

1 Introduction
The water-gas shift reaction (CO + H2O → CO2 + H2) is generally operated according to a two-step process, namely at high (HTS, 350-450 °C) and low (LTS, 250–300 °C) temperatures, over two main types of catalysts, Fe-Cr and Cu-Zn systems, respectively. Iron oxide based HTS catalysts, Fe3O4 [1,2] or Fe2O3 [3], are generally doped with Cr2O3 [4–7]. The recognized role of chromium is preventing iron oxide from sintering as well as catalyzing the WGS reaction. However, catalysts based on Fe-Cr-O have ecological and safety problems linked to chromium compounds. For this reason, development of chromium-free iron-based catalysts with high HTS performance is suitable [3,8,9].
The WGS reaction is thought to occur principally through two main types of mechanisms, the regenerative or redox mechanism [1,10] and the associative mechanism [1]. In the regenerative mechanism the catalyst surface is successively oxidized by H2O and reduced by CO. In turn, the associative mechanism assumes that CO and H2O interact to form a reaction intermediate, such as an adsorbed formate species, which then decomposes to CO2 and H2. It was found by Rethwisch and Dumesic [1] that Fe3O4 catalyzed the water-gas shift reaction via the regenerative mechanism while MgFe2O4, ZnO, MgO, TiO2, SiO2 catalyzed the water-gas shift reaction mainly via the associative mechanism. It was also established that basic oxides were more active than acidic or amphoprotic ones. This can be related to the weaker metal-oxygen bonding in basic oxides.
The associative mechanism generally considers a kind of bifunctional sequence with a specific role of metal and support. Thus, the formate can form from the surface reaction of CO bonded to the metal with OH groups located at the metal-support interface, as follows: COad + OHad → HCOOad. The formate bonded on the metal then spills over to the support where it decomposes to CO2 and H2 [11].
In the present paper, we give the effect of the acid/base properties of various supports on chromium-free iron-based catalyst in the WGS Reaction. Besides the determination of catalytic activity and acid/base properties, catalysts were subjected to in situ DRIFT measurements in order to obtain the type of active sites and the part of these interfaces in the water gas shift reaction.
2 Experimental
2.1 Catalysts preparation
Fe2O3/SiO2, Fe2O3/TiO2 and Fe2O3/MgO systems were prepared by the incipient wetness impregnation method. The 30 wt.% Fe2O3/support catalysts were prepared by impregnation of SiO2, TiO2 or MgO with an appropriate concentration of Fe(NO3)3.9 H2O. After drying at 80 °C overnight, the resulting solid was calcined in air at 400 °C for 2.5 h [12].
Bulk Fe2O3 catalyst was synthesized by the co-precipitation method. The starting reagents of high purity FeSO4.7 H2O and Fe2(SO4)3.5 H2O with a molar ratio (1/1) were dissolved in water to which 100 mL of NH4OH was added as a precipitant agent. The mixture was vigorously stirred for 30 min. The resulting powder was washed several times with deionized water, and dried at 110 °C. Subsequently, the powder was calcined at 400 °C in air (33 mL/min) with a rate of 5 °C/mn for 2.5 h [9].
2.2 Apparatus and procedure
X-ray powder diffraction patterns were obtained with a Philips PW 1050/81 automated powder goniometer, equipped with diffracted-beam graphite monochromatic Cu (Kα) radiation.
H2− TPR profiles were determined using a laboratory-made apparatus equipped with a TCD detector. The catalyst sample (200 mg) was loaded into a U-shape quartz reactor, then heated at 200 °C under argon flow with a rate of 5 °C/min for 1 h and cooled to room temperature. Next, the sample was submitted to a flow of 5% H2/Ar from room temperature to 800 °C. A cold trap was placed downstream of the sample to capture any water that evolved from catalyst prior to entering the TCD detector.
Catalysts were tested under atmospheric pressure between 350 and 450 °C after being activated in situ under a flow of 48% H2/N2 using an amount of catalyst of 250 mg for all runs. After reduction, the reacting gases (H2O/CO = 4.4) were introduced into the reactor at a total flow rate of 33 mL/min. Before each analysis, the effluent passed through a water-trap at 0 °C to remove the water of the reaction. The gas composition was analyzed by a TCD chromatograph containing two 4 m carbosieve B columns (1/8 inch, 100 to 200 meshes).
In situ DRIFT studies were performed on a Nicolet Magna IRTF spectrometer with OMNIC software. The reaction was performed in a high temperature Spectratech cell equipped with a ZnSe window. The sample (∼50 mg) was first treated in a flow of (∼35 mL/min) H2 at 400 °C for 30 min (high-temperature treatment) before each experiment. The heating rate during the treatment was 10 °C/min. After this treatment, the sample was cooled under He and then put in contact with the reacting mixture CO/H2O at increasing temperature (25–450 °C). Unless stated otherwise, infrared (IR) spectra were recorded against a background of the sample at the reaction temperature under flowing He. IR spectra were recorded with co-addition of 64 scans in single-beam spectra or absorbance spectra, by applying a resolution of 4 cm−1.
3 Results and discussion
3.1 XRD and H2− TPR measurements
The X-ray phases of Fe2O3-only and Fe2O3-promoted freshly calcined samples are given in Table 1. α-Fe2O3 hematite is the mainly crystalline phase in Fe2O3, Fe2O3/SiO2 and Fe2O3/TiO2. For the system doped with MgO, at higher Fe2O3 loading (30 wt.%) the band characteristic of MgO decreased while new bands assigned to MgFe2O4 are formed [9]. This indicates that iron oxide is in strong interaction with MgO, leading to a mixed oxide formation.
Characterization results and CO conversion of catalysts prepared.
| Sample | S BET (m2/g) | XRDa phases | H2− TPR Tmax (°C) | Acid/base propertiesb | Rate of CO conv. to CO2 at 450°C (mol/m2cat. s × 105)c | |
| SC3H6O (%) | SC3H6 (%) | |||||
| Fe2O3 | 25.0 | α-Fe2O3 | 446, 607, 684 | 58 | 42 | 0.14 |
| Fe2O3/SiO2 | 90.7 | α-Fe2O3, SiO2 | 500, 600, 680 | 18 | 82 | 0.03 |
| Fe2O3/TiO2 | 14.0 | α-Fe2O3, TiO2 | 340, 430, 620 | 82 | 18 | 0.25 |
| Fe2O3/MgO | 02.2 | MgFe2O4,MgO | 368d, 421, 431 | 92 | 8 | 3.00 |
a Freshly calcined catalyst.
b Selectivity after C3H7OH decomposition at a temperature of 250 °C, using 100 mg of reduced catalyst.
c At a temperature reaction (CO + H2O) of 450 °C.
d Shoulder.
H2− TPR results of the four catalysts are reported in Fig. 1 and their peak maxima are summarized in Table 1. In all cases, three peaks of hydrogen consumption in a temperature range of 25–800 °C are observed [13,14]. For Fe2O3-only, the lower reduction temperature region (446 °C) of Fe2O3 is attributed to the reduction of Fe2O3 to Fe3O4 while the higher temperature region (606–684 °C) is credited to the reduction of Fe3O4 to FeO and Fe. The low temperature peaks of Fe2O3/TiO2 and Fe2O3/MgO shifted to 340 and 368 °C respectively. On the other hand, MgO− promoted catalyst involved a shift of the reduction temperature to lower values as depicted in Fig. 1, curve c. XRD studies showed that MgFe2O4 crystalline phase is present in Fe2O3/MgO. Indeed, the presence of this phase improves the reducibility of Fe2O3/MgO. More detailed studies were reported elsewhere [9].
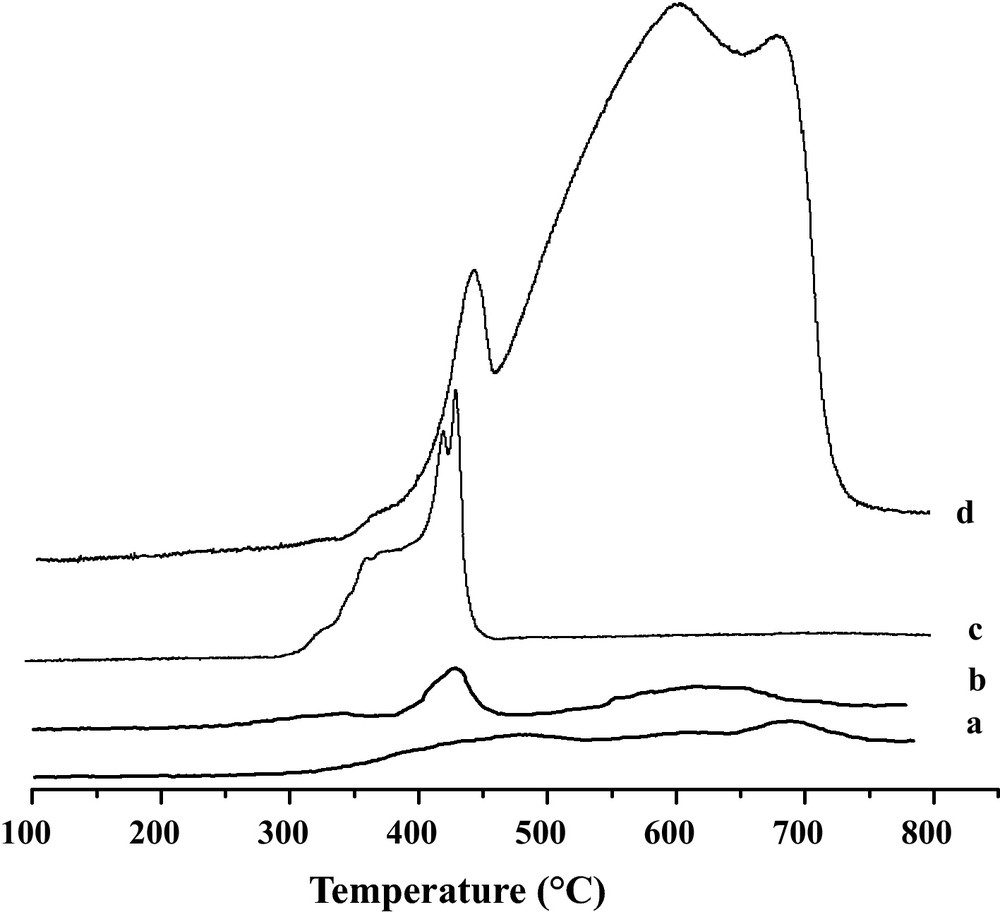
H2− TPR Profiles of freshly calcined samples: a: 30 wt.% Fe2O3/SiO2; b: 30 wt.% Fe2O3/TiO2; c: 30 wt.% Fe2O3/MgO and d: Fe2O3.
3.2 Catalytic properties in CO + H2O reaction
The only products of the WGS reaction were found to be carbon dioxide and hydrogen, no methane being detected. No reaction occurred on every support alone. The WGS reaction conversion of the tested catalysts (unsupported and supported Fe2O3) was expressed by the conversion of CO to CO2, measured at 450 °C, and expressed per surface area unit, as presented in Table 1. The specific activity appears to span over a large range, the most active system (Fe2O3/MgO) displaying a rate 100 times higher more than the least active (Fe2O3/SiO2). The four tested systems were ranked as follows: Fe2O3/MgO >> Fe2O3/TiO2 > Fe2O3 >> Fe2O3/SiO2. The activation energy was estimated to be 97.6 kJ/mol in the temperature range of 350–450 °C over the Fe2O3/MgO catalyst. This value is in accord with the value reported by Rethwisch et al. (106 kJ/mol) [1].
As can be seen in Table 1 and Fig. 2, a direct relationship can be observed between catalytic activity and acid/base properties of our catalysts: Fe2O3/MgO >> Fe2O3/TiO2 > Fe2O3 >> Fe2O3/SiO2. Basic properties of catalysts are given here in terms of dehydrogenation of isopropanol: CH3-CHOH-CH3 → CH3-CO-CH3 + H2 at a temperature reaction of 250 °C [12]. The dependence found that oxides which are basic were more active than those which are acidic or amphoprotic and can be explained in terms of the higher activity of surface species like formates [1]. It is suggested, by these authors, that formate species readily formed and dehydrogenated to provide CO2 and H2.
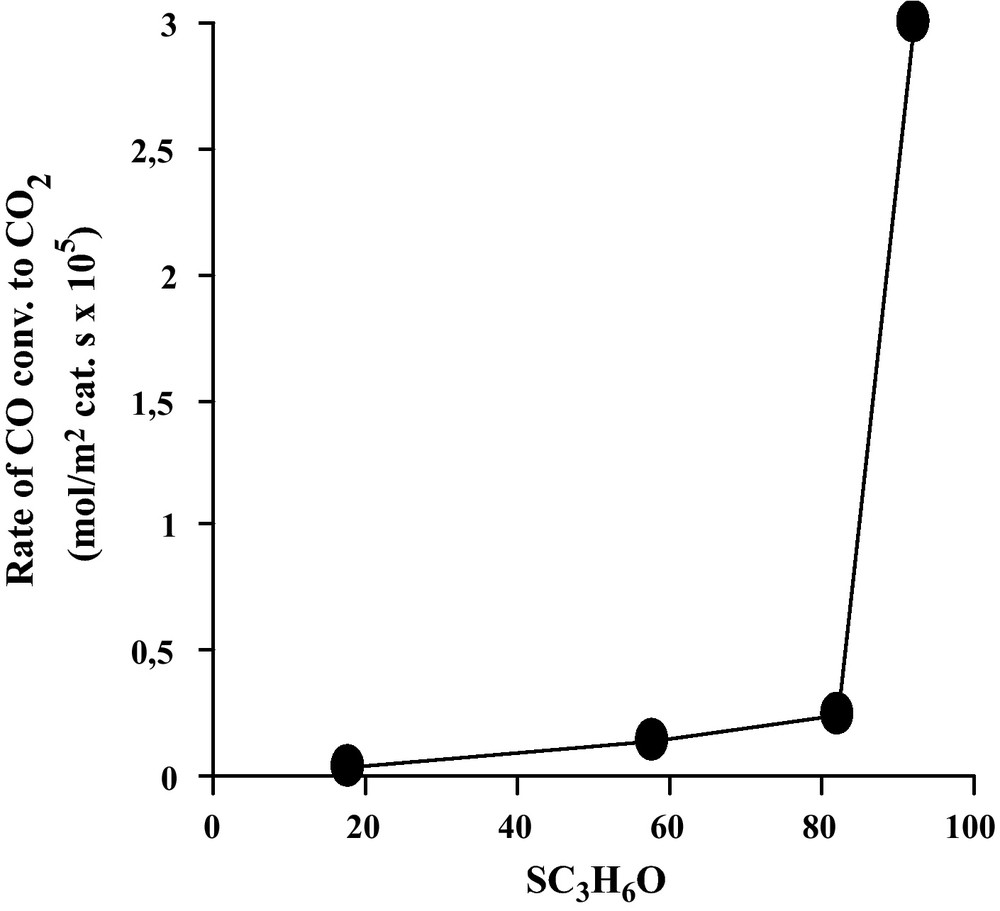
Evolution of the rate of CO conversion to CO2 as a function of basic properties of our prepared catalysts (expressed in term of dehydrogenation of isopropanol C3H7OH at 250 °C. Pisopropanol = 1.080 kPa [12]).
One can see that the increase in intensity of the 2420–2330 cm−1 band (Fig. 3b–d), as well as the decrease of formate species bands with raising the temperature to 450 °C illustrated the decomposition of adsorbed HCOO species to CO2 and H2 [11]. This decomposition is facilitated by the weaker metal-oxygen bond in the basic oxides. In the opposite, on an acidic sample, such as Fe2O3/SiO2, no formate intermediates were detected and the intrinsic rate was the lowest of the four systems.
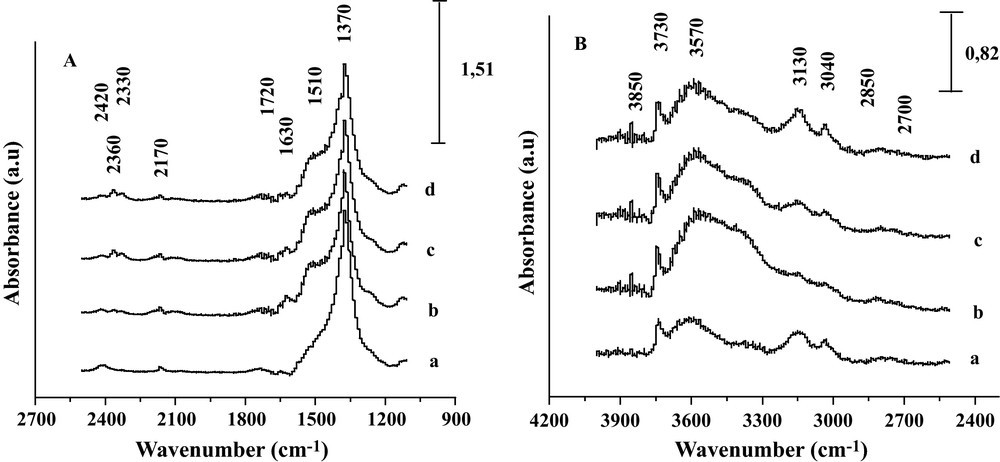
DRIFTS spectra of the Fe2O3/MgO system recorded (a) at RT after reduction, (b) under CO/H2O reaction conditions at 350 °C, (c) 400 °C and (d) 450 °C.
3.3 In situ DRIFT study of surface species
Fig. 3 shows the change in DRIFT spectra recorded for the Fe2O3/MgO system after reduction and after contact with the CO/H2O gas mixture at increasing reaction temperatures. The spectrum (b), recorded after exposure of the reduced catalyst to CO/H2O at 350 °C, showed a very prominent broad OH adsorption band with a maximum at about 3570 cm−1; as expected from H2O interaction with mild basic OH groups of MgO surface along with a sharp band at 3730 cm−1 typical of isolated basic Mg(OH) groups over the surface. Other OH bands are observed at 3130, and 3040 cm−1 which might be assigned to acidic FeOH groups. Moreover, small bands are seen around 2170–2080 cm−1 and in the 2420–2330 cm−1 region. The 2170–2080 cm−1 bands are characteristic of the CO gaseous while the doublet at the 2400–2330 cm−1 region corresponds to adsorbed and/or gaseous CO2 rotation-vibration P and R bands. The increase of the latter with temperature attests that the shift reaction does proceed in the DRIFT reactor, in line with the outlet gas analysis. The inlet of CO/H2O on reduced Fe2O3/MgO produces, as well as at 350 °C, bands at 1720, 1630, and 1370 cm−1, which diminished synchronously by increasing temperature from 350 to 450 °C. The synchronic dynamics of these 3 bands tends to indicate that they arose from the same OCO containing species. By observing simultaneously some C-H stretching vibrations at 2850–2700 cm−1 associated with the 1720, 1630, and 1370 cm−1 OCO region bands, we propose, in agreement with Tabakova et al. [15], that the diminution of the band at 2170 cm−1 as well as the decrease in intensity of the OH band at 3570 cm−1 (from 350 to 450 °C) can be correlated with the production of formate species. As a matter of fact, formate species involve symmetric and asymmetric OCO band vibrations on the surface of the catalyst. The asymmetric OCO band is expected at 1580 cm−1 [15,16], and the symmetric one at 1375 cm−1. The vibrations of the OCO bands are accompanied by C-H stretching vibrations at 2850–2700 cm−1.
In other terms, it is proposed that the carbonyl adspecies bonded to iron sites might react with the magnesia basic OH groups located at the metal-support interface to produce formate intermediate species. Formate adspecies are often considered as active intermediates in the water-gas shift reaction over supported metals and also on support alone [1,11,17,18] and more precisely over MgFe2O4 in [1] or over pure MgO in [19].
Fig. 4 reports the DRIFT spectra of the reduced Fe2O3/TiO2 system after exposure to the CO/H2O gas mixture at increasing temperature. At variance with the previously reported system, no significant IR contribution from formate species was observed on the reduced Fe2O3/TiO2 catalyst after being contacted with CO/H2O. This can be deduced from the absence of vibration characteristic of C-H bonds in the 2850–2700 cm−1 range. In line with this observation, only weak formate bands were observed on TiO2 by Rethwisch and Dumesic [1]. The broad band at 3410 cm−1 observed essentially at 350 °C, and then decreasing at higher temperature, is characteristic of Ti-OH hydroxyl groups. After contacting Fe2O3/TiO2 at 350 °C with CO/H2O gases, two new bands are detected in the carbonate/carboxylate species region, specifically at 1630 and 1550 cm−1 [20]. In addition, Fig. 4 (curve a) shows the doublet at 2170–2080 cm−1 characteristic of the CO gaseous together with the doublet at ∼ 2370 and 2320 cm−1 characteristic of adsorbed and/or gaseous CO2 [20,21]. By increasing temperature the intensities of the bands at 3410 cm−1, at 2080–2170 cm−1, at 1630 and at 1550 cm−1 decreased but the intensity of the bands at 2370–2320 cm−1 increased. These trends indicate that while WGS proceeds, the surface is progressively depleted in adsorbed species while the gaseous CO concentration increases. Therefore, no clear evidence of a WGS route via formate intermediates was obtained for this case of Fe2O3/TiO2 catalyst. The weaker basicity of the TiO2 surface and/or an inadequate interface between metal and oxide phases might be the origin of this statement.
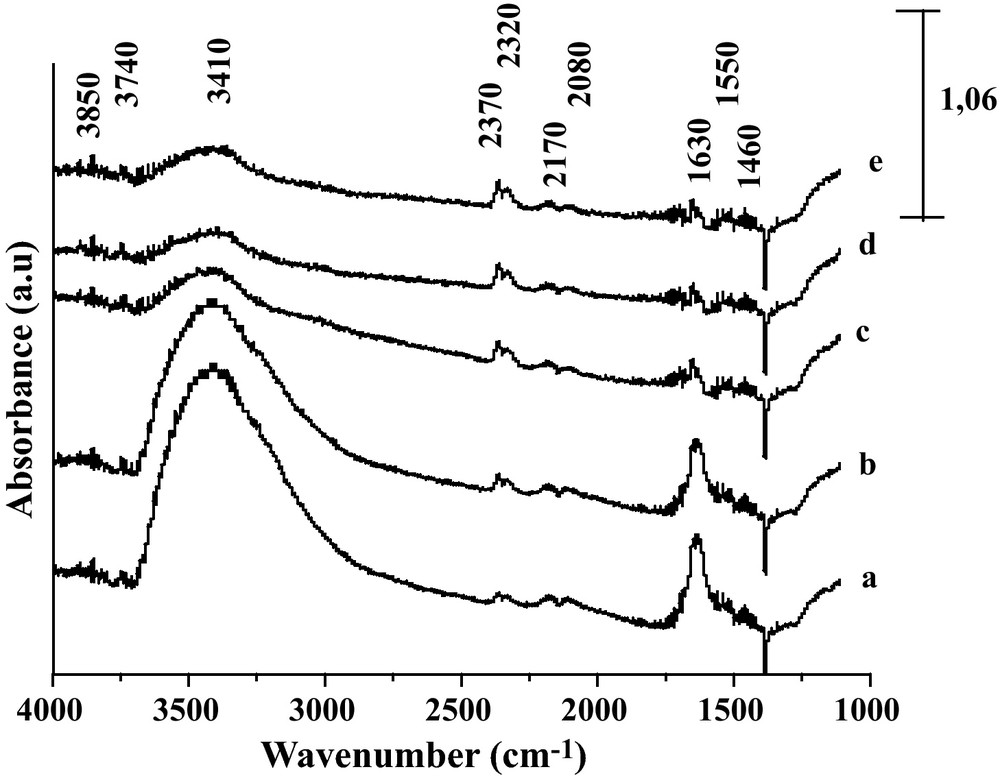
DRIFT spectra of the reaction gaseous CO/H2O on Fe2O3/TiO2 at various reaction temperatures, a: 350 °C; b: 370 °C; c: 400 °C; d: 420 °C and e: 450 °C.
Reduced Fe2O3 and Fe2O3/SiO2 (Fig. 5) catalysts produced spectroscopic features significantly different from those of Fe2O3/MgO and Fe2O3/TiO2, since no formate or carbonate bands were observed either on the unsupported system or on the silica supported one. This statement quite agrees with the expected feature that no formate/carbonate species can form on an acid oxide (silica or magnetite), under any conditions, in line with [1,11]. Therefore, it might be inferred that a redox or regenerative shift mechanism is likely to apply for these two cases, as proposed over magnetite in [1]. We note in addition that no sufficiently resolved IR spectra could be recorded for this last case due to sample black color after reduction.
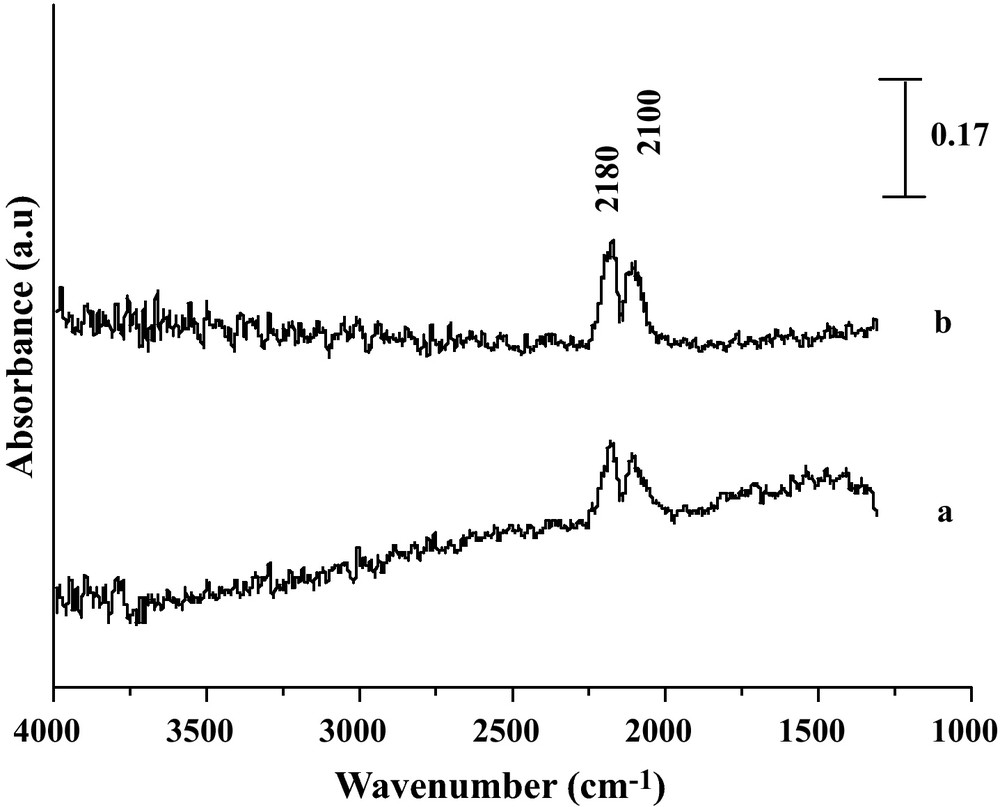
DRIFT spectra of the reaction gaseous CO/H2O on (a) Fe2O3 and (b) Fe2O3/SiO2 under 5% CO at 450 °C.
4 Conclusions
Fe2O3/MgO, Fe2O3/TiO2, Fe2O3 and Fe2O3/SiO2 are active in high temperature WGS Reaction.
A direct correlation between catalytic activity and acid/base properties of the prepared catalysts is found.
The Fe2O3/MgO basic catalyst is 100 times more active than the acidic catalyst Fe2O3/SiO2.