).
4.9 3-N-(2-Methylphenylamino)-1-phenyl-1N-(2-methylphenylimino)but-2-ene (7)
From 2-methylphenylaniline (46 mmol; 5 mL), benzoylacetone (23 mmol; 3.8 g) and paratoluene sulfonic acid (30 mmol; 1.3 equiv) were obtained 6.5 g of 7 as a yellow solid. Yield = 82%. M.p. (°C): 132–133. IR (ATR): νCN = 1622 cm−1, νN–H = 3674 cm−1. 1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 1.92 (s, 3H, H4; CH3), 2.17 (s, 3H, 2-CH3–C6H4), 2.32 (s, 3H, 2-CH3–C6H4), 5.20 (s, 1H, CHC(C6H5)), 6.32–7.36 (m, 13H, Harom), 12.5 (s, 1H, N–H). 13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 18.64 (2-CH3–C6H4), 19.05 (2-CH3–C6H4), 21.91 (CH3–CN), 101.71 (CC(CH3)), 121.34–148.74 (Carom), 155.28 (C–N), 165.66 (CN).
4.10 3-N-(2-Methoxyphenylamino)-1-phenyl-1N-(2-methoxyphenylimino)but-2-ene (8)
From o-methoxyphenylaniline (4.55 g; 37 mmol), benzoylacetone (3 g; 18.5 mmol) and paratoluene sulfonic acid (24.5 mmol; 1.3 equiv) were obtained 3 g of 8 as a yellow solid. Yield = 85%. M.p. (°C): 139–140. IR (ATR): νCN = 1622 cm−1, νN–H = 3380 cm−1. 1H NMR (300 MHz, CDCl3, 25 °C, δ [ppm]): 1.97 (s, 3H, CH3), 3.73 (s, 3H, 2-OCH3), 3.81 (s, 3H, 2-OCH3), 5.17 (s, 1H, CHC(C6H5)), 6.28–7.41 (m, 13H, Harom), 12.69 (s, 1H, N–H). 13C NMR (75.5 MHz, CDCl3, 25 °C, δ [ppm]): 21.84 (CH3–CN), 55.45 (2-OCH3), 55.76 (2-OCH3), 102.10 (CC(CH3)), 110.41–150.53 (Carom), 154.21 (C–N), 166.23 (CN).
The supplementary material has been sent to the Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK (CCDC No. 272755) and can be obtained by contacting the CCDC.