

1 Introduction
Les systèmes catalytiques à base de fer sont habituellement utilisés pour catalyser plusieurs réactions telles que la réaction de Fischer–Tropsch [1], les alkylations de Friedel–Crafts [2], l'oxydation de H2S [3,4], la réaction de conversion du gaz à l'eau [5–7], etc.
La réaction de conversion du gaz à l'eau : CO + H2O → CO2 + H2 est l'un des procédés les plus développés ces dernières années [8]. Il s'agit d'un procédé catalytique qui est réalisé en deux étapes pour optimiser la production en hydrogène lors du vaporeformage du gaz naturel. La première étape est effectuée, dans le domaine de température 350–450 °C, sur catalyseurs à base d'oxyde de fer communément appelés catalyseurs hautes températures [5]. Dans le domaine 150–250 °C, la réaction CO + H2O est catalysée par des systèmes mixtes à base de cuivre modifiés par le zinc (CuO/ZnO/Al2O3) habituellement appelés catalyseurs basses températures [9].
L'utilisation de matériaux à base d'oxydes de fer efficaces aux hautes températures de conversion du gaz à l'eau nécessite un milieu réactionnel CO + H2O très riche en vapeur d'eau afin d'éviter la réduction des particules de Fe3O4, par le monoxyde de carbone, en fer métallique Fe0 [10]. Néanmoins, l'inconvénient majeur, des conditions où la réaction exige un excèdent d'eau, est le risque de vieillissement par frittage de la phase active Fe3O4. Il est alors préconisé de doper l'oxyde de fer pour éviter ce phénomène de frittage des particules Fe3O4 [5,6,11]. Jedynak-Koczuk et Kowalczyk [12] se sont intéressés au catalyseur Fe3O4/C sur lequel ils ont dispersé l'oxyde de chrome Cr2O3. Il a été trouvé par ces chercheurs [12] que le chrome, ex-nitrates, est trois fois plus performant, entre 330 et 370 °C, que le dopant Cr2O3 ajouté à l'état oxyde. Par ailleurs, la substitution du chrome par le thorium a été étudiée par Costa et al. [13]. Ces catalyseurs exempts de chrome sont donc moins toxiques. De plus, ces systèmes catalytiques présentent l'intérêt de mieux catalyser la réaction de conversion du gaz à l'eau CO + H2O. Araujo et Rangel [14] ont étudié l'effet de l'association des particules de cuivre au catalyseur Fe–Cr à une température de 370 °C et ont montré que ce nouveau système a une activité catalytique comparable au catalyseur commercial Fe–Cr. Lors de la promotion de Fe2O3–Cr2O3 par l'oxyde de cérium CeO2, Hu et al. [15] ont mentionné l'effet bénéfique de cet ajout sur la réduction de ces catalyseurs. C'est la présence de CeO2 qui balise la réductibilité de Fe2O3–Cr2O3 et augmente donc son activité catalytique. Dans une étude récente, Boudjemaa et al. [5] ont montré que l'activité catalytique des systèmes exempts de Cr2O3 dépend non seulement des espèces de fer exposées au mélange réactionnel CO + H2O mais aussi des interactions fer-support.
Lors de cette étude, une série de catalyseurs à base d'oxyde de fer supportés a été sélectionnée et étudiée en réaction CO + H2O dans le domaine de températures 350–450 °C et à la pression atmosphérique. L'accent est mis ici sur l'influence des propriétés acido–basiques et redox des systèmes élaborés sur leur activité catalytique.
2 Partie expérimentale
2.1 Préparation des systèmes catalytiques
Les catalyseurs à isoteneur en Fe2O3 (30% en poids) : Fe2O3/SiO2 (SiO2 : Degussa, surfaceBET = 196,0 m2 g−1), Fe2O3/MgO (MgO : Merck, surfaceBET = 7,3 m2 g−1) et Fe2O3/TiO2 (TiO2 : Merck, surfaceBET = 8.4 m2 g−1) ont été préparés par imprégnation à sec. Cette méthode consiste à exposer le support SiO2, MgO ou TiO2 à un dépôt de solution aqueuse appropriée de Fe(NO3)3, 9 H2O (Riedel-de Haën). Le mélange ainsi obtenu est ensuite séché, à l'étuve, à 80 °C puis calciné en présence d'air à 400 °C pendant 2 h 30 min. Le système Fe2O3/MgO est calciné aussi pour les besoins de l'étude à 200 et 300 °C.
2.2 Caractérisations des catalyseurs
Les aires spécifiques ont été déterminées par la méthode BET. Les échantillons sont préalablement dégazés sous vide à 200 °C à raison de 5 °C min−1 pendant 1 h. Les phases cristallines de nos solides, calcinés à 400 °C, ont été obtenues par diffraction des rayons X à l'aide d'un goniomètre de poudre de type Philips PW 1050/81.
Les profils de réduction en température programmée des solides catalytiques ont été obtenus en utilisant la méthode de flux gazeux continu dans un appareil TPDRO 1100 ThermoFisher. La charge catalytique est activée sous atmosphère inerte (argon, 30 ml min−1) à 200 °C durant 30 min, en utilisant une vitesse de chauffage de 5 °C min−1, puis refroidie jusqu'à 40 °C. Le gaz de réduction (5% vol. H2/Ar) est alors acheminé dans le réacteur à un débit de 20 ml min−1. Par la suite, la charge catalytique subit, sous H2/Ar, un chauffage de 40 °C à 800 °C à raison de 5 °C min−1. Les résultats sont ensuite collectés et traités à l'aide d'un logiciel adéquat.
La transformation de l'isopropanol en propène et/ou en acétone a été utilisée comme test de mesure des propriétés acides ou basiques des solides catalytiques réduits Fe3O4, Fe3O4/SiO2, Fe3O4/MgO et Fe3O4/TiO2. La réaction est réalisée dans un réacteur en pyrex, à 250 °C, dans lequel est déposée une masse de 100 mg. L'isopropanol gazeux, à la pression de 1,080 kPa, est obtenu par barbotage de N2 dans l'isopropanol liquide maintenu à une température constante de 0 °C. Les réactifs et les produits de réaction sont analysés par chromatographie en phase gazeuse à l'aide d'un détecteur à ionisation de flamme (FID) équipé d'une colonne, 15% carbowax 20 M déposé sur chromosorb W 80–100 mesh, de 1,8 m de longueur. Les temps de rétention et les aires des pics intégrés sont collectés par un intégrateur calculateur Shimadzu (CR8A).
2.3 Réaction CO + H2O
La réaction de conversion du gaz à l'eau est réalisée dans un réacteur en quartz à lit fixe, à la pression atmosphérique et à des températures de réaction comprises entre 350 et 450 °C sur 250 mg, avec un débit de H2O/CO de 33 ml min−1 après réduction, au préalable, sous hydrogène dilué (48% vol. H2/N2). La vapeur d'eau, utilisée comme réactif, est produite à l'aide d'un saturateur de type T Lauda rempli d'eau, deux fois distillée, et placé dans un bain thermostaté. L'analyse de CO, CO2 et de H2 est assurée par chromatographie en phase gazeuse (TCD) IGC 121 ML. Les temps de rétention et les aires des pics intégrés sont collectés par un intégrateur calculateur Shimadzu (CR8A). Avant toute analyse, l'effluent gazeux passe par un piége à eau (glace à 0 °C et tamis moléculaire).
3 Résultats et discussion
3.1 Déshydratation et déshydrogénation de l'isopropanol
La décomposition de l'isopropanol (C3H7OH) en propène et/ou en acétone est utilisée ici comme test de caractérisation des propriétés acido–basiques des solides catalytiques réduits Fe3O4, Fe3O4/SiO2, Fe3O4/TiO2 et Fe3O4/MgO. Le but de cette étude est de corréler les propriétés acides et/ou basiques de ces solides à l'activité catalytique. Les résultats obtenus à 250 °C sur 100 mg de catalyseur sont illustrés par la Fig. 1 et le Tableau 1. Sur cette figure, sont portées les sélectivités en propène et en acétone après 4 h de réaction à 250 °C.
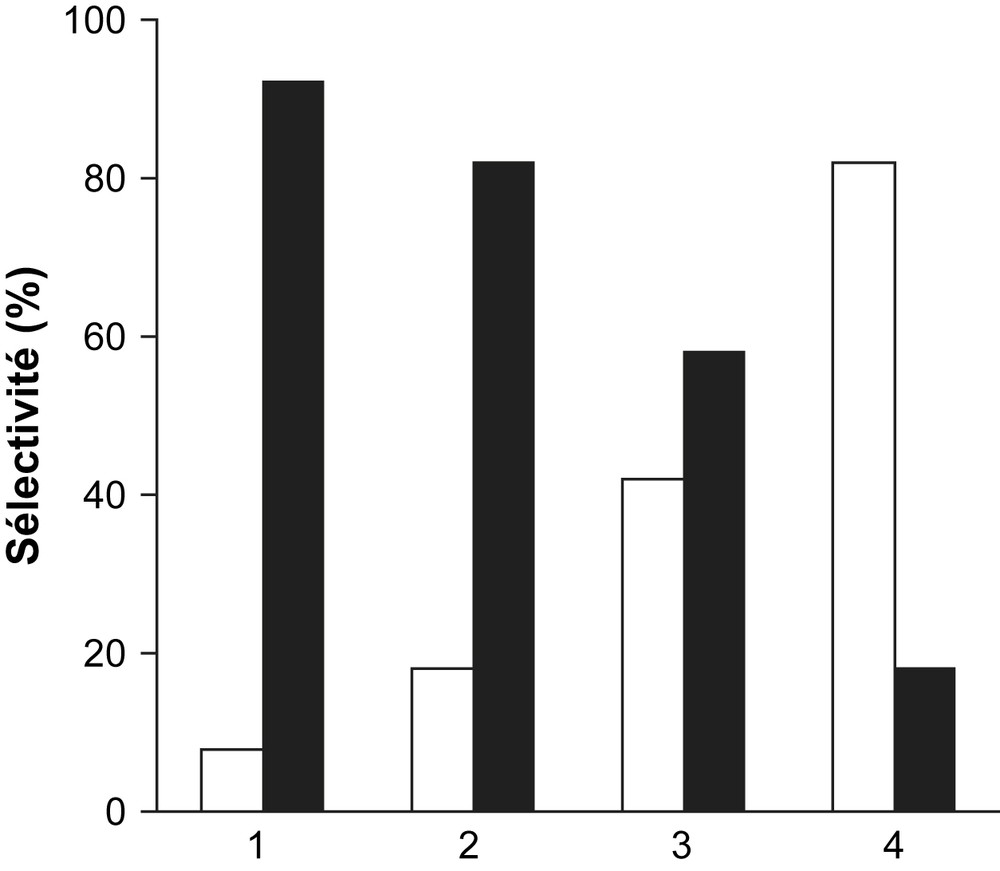
Sélectivités en propène (□) et en acétone (■) des systèmes Fe3O4/MgO (1), Fe3O4/TiO2 (2), Fe3O4 (3) et Fe3O4/SiO2 (4) après 4 h de décomposition de C3H7OH à 250 °C. Pisopropanol = 1.080 kPa.
Décomposition de C3H7OH et résultats de la réaction CO + H2O
| Phases DRXa | SBET m2/g | Conv. de C3H7OHb mol/(g s) × 105 | Conversion de CO en CO2 | |||||||||
| 350 °C | 400 °C | 450 °C | ||||||||||
| % | mol/(g s) × 105 | mol/(m2 s) × 105 | % | mol/(g s) × 105 | mol/(m2 s) × 105 | % | mol/(g s) × 105 | mol/(m2 s) × 105 | ||||
| Fe3O4 | Fe3O4 | 25,0 | 0,9 | 33,0 | 3,2 | 0,12 | 35,0 | 3,4 | 0,13 | 37,0 | 3,6 | 0,14 |
| Fe3O4/SiO2 | SiO2, Fe3O4 | 90,7 | 11,5 | 29,0 | 2,8 | 0,03 | 28,5 | 2,8 | 0,03 | 28,0 | 2,7 | 0,03 |
| Fe3O4/TiO2 | TiO2, Fe3O4 | 14,0 | 15,9 | 36,0 | 3,5 | 0,25 | 36,5 | 3,6 | 0,25 | 38,0 | 3,7 | 0,26 |
| Fe3O4/MgOc | MgO, Fe–O–Mg | 2,2 | 11,0 | 27,0 | 2,7 | 1,23 | 40,0 | 3,9 | 1,77 | 68,0 | 6,6 | 3,00 |
| Fe3O4/MgOd | MgO, Mg(OH)2 | 3,7 | – | 16,0 | 1,5 | 0,41 | 23,0 | 2,3 | 0,62 | 37,0 | 3,6 | 0,97 |
| Fe3O4/MgOe | MgO | 5,3 | – | 04,0 | 0,4 | 0,07 | 15,0 | 1,5 | 0,28 | 25,0 | 2,4 | 0,45 |
a Après réduction à 350 °C.
b Après 4 h de décomposition de C3H7OH à 250 °C. Charge de 100 mg et .
c Calciné à 400 °C.
d Calciné à 300 °C.
e Calciné à 200 °C.
Dans le cas du catalyseur Fe3O4/SiO2, la sélectivité en propène est beaucoup plus importante que la sélectivité en acétone, ce qui se traduit, en somme, par un nombre de sites déshydratants (CH3–CHOH–CH3 → CH2CH–CH3 + H2O) bien plus grand que les sites déshydrogénants (CH3–CHOH–CH3 → CH3–CO–CH3 + H2). Le catalyseur Fe3O4/MgO présente la plus grande sélectivité en acétone (92%) contre une sélectivité en propène de l'ordre de 8%. Le système catalytique non supporté Fe3O4, de loin moins actif que les autres catalyseurs, semble exhiber une sélectivité en acétone modérée. Fe3O4/MgO, sélectif en acétone et moins actif que Fe3O4/TiO2 (Tableau 1), confère au catalyseur des propriétés basiques. A l'issue de cette étude à 250 °C, pour des taux de conversion d'isopropanol assez proches, la basicité est donnée par la séquence : Fe3O4/MgO > Fe3O4/TiO2 > Fe3O4 ≫ Fe3O4/SiO2.
3.2 Effet du support en réaction CO + H2O
L'étude de l'effet du support sur l'activité catalytique du système, fraîchement calciné, Fe2O3 exempt d'oxyde de chrome Cr2O3 a été réalisée, à la pression atmosphérique, dans le domaine de températures allant de 350 °C à 450 °C en utilisant un mélange réactionnel H2O/CO = 4,4. Même s'il est bien établi que Cr2O3 stabilise l'espèce active à base de fer en jugulant le frittage des particules d'oxyde de fer [6,16–18] et la formation du fer métallique Fe0, le développement de catalyseurs exempt de chrome est plus que nécessaire car, il est connu que les ions Cr6+ sont nocifs pour l'homme et peuvent dans certains cas causer de sérieux problèmes de santé. Cette situation nous a conduit, dans un premier temps, à préparer une série de catalyseurs à base d'oxyde de fer supportés et exempt de Cr2O3. Les supports sélectionnés sont : MgO (oxyde à caractère basique), SiO2 (oxyde plutôt acide) et TiO2 (amphotère). MgO a été utilisé, en réaction de conversion du gaz à l'eau, pour supporter le platine [19], le ruthénium [20,21].
Une charge catalytique de 250 mg de Fe2O3/support et à titre de comparaison une même masse de Fe2O3 pur, sont préalablement prétraitées, à 350 °C, dans un flux d'hydrogène (48% vol. H2/N2). Une fois le catalyseur porté à la température de travail, les analyses chromatographiques sont effectuées à intervalle de temps régulier. L'activité catalytique est exprimée par la conversion de CO en CO2 (Tableau 1 et Fig. 2). Les résultats consignés dans ce tableau révèlent que le support MgO (cas de Fe3O4/MgO cal.400 °C) améliore nettement les performances catalytiques de la phase active Fe3O4. Ce catalyseur apparaît donc sensible à la réaction : CO + H2O → CO2 + H2. Bien plus, dans nos conditions où la thermodynamique est non limitante, la conversion de CO en CO2 de Fe3O4/MgO cal.400 °C augmente crescendo avec la température ; elle passe de 27% (350 °C) à 68% (450 °C) (Fig. 2). A 450 °C, le catalyseur Fe3O4/MgO cal.400 °C est deux fois plus performant que le système catalytique non supporté, en l'occurrence Fe3O4. En revanche, le catalyseur Fe3O4/MgO cal.200 °C parait le moins actif au cours de la montée en température. A 450 °C, par exemple, la vitesse de conversion spécifique est donnée par la séquence : Fe3O4/MgO cal.400 °C > Fe3O4/TiO2 > Fe3O4/MgO cal.300 °C, Fe3O4 > Fe3O4/SiO2 > Fe3O4/MgO cal.200 °C. De même, la vitesse de conversion intrinsèque de Fe3O4/MgO cal.400 °C est ∼10, 17, 100 fois plus grande que celles des catalyseurs Fe3O4/TiO2, Fe3O4, Fe3O4/SiO2 respectivement (Tableau 1). Rethwisch et Dumesic [22] ont étudié la réaction de conversion du gaz à l'eau sur une série d'oxydes non supportés au voisinage de 377 °C. Selon ces deux auteurs, c'est Fe3O4 qui montre la meilleure activité. Ce catalyseur est à juste titre 30, 1000, 7500 fois plus actif que les catalyseurs ZnFe2O4, MgO, TiO2 respectivement. Il est établi par plusieurs auteurs que la réaction de conversion du gaz à l'eau se produit suivant deux schémas mécanistiques différents [17,19,22–25]. Dans un premier mécanisme, la surface du catalyseur est successivement oxydée par H2O puis réduite par CO. Dans un deuxième mécanisme, l'adsorption de CO et H2O à la surface du catalyseur donne lieu à la formation d'un intermédiaire réactionnel formiate HCOO−. Par ailleurs, Ai [26] décompose l'acide formique sur une série d'oxydes et constate que les matériaux à caractère acide favorisent la déshydratation (HCOOH → CO + H2O) alors que les oxydes basiques favorisent la déshydrogénation (HCOOH → CO2 + H2). Les oxydes amphotères catalysent les deux réactions. En tentant de corréler les résultats des travaux de Rethwisch et al. [22] et de Ai [26], il apparaît clairement que se sont les catalyseurs à caractère basique qui favorisent la réaction de conversion du gaz à l'eau : CO + H2O → CO2 + H2 dans le cas du mécanisme faisant intervenir l'intermédiaire réactionnel HCOO−. En revanche, les catalyseurs à caractère acide orientent la réaction dans le sens inverse.
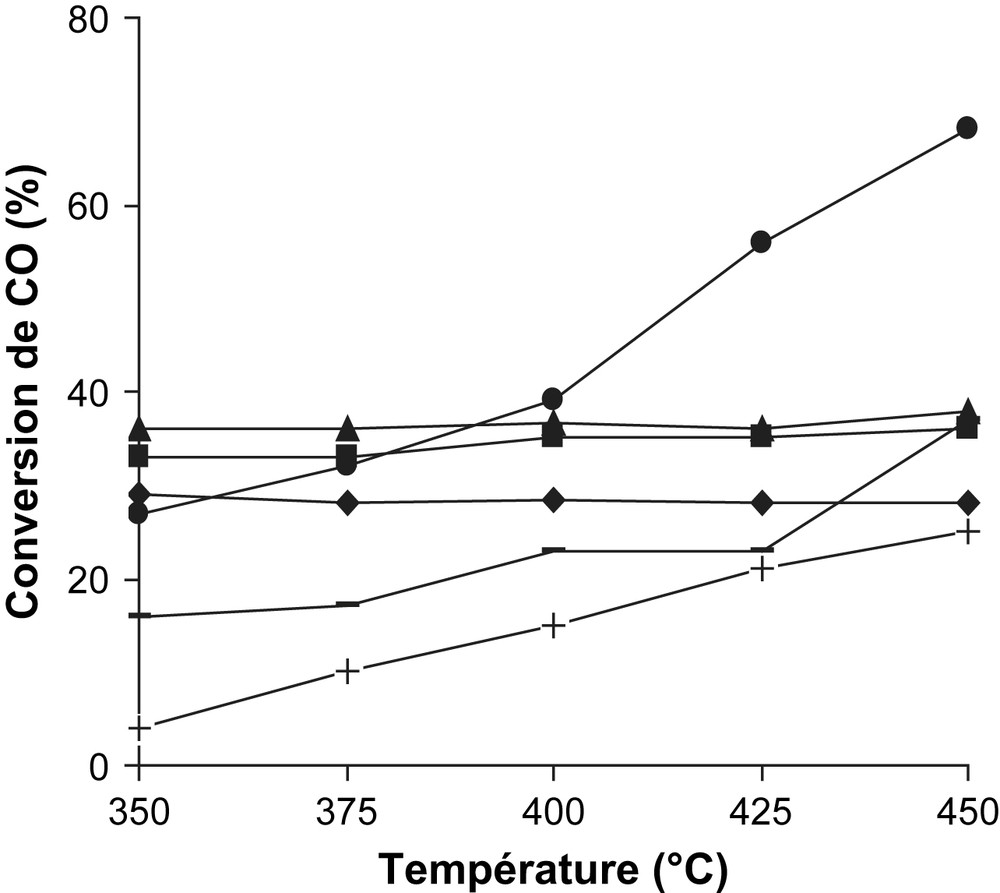
Conversion de CO au cours de la montée en température de : 30% Fe3O4/MgO (●), 30% Fe3O4/TiO2 (▴), 30% Fe3O4 (■), 30% Fe3O4/SiO2 (♦), tous calcinés à 400 °C ainsi que 30% Fe3O4/MgO calciné à 300 °C (−) et 30% Fe3O4/MgO calciné à 200 °C (+).
Par ailleurs, l'étude de diffraction des rayons X (Fig. 3) des échantillons non réduits révèle, dans le cas des catalyseurs supportés par MgO, la présence des phases cristallines Mg(OH)2, MgO et Fe–O–Mg. La phase Mg(OH)2 serait formée par interaction de MgO avec l'air ambiant chargé de vapeur d'eau lors du refroidissement de nos solides catalytiques [5]. Aucun pic caractéristique de Fe2O3 n'a été mis en évidence sur les systèmes supportés par MgO. Sur la Fig. 4 sont rapportés les profils RTP des systèmes catalytiques élaborés. L'oxyde Fe2O3 massique présente trois régions de consommation de H2 avec des Tmax de 445, 613 et 648 °C. Le pic apparaissant vers 445 °C exhibe un épaulement à 368 °C. Ces pics sont attribués à la réduction de Fe2O3α selon le processus : Fe2O3 → Fe3O4 → FeO → Fe [13]. L'ajout de MgO change substantiellement le profil de Fe2O3. Le pic à 420–450 °C avec son épaulement à 370 °C correspond à la réduction de Fe3+, bien dispersé et n'apparaissant pas en DRX. De plus, il apparaît nettement à partir des résultats de DRX (Fig. 3) que l'intensité des pics assignés à la phase MgO augmente lorsque la température de calcination de Fe3O4/MgO passe de 200 °C à 400 °C. En calcinant le catalyseur Fe3O4/MgO à 400 °C, nous avons donc généré un réservoir MgO susceptible d'alimenter la surface du catalyseur en espèce formiate lors de la réaction de conversion du gaz à l'eau.
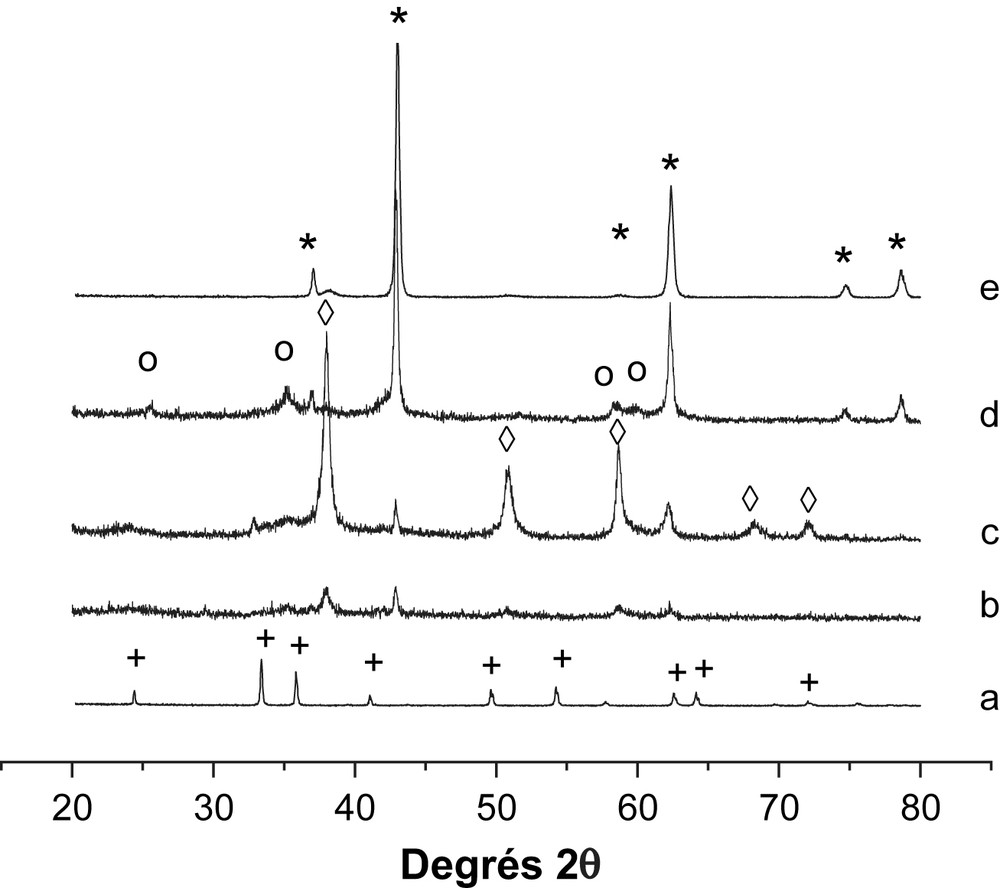
Diffractogrammes des échantillons avant réduction : (a) α-Fe2O3, (b) 30% Fe2O3/MgO calciné à 200 °C, (c) 30% Fe2O3/MgO calciné à 300 °C, (d) 30% Fe2O3/MgO calciné à 400 °C et (e) MgO. (+) Fe2O3, (*) MgO, (○) Fe–Mg–O et (♢) Mg(OH)2.
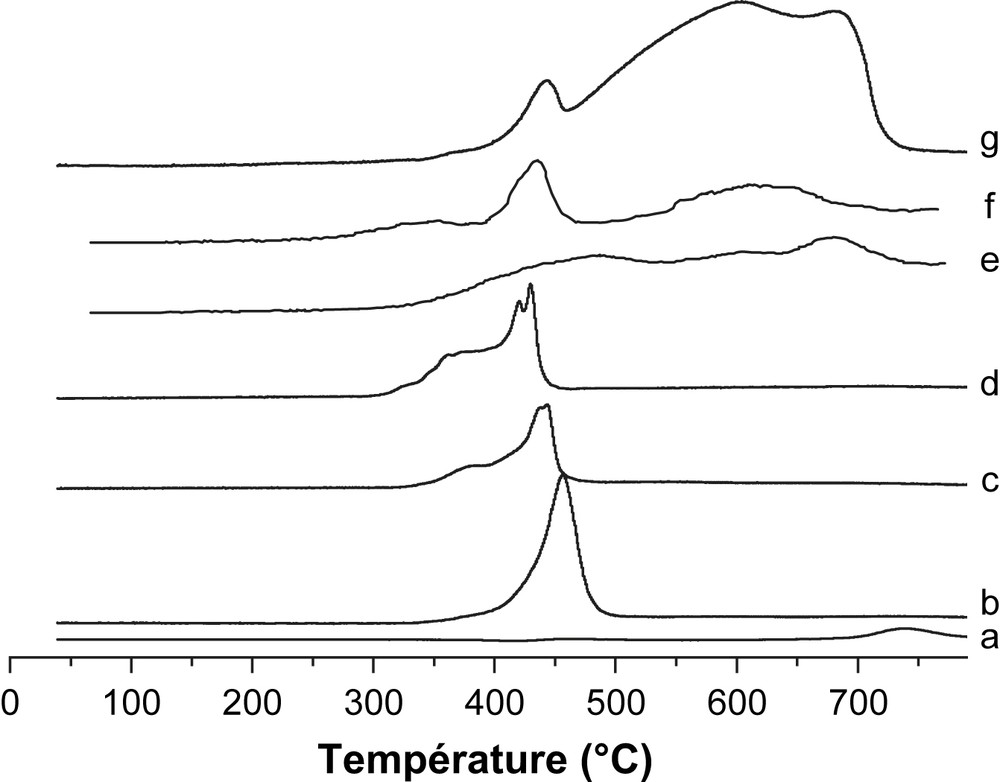
Profils H2- RTP de : (a) MgO, (b) 30% Fe2O3/MgO calciné à 200 °C, (c) 30% Fe2O3/MgO calciné à 300 °C, (d) 30% Fe2O3/MgO calciné à 400 °C, (e) 30%Fe2O3/SiO2 calciné à 400 °C, (f) 30% Fe2O3/TiO2 calciné à 400 °C et (g) Fe2O3.
4 Conclusions
Les catalyseurs à base de fer exempts d'oxyde de chrome Cr2O3 ont été étudiés en réaction CO + H2O. Les propriétés acides et/ou basiques de ces catalyseurs ont été corrélées à la conversion de CO en CO2. Le caractère basique de Fe3O4/MgO calciné à 400 °C a été montré par l'étude de la décomposition de l'isopropanol à 250 °C. Les résultats de l'analyse par DRX ont révélé la présence d'une forte fraction cristalline de MgO dans le catalyseur Fe3O4/MgO calciné à 400 °C. Cette phase alimente la surface du catalyseur en intermédiaire formiate et favorise la réaction CO + H2O.