

1 Introduction
The oxalate group (dianion of the ethanedioic acid, H2ox) is a classical ligand in coordination chemistry and in magneto-structural studies due to the great number of coordination modes that it exhibits in its metal complexes [1–3] together with its remarkable ability to mediate strong magnetic interactions between the paramagnetic metal ions when acting as a bis-bidentate bridge, the metal-metal separation being larger than 5.4 Å [4]. In this respect, magneto-structural studies of its dicopper(II) complexes constitute textbook examples of tunable magnetic coupling, the plasticity of the coordination sphere of the copper(II) making it possible to vary in a prefixed manner the symmetry and orientation of the interacting magnetic orbitals [5]. The polydentate nitrogen donor used as peripheral ligand determines the type of magnetic orbital and avoids the unwanted precipitation of the oxalato-bridged copper(II) chain Cu(ox) · 1/3H2O [6]. So, the use of chelating ligands such as N,N′,N″,N′′′-tetramethylethylenediamine (tmen), 2,2′-bipyridine (bipy), 1,10-phenanthroline (phen), 2,2′-bipyrimidine (bpym), 2-(2-pyridyl)imidazole (pyim) or N,N′,N″-diethylenetriamine (dien) affords the mononuclear species [Cu(tmen)(ox)(H2O)] · 3H2O [7], [Cu(bipy)(ox)(H2O)] · 2H2O [8], [Cu(phen)(ox)(H2O)] · H2O [9], [Cu(bpym)(ox)] · 5H2O [10], [Cu(pyim)(ox)(H2O)] · 2H2O [11] and [Cu(dien)(ox)] · 4H2O [12] that could be the used as ligands towards preformed complexes whose coordination sphere is unsaturated. As an illustrative example, one can point out the neutral two-dimensional networks of formula [Cu2(bpym)(ox)2]n · 5nH2O [10] and [Cu2(bpym)(ox)Cl2]n [13] which have in common the occurrence of bis-bidentate bpym and ox ligands, the chloro groups acting also as bridges in the last compound.
Another strategy that avoids the formation of the above mentioned oxalato-bridged copper(II) chain in the synthetic process is to use a 2:1 oxalate to copper(II) molar ratio in order to favour the formation of the dianionic unit [Cu(ox)2]2−, an entity which has been isolated as both hydrated (cat)2[Cu(ox)2]·2H2O (cat+ = Na+, K+, NH4+ and 4-Hampy+) and anhydrous (Hpy)2[Cu(ox)2] and (H2pda)[Cu(ox)2] complexes (Hampy+, Hpy+ and H2pda2+ are 4-aminopyridinium, pyridinium and propylenediammonium cations, respectively) [14,15]. Curiously, the reaction of the (NH4)2[Cu(ox)2]2 derivative with the 1-(2-carboxyethyl)-4,4′-bipyridium chloride (H2cebpyCl) in aqueous solution afforded the one-dimensional copper(II) compound (H2cebpy)2[Cu(ox)2] · 2H2O which exhibits a weak ferromagnetic coupling (J = +0.62 cm−1, the Hamiltonian being defined as H = –J ∑nm Si · Sj) across the bidentate/inner monodentate oxalate bridge [16]. Such a μ-1,2,2 bridging mode of the oxalate group is quite unusual in the literature [15b,17].
In this work, we describe how the copper(II)-asssisted hydrolysis of basic methanolic solutions of the ligand (2-pyrimidyl)oxamic acid ethyl ester (HEtpmo) constitutes a new synthetic strategy to prepare single crystals of oxalato-bridged copper(II) chains with intrachain ferromagnetic coupling. This is illustrated by the zig-zag copper(II) chain of formula {[(CH3)4N]2[Cu(C2O4)2] · H2O}n (1) (Me4N+ = tetramethylamonium cation) whose preparation, crystal structure and variable-temperature magnetic study are included here.
2 Experimental
2.1 Materials
Copper(II) chloride dihydrate, tetramethylammonium hydroxide, triethylamine, ethyl oxalyl chloride and 2-aminopyrimidine were purchased from commercial sources and used as received. The proligand (2-pyrimidyl)oxamic acid ethyl ester (HEtpmo) was prepared by reacting ethyl oxalyl chloride with 2-aminopyrimidine and triethylamine in 1:1:1 molar ratio under reflux at 80 °C during 4 h, the general procedure previously described for these type of ligands being followed [18]. Elemental analysis (C, H, N) were conducted by the Microanalytical Service of the Federal University of Goiás and the copper contents was determined by spectrophotometry with a Hitachi-Z8200 spectrophotometer.
2.2 Preparation of {[(CH3)4N]2[Cu(C2O4)2] · H2O}n (1)
Teramethylammonium hydroxide (0.441 cm3, 4.4 mmol) was added to a methanolic solution (10 cm3) of HEtpmo (0.20 g, 1.1 mmol) under continuous stirring. After 10 min, an ethanolic solution (5 cm3) of copper(II) chloride dihydrate (0.093 g, 0.55 mmol) was added dropwise. The small amount of a bluish solid that resulted was removed by filtration and the remaining deep blue solution was allowed to slowly evaporate in a hood at room temperature. X-ray quality turquoise crystals of 1 as blue prisms were grown after several weeks. They were collected by filtration, washed with a small amount of cold water and dried over filter paper. Yield: 45.0%. Anal. found: C, 35,37; H, 6.35; Cu, 15.59; N, 6.80%. Calc. for C12H26CuN2O9: C, 35.48; H, 6.40; Cu, 15.66; N, 6.90%. Main IR features (values in cm−1, KBr pellet): 3326 m [ν(O–H)], 3020w, 2914w and 2837w [ν(C–H)], 1673s, 1652s and 1637s] [νas(OCO)], 1488 and 1418 [νs(OCO)], 1289 m [νs(C–O)] and 808w [δ(OCO)].
2.3 Physical measurements
An infrared spectrum of 1 was recorded with a Perkin Elmer Precesily Spectrum 400 FT-IR/FT-FIR spectrophotometer as a KBr pellet in the range 4000–400 cm−1. Magnetic susceptibility measurements on a polycrystalline sample of 1 were carried out in the temperature range 1.9–300 K with a Quantum Design SQUID susceptometer, using applied magnetic dc fields of 0.1 T (T ≥ 50 K) and 500 G (T < 50 K). The corrections for the diamagnetism of the constituent atoms were estimated from Pascal's constants [19] as –204 × 10−6 cm3 mol−1 [per mol of copper(II) ions]. Corrections for the temperature-independent paramagnetism [60 × 10−6 cm3 mol−1 per Cu(II)] and for the simple holder were also applied.
2.4 Crystal structure determination and refinement
A prism-shaped single crystal of 1 with dimensions 0.12 × 0.08 × 0.07 mm3 was selected for the X-ray data collection. It was mounted on a glass fiber and afterwards positioned on the goniometer. Room temperature X-ray diffraction intensities (298 K) were measured using graphite-monochromated Mo Kα line (λ = 0.71073 Å), Enraf-Nonius Kappa-CCD diffractometer equipped with a 95 mm CCD camera on a κ-goniostat operating in φ-ω scans mode with κ offsets. All reflections were used to index the final unit cell. The acquisition of the X-ray diffraction frames was monitored by using the program COLLECT [20] and the raw dataset was treated with the HKL Denzo-Scalepack [21]. Because of the small size (medial crystal size x = 0.08 mm) and the not too high absorption coefficient (μ = 1.275 mm−1), no absorption correction was applied. According to the International Union of Crystallography (IUCr) [22], there is no need of absorption correction if the product μx is not above 0.1. This product was 0.102 for the measured crystal of 1 by using Mo Kα radiation. Therefore, we have preferred to comply with IUCr recommendations even if the refinement statistic was satisfactory (Table 1).
Crystal data and structure refinement for {[(CH3)4N]2[Cu(C2O4)2] · H2O}n (1).
| Empirical formula | C12H26CuN2O9 |
| Formula weight | 405.90 |
| Temperature, K | 298(2) |
| Wavelength, Å | 0.71073 |
| Crystal system | Orthorhombic |
| Space group | Pbca |
| Unit cell parameters, Å | a = 11.2757 (2) |
| b = 10.1540 (2) | |
| c = 31.0566 (7) | |
| Volume, Å3 | 3555.8 (1) |
| Z | 8 |
| ρcalcd, Mg/m3 | 1.516 |
| Absorption coefficient, mm−1 | 1.275 |
| Crystal size, mm | 0.12 × 0.08 × 0.07 |
| Theta range for data collection | 3.00 – 25.68° |
| F(000) | 1704 |
| Index ranges | –13 ≤ h ≤ 13, –12 ≤ k ≤ 12, –37 ≤ l ≤ 37 |
| Reflections collected | 6221 |
| Independent reflections | 3322 [R(int) = 0.0292] |
| Completeness to θ = 25.68° | 98.2% |
| Data/restraints/parameters | 3322/0/223 |
| Goodness-of-fit on F2 | 1.066 |
| R1 [I > 2σ(I)] | 0.0400 |
| wR2 (all data) | 0.1138 |
| Largest diff. peak and hole, e Å−3 | 0.431 and –0.538 |
The structure was solved by direct methods with SHELXS-97 [23]. All non-hydrogen atoms were readily found from the electronic density map constructed by Fourier synthesis, in which the Cu, C, O and N atoms were clearly identified. The initial model was refined by the full-matrix least squares method on F2 with SHELXL-97 [23] adopting anisotropic thermal parameters for all non-hydrogen atoms. The hydrogen atoms form the NMe4+ cation were positioned stereochemically and they were refined with fixed individual isotropic displacement parameters [Uiso(H) = 1.5Ueq of both methyl carbons and water oxygen] by using a riding model with CH bond lengths of 0.96 Å. The positions of the hydrogen atoms from the water molecules were assigned from the electronic density map generated by Fourier difference and they were refined freely.
The ORTEP-3 program [24] was used within the WinGX software package [25] to deal with the processed crystallographic data and artwork representations. After the end of the refinement, the molecular structure of 1 was checked with MOGUL program [26]. This is a valuable program for analyzing geometric features of any structure. It searches for substructures of compounds deposited at the Cambridge Crystallographic Data Centre (CCDC) that are similar to those of a target. Comparisons of bond angles and lengths of 1 with the corresponding parameters of similar structures deposited in the Cambridge Structural Database (CSD, version 5.32 of November 2010 with August 2011 update) [27] were useful to strengthen interesting intramolecular features of the complex studied. Bond lengths and angles of 1 together with those for structurally related complexes are listed in Tables 2 and 3 whereas the X-H O (HC and Ow) intermolecular interactions of 1 are given in Table 4.
Bond lengths (Å) for 1 and for structurally related compounds deposited in the Cambridge Structural Database (CSD).a
| Bond | 1 | Mean value in the CSDb | No. of entries in the CSD |
| Cu1O1A | 2.323(2) | 1.97(5) | 22 |
| Cu1O1B | 1.983(2) | 2.0(1) | 17 |
| Cu1O2A | 1.973(1) | 1.97(5) | 22 |
| Cu1O2B | 1.957(2) | 2.0(1) | 17 |
| Cu1O3Aii | 2.437(2) | – | – |
| Cu1O4Aii | 1.978(2) | – | – |
| C1AO1A | 1.234(2) | 1.25(2) | 170 |
| C1BO1B | 1.269(3) | 1.28(1) | 76 |
| C1AO4A | 1.262(3) | 1.26(3) | 16 |
| C1BO4B | 1.236(3) | 1.21(2) | 10000 |
| C2AO2A | 1.266(3) | 1.25(2) | 170 |
| C2AO3A | 1.236(3) | 1.26(3) | 16 |
| C2BO2B | 1.269(3) | 1.28(1) | 76 |
| C2BO3B | 1.227(3) | 1.21(2) | 10000 |
| C1AC2A | 1.566(3) | 1.54(2) | 868 |
| C1BC2B | 1.545(4) | 1.54(2) | 1485 |
| C1A′N1A | 1.501(3) | 1.48(5) | 7875 |
| C1B′N1B | 1.500(3) | 1.48(5) | 7875 |
| C2A′N1A | 1.497(3) | 1.48(5) | 7875 |
| C2B′N1B | 1.470(3) | 1.48(5) | 7875 |
| C3A′N1A | 1.486(3) | 1.48(5) | 7875 |
| C3B′N1B | 1.494(3) | 1.48(5) | 7875 |
| C4A′N1A | 1.487(3) | 1.48(5) | 7875 |
| C4B′N1B | 1.491(3) | 1.48(5) | 7875 |
a Results from the MOGUL intramolecular analysis.
b Symmetry code: (ii) = ½ – x; – ½ + y; z.
Bond angles (°) for 1 and for structurally related complexes deposited in the Cambridge Structural Database (CSD).a
| Angle | 1 | Mean value in the CSDb | No. of entries in the CSD |
| Cu1O1AC1A | 107.7(1) | – | – |
| Cu1O1BC1B | 111.9(2) | – | – |
| Cu1O2AC2A | 118.6(1) | – | – |
| Cu1O2BC2B | 112.3(1) | – | – |
| Cu1iO3AC2A | 106.1(1) | – | – |
| Cu1iO4AC1A | 120.3(1) | – | – |
| O1ACu1O1B | 90.89(7) | – | – |
| O1BCu1O2A | 91.84(7) | – | – |
| O1ACu1O2A | 77.57(6) | – | – |
| O1ACu1O2B | 99.85(6) | – | – |
| O1BCu1O2B | 83.93(7) | – | – |
| O1ACu1O3Aii | 170.72(5) | – | – |
| O1BCu1O3Aii | 96.55(6) | – | – |
| O1ACu1O4Aii | 97.44(6) | – | – |
| O1BCu1O4Aii | 170.57(7) | – | – |
| O2ACu1O2B | 175.04(7) | – | – |
| O2ACu1O3Aii | 96.61(6) | – | – |
| O2ACu1O4Aii | 94.24(7) | – | – |
| O2BCu1O3Aii | 86.48(6) | – | – |
| O2BCu1O4Aii | 90.28(7) | – | – |
| O3ACu1iO4A | 75.61(6) | – | – |
| C1AC2AO2A | 116.7(2) | 117(2) | 168 |
| C1AC2AO3A | 118.5(2) | 117(2) | 15 |
| C1BC2BO2B | 115.5(2) | 115(1) | 38 |
| C1BC2BO3B | 119.4(2) | 120(2) | 2879 |
| C2AC1AO1A | 117.4(2) | 117(2) | 168 |
| C2AC1AO4A | 117.0(2) | 117(2) | 15 |
| C2BC1BO1B | 115.3(2) | 115(1) | 38 |
| C2BC1BO4B | 118.6(2) | 120(2) | 2879 |
| C1A′N1AC2A′ | 109.7(2) | 109(6) | 10000 |
| C1A′N1AC3A′ | 108.5(2) | 109(6) | 10000 |
| C1A′N1AC4A′ | 109.4(2) | 109(6) | 10000 |
| C1B′N1BC2B′ | 109.5(2) | 109(6) | 10000 |
| C1B′N1BC3B′ | 109.3(2) | 109(6) | 10000 |
| C1B′N1BC4B′ | 109.8(2) | 109(6) | 10000 |
| C2A′N1AC3A′ | 109.5(2) | 109(6) | 10000 |
| C2A′N1AC4A′ | 110.4(2) | 109(6) | 10000 |
| C2B′N1BC3B′ | 110.4(2) | 109(6) | 10000 |
| C2B′N1BC4B′ | 109.8(2) | 109(6) | 10000 |
| C3A′N1AC4A′ | 109.3(2) | 109(6) | 10000 |
| C3B′N1BC4B′ | 108.0(2) | 109(6) | 10000 |
| O1AC1AO4A | 125.5(2) | 126(2) | 16 |
| O1BC1BO4B | 126.1(2) | 126(2) | 76 |
| O2AC2AO3A | 124.8(2) | 126(2) | 16 |
| O2BC2BO3B | 125.1(2) | 126(2) | 76 |
a Results from the MOGUL intramolecular analysis.
b Symmetry code: (i) = ½ – x, ½ + y, z; (ii) = ½ – x; – ½ + y, z.
Intermolecular XHA [XC or Ow; YOox or Ow)] type interactions in 1.a
| XHY | XH, Å | HY, Å | XY, Å | XHY, degree |
| C1A′H1A1O1Wv | 0.96 | 2.69 | 3.622(4) | 163 |
| C2A′H2A3O1w | 0.96 | 2.63 | 3.510(3) | 152 |
| C3A′H3A3O1w | 0.96 | 2.70 | 3. 561(3) | 150 |
| O1wH1wO4B | 0.91(3) | 1.88(3) | 2.783(3) | 173(3) |
| O2wH2wO3Bvi | 0.92(3) | 2.57(3) | 3.343(3) | 142(2) |
| O2wH2wO4Bvi | 0.92(3) | 2.17(3) | 2.981(3) | 147(3) |
a Symmetry code: (v) = – ½ + x, y, ½ – z + 2; (vi) = 1.5 – x, ½ + y, z.
3 Results and discussion
3.1 Synthesis and IR characterization
It is known that oxamato and oxamidato groups can undergo a copper(II)-assisted hydrolysis in basic media yielding oxalate [18,28]. This hydrolytic reaction transforms HEtpmo into oxalate and it accounts for the slow formation of single crystals of 1 from the basic aqueous solution containing copper(II) and the oxamate ligand (Scheme 1). When the synthesis was carried out by using oxalic acid instead of the monoxamic derivative, a blue powder resulted whose analytic data indicate that a different species has been obtained. It seems that the hydrolytic process can be considered as a suitable way to get X-ray quality crystals of oxalate-bridged copper(II) complexes that could not be obtained by the direct reaction between oxalate and the copper(II) ion.

Reaction route affording compound 1.
The IR absorptions of the oxalate group in the spectrum of 1 [1673,1652 and 1637 cm−1, (νas(COO) stretching vibrations), 1488 and 1418 cm−1 (νs(COO) stretching vibrations) and 1289 and 808 cm−1 (νs(CO) and δ(OCO) vibrations, respectively] suggest the presence of chelating and bis-chelating oxalato [29], a feature that has been demonstrated by the X-ray structure of this compound (see below).
3.2 Description of the structure
Compound 1 crystallizes in the centrosymmetric orthorhombic space group Pbca, with one copper(II) ion, two oxalate ligands, two tetramethylammonium counterions, and one free water molecule in the asymmetric unit (Fig. 1). One of the two oxalate groups in the asymmetric unit acts as a bis-bidentate ligand coordinating two b-glide plane symmetry-related copper(II) ions through its four oxygen atoms [O1A and O2A at Cu1 and O3A and O4A at Cu1i [symmetry code: (i) = ½ – x, ½ + y, z]. The other oxalate group adopts the bidentate coordination mode through the O1B and O2B atoms completing the six-fold coordination-sphere around the copper atom. The resulting [Cu(ox)2]n2n− anionic unit grows along the crystallographic b axis as a zigzag copper(II) chain (Fig. 2). The copper(II)-copper(II) separation across the bridging oxalate is 5.5898(3) Å [Cu1 Cu1i] whereas the distances between copper(II) ions belonging to neighbouring chains are much longer: 8.9746(4) Å for the distance between chains packed parallel to the crystallographic c axis and related by 21 screw axis along the [100] direction [Cu1Cu1iii; symmetry code: (iii) = –½ + x, ½ – y, – z]. It is striking to notice that these chains are not mirror images. They are only running on opposite directions parallel to the crystallographic b axis (Fig. 2). In addition, a-glide plane symmetry-related chains are also packed parallel to the crystallographic c axis. In this case, there is a separation of 10.2382(4) Å between the copper(II) ions on adjacent mirror chains [Cu1Cu1iv; symmetry code: (iv) = –½ + x, y, ½ – z]. Finally, the interchain distances between translation symmetry-related ribbons packed along the crystallographic a axis are the largest ones, 10.2783(3) and 11.2757(4) Å for Cu1Cu1v and Cu1Cu1vi, respectively [symmetry code: (v) = 5 – x, ½ + y, z; (vi): 1 + x, y, z].

ORTEP diagram of the asymmetric unit of 1 showing the atom numbering. Thermal ellipsoids are at the 50% probability level and the hydrogen atoms are shown as spheres of arbitrary radii. Symmetry code: (ii) = ½ – x, – ½ + y, z.
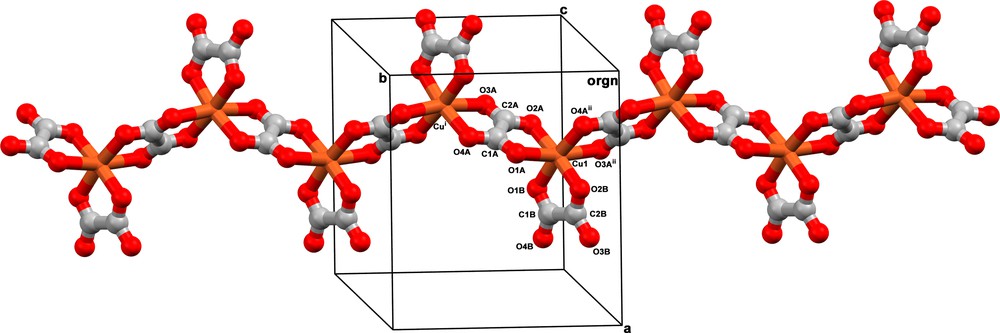
View of a fragment of the helical oxalate-bridged copper(II) chain in 1. Symmetry code: (i) = ½ – x, ½ + y, z; (ii) = ½ – x, – ½ + y, z.
Each copper(II) ion in 1 adopts a distorted octahedral coordination geometry, a feature that contrast with the preferential four- or five-coordination of this metal ion in its oxalato-bridged complexes [5]. In the light of the CuO bond lengths from Table 2, the geometry around the copper(II) ion in 1 is better described as an elongated and statically distorted octahedron because of four short CuO distances [1.983(2), 1.973(2), 1.957(2) and 1.978(2) Å for Cu1O1B, Cu1–O2A, Cu1O2B and Cu1O4Aii, respectively; symmetry code: (ii) = ½ – x, – ½ + y, z] and two weaker CuO interactions [2.323(2) and 2.437(2) Å for Cu1O1A and Cu1O3Aii]. Distances similar to those observed in 1 are also observed for a tris(oxalato)copper(II) unit in a previous report where an elongated octahedral geometry around the copper(II) ion with four equatorial oxygen atoms and two axially bound oxygens at 1.99 and 2.32 Å, respectively, from the copper atom were suggested on the basis of EXAFS data [30]. In the related complex [Cu(en)3]SO4 [31]. EPR and XAS results demonstrated that the copper(II) ion geometry is a dynamically distorted octahedron at room temperature due to four short CuN lengths and two enlarged CuN oscillating distances, adopting a 4 + 2 coordination. Both geometry distortions of the copper(II) ion were attributed to the Jahn-Teller effect [30,31]. In this way, one can conclude that such an effect is responsible for the elongated and distorted octahedron geometry of 1. Moreover, it is important to note that the 4 + 2 octahedron geometry in 1 is a statically distorted octahedron rather than the dynamically one reported for the related complexes aforementioned, the two longer Cu1O1A and Cu1O3Aii bond distances being clearly different in 1.
In the coordination sphere of copper(II) atom of 1, two of the four equatorial oxygen-atoms belong to two oxalate bridges related by b-glide plane symmetry (O2A and O4Aii) and the two others (O1B and O2B) are from the oxalate which does not bridge between two copper(II) atoms. All these four equatorial oxygens deviate considerably from the least-squares plane passing through them [O1B, O2A, O2B and O4Aii deviate –0.090(2) Å, 0.076(2) Å, 0.091(2) Å and –0.073(2) Å; rmsd of the four fitted atoms is 0.0063 Å]. The Cu1 atom is shifted by 0.0446(2) Å from the least-squares basal plane. The cis OeqCuOeq bond angles vary in the range 90.28(7)–94.24(7)° whereas the trans ones are 170.57(7)° (O1BCu1O4Aii) and 175.04(7)° (O2ACu1O2B). The OeqCuOax bond angles span in the range 75.61(6) to 99.85(6)° and the axial O1ACu1O3Aii one is 170.72(5)° (Table 3). The short values of the bite of the chelating and bis-chelating oxalate ligands are at the origin of the main deviation from the ideal 90° or 180° bond angles at the copper environment in 1. The shortening of the C1AO1A (1.234(2) Å) and C2AO3A (1.236(3) Å) bond distances when compared to the related equatorial carbon-oxygen bonds [values in the range 1.262(3)–1.269(3) Å] is due to the strongest electrostatic interactions in the equatorial oxalate-oxygen to copper(II) bonds. Therefore, C1AO1A and C2AO3A bonds exhibit an increased CO double bond character rather than a major CO single bond feature of the enlarged bonds C1AO4A (1.262(3) Å), C1BO1B (1.269(3) Å), C2AO2A (1.266(3) Å) and C2BO2B (1.269(3) Å). Mean values of 1.25(2) Å and 1.26(3) Å were averaged by MOGUL in the Cambridge Structural Database (CSD) [27] on 170 and 16 structures with bonds equivalent to C1AO1A and C2AO3A of 1, respectively. MOGUL is a knowledge database of molecular geometry derived from CSD [27] that provides access to information on the preferred values of bond lengths, valence angles and torsion angles. Besides highlighting interesting geometrical features such as these, MOGUL searches have showed that all bond lengths and bond angles are in agreement with the expected values for a good X-ray diffraction structure refinement (Tables 2 and 3). In addition, MOGUL surveys have revealed an elongation of the bond distance between the two sp2-hybridized carbons of the oxalate bridge, which is rationalized by taking into account that this oxalate unit is four-coordinated to copper(II) and then there is a notable tension about the C1AC2A bond. This bond measures 1.566(3) Å in I whereas a mean value of 1.54(2) Å was averaged on 868 1-like compounds searched by MOGUL in the CSD [27]. On the other hand, the carbon-carbon equivalent bond in the non-bridging oxalate is 1.545(4) Å (C1BC2B bond length). This value matches to the MOGUL mean value averaged on 1485 structures similar to 1 (1.54(2) Å). Based on this MOGUL matching, it is possible to state that elongation of the C1BC2B bond length does not occur, which is agreement with the fact that the non-bridging oxalate is bidentate towards copper(II) and thus, it undergoes less tension on its Csp2Csp2 bond than the bis-bidentate oxalate.
The three oxalate ligands are not coplanar: the octahedral coordination geometry around the CuII atom forces each oxalate ligand to lie almost perpendicular to each one. The least-squares planes through two b-glide plane symmetry-related oxalate bridges [the root-mean-square deviation (rmsd) of the six fitted atoms C1A, C2A, O1A, O2A, O3A and O4A is 0.0101 Å] form an angle of 68.38(3)°, while the least-squares plane of the other non-bridging oxalate fragment (rmsd of the six fitted atoms C1B, C2B, O1B, O2B, O3B and O4B is 0.0121 Å) forms angles of 72.33(5)° and 88.01(6)° with the corresponding planes of the bridging oxalate groups.
It is striking to note that the synthesis of a complex such as 1 with the CuII ion surrounded by three coordinated oxalate ligands in which the transition metal ion undertakes an octahedral coordination geometry is unpredicted because only three structures loading this framework are found in the CSD [27]. Likewise, a one-dimensional zigzag coordination polymer of six-coordinated copper(II) assembled with oxalate ligands is unlikely to be prepared using common inorganic preparative routes since no example of this is reported. In this work, such a structure is described for the first time. Oxalate bridges intercalates copper(II) ions along the [010] direction, resulting in a zigzag chain grown along the crystallographic b axis (Fig. 2).
Tetramethylammonium cations are also packed along the b axis, even though they are further arranged parallel to the a axis. Only van der Waals interactions keep them in contact along both directions. Therefore, 2D columns of tetramethylammonium running parallel to the [100] and [010] directions are assembled in the structure of 1. These columns intercalate between two copper(II) chains along the c axis, (Fig. 3). Moreover, the organic cations contribute to stabilize the crystal packing through weak CHO type interactions with both oxalate-oxygens and the crystallization water molecule (Table 4). These contacts connect the chains along the a and c axes, giving rise to a supramolecular 3D network. Likewise, water plays a dual role in the crystal assembly of 1. It is a non-classical hydrogen bonding acceptor from a given (CH3)4N+ cation in the C1A′H1A1O1Wv type interactions [symmetry code: (v) = –½ + x, y, ½ – z], C2A′–H2A3O1W, and C3A′H3A3O1w (Fig. 4). At the same time, each water molecule forms hydrogen bonds with the non-coordinated oxygens of two b-glide plane symmetry-related non-bridging oxalate units. The O1wH1WO4B hydrogen bond and the bifurcated O1w–H2WO4Bvi [symmetry code: (vi) = 1.5 – x, ½ + y, z] and O1wH2WO3Bvi ones connect water molecule to two coordination ribbons, acting as a cross-linker between two anionic chains parallel to the [100] direction (Fig. 4 and Table 4). Therefore, the non-coordinated oxalate-oxygen atoms are involved in hydrogen bonding interactions, which contribute to the stabilization of the supramolecular three-dimensional structure of 1. This reveals that there is a fine tuning between the formation of coordination bonds and intermolecular contacts in this noteworthy structure.

A view of the packing in 1 down the crystallographic a axis, showing the segregated stacking of the anionic oxalate-bridged copper(II) chains and tetramethylammonium counterions.

A view onto the (001) plane of the intermolecular interactions in 1 involving the crystallization water molecule, the tetramethylammonium cations and the free oxalate-oxygens from the anionic oxalate-bridged copper(II) chain. Symmetry code: (vi) = 1.5 – x, ½ + y, z; (vii) = ½ + x, y, ½ – z.
3.3 Magnetic properties
The magnetic properties of 1 in the form of χMT against T plot [χM is the magnetic susceptibility per copper(II) ion] are shown in Fig. 5. At room temperature, χMT is equal to 0.40 cm3 mol−1 K, a value which is as expected for a magnetically non-interacting spin doublet. This value remains practically constant upon cooling until T = 150 K and it further increases continuously to reach 0.69 cm3 mol−1 K at 1.9 K. Because of the one-dimensional character of 1, we have analyzed its magnetic data through the numerical expression proposed by Baker and Rushbrooke [32] for a ferromagnetically coupled uniform chain of spin doublets [Eq. (1)]
| (1) |

Thermal variation of the χMT product for 1: (o) experimental; (–) best-fit curve through Eq. (1) (see text).
In such an expression, J is the exchange coupling between adjacent copper(II) ion of the chain parameter, g is the average Landé factor and N, β and k have their usual meaning. Least-squares fit of the susceptibility data lead to the following values: J = +1.14 cm−1, g = 2.08 and R = 1.6 × 10−5 (R is the agreement factor defined as . The calculated plot matches well the magnetic data in the whole temperature range explored.
The ferromagnetic coupling found in 1 is weak and it corresponds to a case of accidental orthogonality between the interacting magnetic orbitals through the out-of-plane exchange pathway, a situation analogous to that previously observed in μ-chloro, di-μ-chloro and μ-oxamato dicopper(II) complexes where this exchange pathway is involved [33,34]. Simple orbital symmetry considerations allow to visualize the origin of this ferromagnetic interaction across the bridging oxalato in 1. Let us focus on a dicopper(II) fragment of this chain. The unpaired electron on each copper(II) ion in 1 is described by a d(x2–y2) type metal centered magnetic orbital [the x and y axes being roughly defined by the Cu1O2A and Cu1O1B bonds] which is located in the equatorial plane and the two magnetic orbitals are parallel to each other and perpendicular to the oxalate plane (Scheme 2).
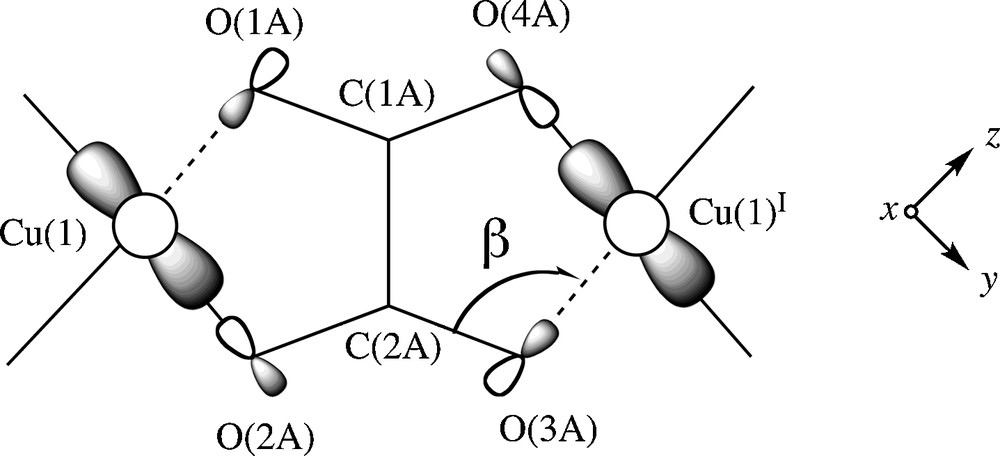
A view of the interaction between the magnetic orbitals in the oxalate-bridged dicopper(II) fragment of 1.
The exchange pathway involves the oxalate O2AC2AO3A and its symmetry-related O1AC1AO(4a) set of atoms. According to Kahn's model [35], the coupling constant J in a dicopper(II) unit can be decomposed into two terms, one positive (ferro-, JF) and the other negative (antiferromagnetic, JAF), the expression being J = JF + JAF. In such a model, the value of the negative term is proportional to the square of the overlap integral (S2) between the two metal centered magnetic orbitals. The poor overlap between the two parallel magnetic orbitals through the two OCO oxalate set of atoms would lead to a weak antiferromagnetic coupling. However, in the case where this overlap is zero (the accidental orthogonality occuring in 1), a weak ferromagnetic coupling is predicted.
In that respect, the magneto-structural data of the oxalato-bridged dicopper(II) complexes listed in Table 5 show that the values of the bond angle at the axial copper to oxalate-oxygen (β) and the axial copper to oxalate-oxygen bond length are the main structural factors governing the nature of the magnetic coupling for the topology shown in Scheme 2. In the light of this Table, one can conclude that the frontier between the ferro- and antiferromagnetic coupling in this family of complexes corresponds to a value of β equal to 109.5°. Values of β greater than this one will favour the antiferromagnetic coupling whereas smaller ones will give rise to ferromagnetic interactions. Finally, it deserves to be pointed out that this magneto-structural result was first observed by Castillo and García-Coucerio during their doctoral work [42].
Selected magneto-structural data for oxalato-bridged copper(II) complexes having the out-of-plane exchange pathway.
| Compounda | Donor setb | dCuO(ax)c (Å) | hCud (Å) | βe (°) | γf (°) | dCu···Cug (Å) | Jh (cm−1) | Ref. |
| [Cu2(bpca)2(ox)] | N3O2 | 2.26 | 0.16 | 107.5 | 92.0 | 5.44 | +1.1 | [35] |
| [Cu2(bpca)2(H2O)2(ox)]·2H2O | N3O2O′ | 2.41 | 0.05 | 106.9 | 80.7 | 5.63 | +1.0 | [36] |
| {[(CH3)4N]2[Cu(ox)2]·H2O}n | O6 | 2.38 | 0.04 | 106.9 | 79.1 | 5.59 | +1.14 | This work |
| [Cu2(bpcam)2(H2O)2(ox)] | N3O2O′ | 2.44 | 0.00 | 106.6 | 101.8 | 5.68 | +0.75 | [37] |
| {[Cu(bipy)(ox)] · 2H2O}n | N2O4 | 2.31 | 0.034 | 108.3 | 86.8 | 5.56 | +2.4 | [38] |
| [Cu(py)2(ox)]n | N2O4 | 2.27 | 0.00 | 108.0 | 80.0 | 5.46 | +1.4 | [39] |
| [Cu(2-ampy)2(ox)]n | N2O4 | 2.38 | 0.02 | 107.8 | 75.6 | 5.63 | +2.0 | [40] |
| [Cu(isq)2(ox)]n | N2O4 | 2.23 | 0.06 | 109.5 | 88.5 | 5.48 | +0.63 | [41] |
| [Cu(4-ampy)2(ox)]n | N2O4 | 2.35 | 0.00 | 109.7 | 88.7 | 5.66 | –1.1 | [40] |
| [Cu(3-ampy)2(ox)]n | N2O4 | 2.17 | 0.00 | 111.0 | 85.2 | 5.46 | –1.3 | [40] |
a Abbreviations of the ligands: bpca: bis(2-pyridylcarbonyl)amidate; ox: oxalate; bpcam: bis(2.pyrimidylcarbonyl)amidate; bipy: 2,2′-bipyridine; py: pyridine; 2-ampy: 2-aminopyridine; isq: isoquinoline; 4-ampy: 4-aminopyridine; 3-ampy: 3-aminopyridine.
b The first four atoms form the basal/equatorial plane.
c Value of the axial copper to oxalate-oxygen.
d The height of the copper atom from the mean basal/equatorial plane.
e Bond angle at the axial oxalate–oxygen (CuOC).
f Dihedral angle between the basal/equatorial and oxalate mean planes.
g Copper–copper distance across the bridging oxalato.
h Value of the magnetic coupling.
4 Conclusions
A new one-dimensional oxalate-containing copper(II) complex, namely {[N(CH3)4]2[Cu(C2O4)2] · H2O}n (1), has been synthesized by the hydrolysis of an oxamato proligand under basic conditions in the presence of copper(II) ions. Chiral anionic copper(II) chains with opposite helicities occur in 1 resulting in an achiral structure. The surrounding of each copper(II) ion is elongated octahedral and the intrachain copper-copper separation is 5.5898(3) Å. The magnetic studies reveal the occurrence of a weak ferromagnetic interaction through the oxalate bridge, its nature and magnitude being in a good agreement with available magneto-structural data for oxalate-bridged copper(II) complexes where the same out-of-plane exchange pathway is involved.
5 Supplementary material
All details concerning the unit cell and structure determination of (I) were grouped in a data set. The file containing such data (CIF file), except structure factors, were deposited with the Cambridge Structural Database under deposit code CCDC 863398. Copies of these files may be retrieved free of charge from The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, UK; fax: +44 123 336 033; email: deposit@ccdc.cam.ac.uk or www.ccdc.cam.ac.uk.
Acknowledgements
This research was supported by the Ministério da Educação do Brasil for MCT/CNPQ (Project Universal no 476211/2010-7 and n° 472623/2011-7) and the Ministerio Español de Ciencia e Innovación (Project CTQ2010-15364); R.S.V. and T.L.O. thanks the CAPES for grant. Thanks are due to the Consejo Superior de Investigaciones Científicas (CSIC) of Spain for the award of a license for the use of the Cambridge Structural Database (CSD).