

1 Introduction
Traditionally, the determination of three-dimensional structures of biological macromolecules employs distance and torsion angle constraints extracted from NOE and J-coupling data, respectively 〚1, 2〛. A limitation inherent to the commonly used NMR methodology arises from the strictly local nature of the experimental constraints. Fortunately, despite this disadvantage, structure determination by NMR for globular proteins has been extremely successful, primarily because the numerous short inter-proton distances between amino acids far apart in sequence are highly correlated, rendering them conformationally extremely restrictive.
Nevertheless, the use of only short distance information can limit the accuracy of NMR derived structures, especially for non-globular architectures where the cumulative error may become significant or in cases where only few contacts are available between structural elements. Examples of such systems include modular and multi-domain proteins and linear nucleic acids. Recently, NMR methodology has been developed that exploits weak alignment of molecules in the magnetic field, either caused by the molecule’s own magnetic susceptibility anisotropy or employing very dilute liquid crystalline media 〚3〛. This allows the measurement of residual dipolar couplings, which contain information about the orientation of an inter-nuclear vector relative to the molecular susceptibility anisotropy tensor. These couplings can be used to extract angular restraints for use in NMR based structure calculations. Incorporation of these additional restraints into conventional structure determination algorithms results in remarkable improvements of the resulting NMR structures, both locally and globally.
2 Background
The existence of magnetic field induced residual dipolar couplings for proteins in isotropic solution was known for a number of years, their utility for structural studies, however, was only realized after high field magnets and heteronuclear methods that allowed their precise determination became widely available. Increased field strength was important, since the size of the residual one-bond dipolar coupling scales with the square of the magnetic field and novel experiments had to be devised to extract these extremely small couplings with a precision of a fraction of a Hertz 〚4, 5〛. Given the small degree of alignment of diamagnetic proteins in the magnetic field, resulting in minute residual dipolar couplings of typically < 0.2 Hz, the practicality of extracting such couplings reliably seemed limited. More promising, on the other hand, appeared the magnetic field induced alignment for paramagnetic proteins, nucleic acids and protein/nucleic acid complexes, which exhibit magnetic susceptibility anisotropies greater than –10 × 10–34 m3 per molecule, yielding residual dipolar couplings of ∼0.5 Hz for N–H vectors and ∼0.9 Hz for C–H vectors.
The major breakthrough with respect to any potential routine use of dipolar couplings for biomolecular structure determinations was the demonstration that tunable degrees of molecular alignment could be achieved by placing the molecule under investigation into a dilute, aqueous liquid crystalline phase of dihexanoyl phosphatidylcholine (DHPC) and dimyristoyl phosphatidylcholine (DMPC) 〚3, see 6, 7 for reviews〛. Sufficiently high degrees of alignment were obtained in this manner resulting in one-bond dipolar couplings of 5–40 Hz that are easily detectable by simply measuring the splitting in 2D and 3D coupled HSQC spectra. This opened the door for developing additional experiments to extract other types of residual dipolar couplings and resulted in a flurry of activity exploring possible alignment media with the aim of improving the initial bicelle system as well as discovering novel ones. Several of these media are described below.
Although a number of liquid crystalline media exhibit large macroscopic viscosity when compared to pure water, interestingly, the rotational diffusion of the dissolved biological macromolecule was only marginally affected. This makes it possible to take advantage of high resolution NMR methodology while simultaneously extracting residual dipolar couplings. Therefore, it is generally desired that the order imparted onto the macromolecule be relatively small, typically less than 2 × 10–3. To achieve such low solute alignment, interactions between the medium and solute have to be negligible. This cannot be predicted a priori and it is frequently necessary to empirically determine which medium is best suited for a particular case. If, however, binding to the medium occurs, line broadening is observed and the ordering will be too large to be of practical use. At present there is as yet not a single, universal alignment medium and it is generally advisable to test several for practical purposes.
The degree of alignment in any given liquid crystalline medium can easily be assessed by monitoring the deuterium quadrupolar splitting of the HDO signal in the sample prepared as an aqueous solution containing 90% H2O/10% D2O. This signal arises from exchange between bulk water and water bound to the oriented liquid crystal. Quadrupole splitting ranging from 10 Hz to 30 Hz are generally observed for the different media of variable composition. Interestingly, the degree of alignment of the solute macromolecule under investigation is not directly correlated to the deuterium quadrupole splitting across different liquid crystalline media, although a linear dependence on concentration is frequently observed for each individual medium. This is most likely due to the fact that the distribution and location of bound water molecules on the surfaces of the different large, anisotropic particles (lamellae of varying composition, phages and viruses, and assemblies of other surfactant phases) are very distinct and strongly dependent on the local surface structure. Nevertheless, measuring the deuterium quadrupole splitting of the HDO resonance is generally an excellent method to rapidly assess whether a medium is suitable for alignment purposes.
3 Alignment media
3.1 Liquid crystalline media based on phospholipids
The first medium used for purposes of partial alignment of solute macromolecules with the magnetic field was a lyotropic liquid crystal consisting of binary mixtures of DMPC and DHPC. Binary mixtures of these phospholipids were initially believed to form disc shaped particles 〚8〛, commonly referred to as bicelles 〚9〛. More recently, however, it was demonstrated that the morphology of these mixtures at concentrations and temperatures used for alignment in the magnetic field are stacked lamellar phases that align with their bilayer normal orthogonal to the field direction 〚10〛. Using these dilute phospholipid mixtures, biological macromolecules such as proteins, nucleic acids or complexes thereof can be dissolved in the interstitial aqueous spaces between the layers, rendering their rotational diffusion rates essentially unaffected by the lamellae. Residual dipolar couplings arising from the small degree of molecular alignment imparted onto the solute molecules by the oriented lamellae can therefore be measured. For most applications, the concentration of the phospholipids must be kept small (∼ 5%) to avoid line broadening caused by unresolved dipolar couplings. However, under these dilute conditions the liquid crystal becomes less stable and the temperature range over which a stable nematic phase is observed is limited. In addition, samples may phase separate within the time required to collect the NMR data. Commonly used mixtures contain ∼ 3% to 5% w/v DMPC/DHPC with a DMPC/DHPC ratio of ∼ 3:1. They form stable liquid crystal phases over the temperature interval of 29–45 °C. This somewhat limited temperature range can be extended by using a ternary mixture of DHPC, DMPC and charged amphiphiles such as hexadecyl(cetyl)trimethyl ammonium bromide (CTAB) or SDS 〚11〛 or by using shorter chain phospholipids instead of DMPC 〚12〛. In addition, the long-term stability of the phospholipid based liquid crystal phase exhibits a strong pH dependence, restricting the solute pH to a narrow range around pH 7. Replacement of the diacyl phospholipids by non-hydrolyzable dialkyl analogs overcomes this limitation 〚13〛. Despite all these improvements one has to keep in mind, that the stability of the liquid crystal phase is also affected by the solute, namely the protein solution under investigation. Solubility and stability of the protein naturally influence the choice of solvent conditions for the aqueous component, i.e. concentration, buffer choice and pH, and ionic strength. Each of these parameters affects the stability of the liquid crystalline phase in a complex manner.
3.2 Nematic phases of rod-shaped viruses and filamentous phages
Suspensions of charged, rod-shaped viruses, such as tobacco mosaic virus (TMV) and filamentous bacteriophages fd/M13 and Pf1 were known to undergo a magnetic field induced isotropic-nematic phase transition at moderate concentrations 〚14, 15〛. Solutions of magnetically aligned virus or phage therefore seemed like an attractive alternative to the above described phospholipid phases. Two independent reports of successfully using virus/phage solutions as a medium to measure residual dipolar couplings appeared, employing TMV and fd solutions 〚16〛 or Pf1 〚17〛. The molecular structures of the virus and phages are very similar. They are long, negatively charged rods, in which a cylinder of coat proteins is arranged in a helical fashion around either an RNA or DNA single stranded genome. TMV is approximately 15 nm wide and ∼3000 nm long, while the bacteriophages are both ∼6.6 nm in diameter and ∼880 nm (fd/M13) or ∼1900 nm (Pf1) long. Partial molecular alignment of the dissolved macromolecules arises from collisions with the aligned virus particles, thus imposing a preferred direction of diffusion rather than alignment via transient binding. It appears, that it is possible to align macromolecules over a wide range of temperatures and buffer conditions using dilute colloidal phage suspensions 〚18〛. Indeed, the degree of alignment and the size of the residual dipolar coupling are related to the length of the phage, as expected from Onsager theory for semi-flexible charged rods 〚18〛. Similar to findings for the phospholipid systems, high macroscopic viscosity due to the large viral particle size is observed. The microscopic tumbling rates of the dissolved macromolecules, however, should not be affected in the absence of binding to the particles. This can easily be established by measuring T2 relaxation times in the absence or presence of phage solution.
3.3 Liquid crystal phases of surfactants
The search for other, alternative and robust liquid crystalline media suitable for partially aligning biomolecules lead to investigations of dilute, quasi-ternary systems of surfactant/salt/alcohol, known to form Helfrich lamellar phases 〚19, 20〛. These phases were thought to consist of bilayers, which can be swelled by solvent such that the spacing between the bilayers is much larger than the thickness of the bilayer itself. Thus, Helfrich lamellar phases were potentially another medium for studying partially aligned biological macromolecules. Prosser et al. used a 2% aqueous solution of CPCl/hexanol (1:1) in 200 mM NaCl and demonstrated that residual dipolar couplings up to 15 Hz could be measured on ubiquitin. Our laboratory investigated surfactant phases from CPBr/hexanol and NaBr and found that solutions of 3–6% CPBr/hexanol in 20–30 mM NaBr gave excellent results. Although initially expected to form lamellar phases, further characterization of the CPBr/hexanol/NaBr liquid crystal phase employed by us revealed, that the particle morphology consists of cylinders. Thus, the above quasi-ternary surfactant liquid crystalline phases are most likely cylindrical micelles 〚21〛.
Another lyotropic liquid crystalline phase suitable for alignment purposes is formed by alkyl-poly(ethylene glycol)/alcohol mixtures in water. These mixtures were known to form superstructures of stacked planar bilayers and the application of ∼5% C12E5/hexanol and related phases for measuring residual dipolar couplings was demonstrated 〚22〛. The advantage of these phases is their insensitivity to pH (as compared to phospholipid phases), although, like with any thermotropic liquid crystal, only a limited temperature range is accessible. The major benefit of using these media is their low tendency to interact with proteins.
3.4 Other liquid crystals
In contrast to alignment caused by steric interactions with liquid crystalline media as described above, transient binding to oriented particles can also give rise to residual dipolar couplings. Such data has been reported for proteins interacting with purple membrane fragments 〚23, 24〛. Binding is assumed to occur since significant line broadening and a large decrease in 15N T1ρ and T2 relaxation times of the solute proteins is observed. Obviously, here the degree of alignment depends on the binding properties of the biomolecule under investigation, and any information derived from residual couplings reflects the conformation in the bound state. The weak binding and fast exchange interaction between the medium and solute molecule is reminiscent of the transferred NOE effect.
The use of a suspension of cellulose crystallites for measuring residual dipolar couplings has also been reported 〚25〛. This material can be prepared form wood pulp or filter paper by hydrolysis. Alignment in the magnetic field occurs due to the large negative diamagnetic anisotropy of individual cellulose crystallites. These particles generally have a length of several 100 nm and a width of ca 10 nm, and proteins can be dissolved in suspensions thereof.
Other media and methods for creating weakly aligned states for measuring residual dipolar couplings are still being searched for. It should be pointed out, at this juncture, that for all the media described above, sufficient care must be taken to ensure that the medium is not influencing the structural interpretation of the measured dipolar couplings. Flexible regions of proteins may transiently interact with the media and the bound conformation could become the dominant one, with other, non-binding conformations becoming underrepresented. Typically, electrostatic and hydrophobic interactions between the solute proteins or nucleic acids will be weak enough, but this has to be verified for each individual case.
3.5 Strained gels
Recently, an alternative method for inducing weak alignment has been developed. This approach does not involve any liquid crystalline media, but rather exploits the anisotropy of strained polymeric gels. Either compression 〚26〛, or both vertical and radial squeezing of polyacrylamide gels 〚27〛, can be used. Compression of a gel is achieved by using a susceptibility-matched plunger for pushing onto the gel. Stretching, in general, yields stronger and more uniform alignment. In this respect, stretching usually refers to radial compression accomplished by squeezing a larger diameter gel into a regular sample tube. Proteins are introduced into the gel matrix simply by diffusion. Such gels are extremely stable and inert and even allow the study of proteins under denaturing conditions 〚28〛. In addition, gels may even be useful for aligning detergent solubilized systems that hitherto were not amenable to alignment. The principal disadvantage of using anisotropically compressed gels lies in a decrease in rotational diffusion rate of the dissolved macromolecules, especially for larger systems. Careful attention to the concentration of the gel as well as the degree of cross-linking has to be paid and adjusting these parameters to the system under investigation may be necessary.
3.6 The importance of multiple alignment media
There is no doubt that in the future more materials and methods will be discovered and exploited for imparting anisotropy onto biomolecules in order to extract residual dipolar couplings. There are several important reasons to have different alignment media available. First, not every medium is compatible with the properties of the molecules or systems under investigation. Proteins that interact with membranes are clearly not compatible with phospholipid-based media, and very flexible or partially folded proteins have a tendency to strongly interact with bicelles. Likewise, negatively charged molecules, such as nucleic acids, tend to bind to positively charged lamellae and, positively charged proteins can potentially interact with negatively charged phage particles at neutral pH values. This can result in an increase in the electrostatic component of the alignment, leading to large linewidths or to collapse of the liquid crystalline phase. As an example, the protein ubiquitin with a pI of ∼ 6.5 interacts strongly with pf1, unless high ionic strength is used to screen the charges on the surface of the phage. Second, different alignment media frequently result in different orientations of the solute molecule with respect to the magnetic field, because the alignment tensors in two different alignment media will exhibit different orientations. This is an important property that allows lifting the degeneracies in the orientation of a given inter-atomic bond, inherent in the relationship between dipolar coupling and inter-nuclear vector orientation. A dipolar coupling measured in a given liquid crystalline medium positions the vector between the two coupled partners on one of the two possible, oppositely oriented cones. If the alignment tensor in the second medium has a different orientation relative to the molecular frame of the molecule, the same vector will now reside on two different cones. Thus the true orientation of this particular inter-atomic vector will lie at the intersections between the two cones. Therefore, dipolar couplings measured in several independent media allow uniquely defining the associated vector orientations.
4 Structure refinement using residual dipolar couplings
A key aspect of any NMR structure determination is that the ensemble of calculated structures satisfies all of the experimental NMR constraints, exhibits only very small deviations from idealized covalent geometries, such as bond lengths, bond angles, and planarity, and displays good non-bonded contacts. It therefore is of utmost importance for devising any calculational strategy, that the global minimum of the target function is reliably and efficiently located. The use of residual dipolar couplings, which impose a tight restriction on the orientation of a bond (if measured for directly bonded nuclei) should therefore greatly improve the quality of traditional NMR structures, calculated based on NOE distance restraints, coupling constants and chemical shifts. The simplest way to incorporate the geometric content of the dipolar couplings into a structure calculation is by means of an error function. For each measured dipolar contribution a term
Naturally, using dipolar couplings in NMR structure determination and refinement is predicated on the assumption that motional averaging will not compromise the data. The magnitude of dipolar coupling depends on the generalized order parameter S for internal motions of the inter-atomic vector 〚31〛, thus different contributions have to be considered, at least in principle. Rather than using individual, residue specific S values, it seems reasonable to assume uniform S values for all those residues for which heteronuclear relaxation measurements indicate a well ordered conformation, as evidenced by experimental S2 values of 0.7–0.9 (corresponding to S values of 0.85 to 0.95). Dipolar coupling constraints for residues that experience either slow conformational exchange or low order parameters (S2 < 0.6) need to be excluded from the data set 〚29〛. As an aside, it should be pointed out, that Da and Dr scale with S, rather than S2, thus the assumption of an overall S value introduces at most an error of a few percent in the dipolar couplings for the ordered regions of the molecule, well within the error of the experimental measurements.
In order to use equations (1) and (2) for structure refinement, the values of Da and R have to be determined. They are obtained by either iteratively best fitting the alignment tensor for a given structure during the course of refinement as described above, or directly from the experimental data by examining the distribution of dipolar couplings 〚32〛. A histogram of the ensemble of normalized residual dipolar couplings for the protein cyanovirin-N is illustrated in Fig. 1. It is possible to extract Da and R from this distribution, given that different, fixed-distance inter-nuclear vector types in a molecule are approximately uniformly and isotropically distributed in space relative to the alignment tensor of the molecule. The magnitude of the axial and rhombic components of the molecular alignment tensor are related to the extrema and mode of the coupling histograms, which, in the absence of random errors, look almost like perfect powder patterns. The highest probability dipolar coupling value, therefore, coincides with the magnitude of the bond vector aligned along the x axis of the alignment tensor 〚32〛. Using this approach, the accuracy with which Da and R are determined, clearly depends on the accuracy in the estimates for the two extrema and the maximum of the distribution, which in turn depends on the number of dipolar couplings observed and the degree of anisotropy in the orientation of the inter-nuclear vectors. In cases where it is not possible to measure a large set of dipolar couplings, one can also use a maximum likelihood method for extracting Da and R 〚33〛. Alternatively, singular value decomposition for calculation of the Saupe order matrix allows the determination of the axial and rhombic components of the alignment tensor 〚34〛. Still another way to obtain the alignment tensor exists, if the alignment is purely steric. In this case, the alignment tensor can be predicted based on the shape of the solute molecule using an obstruction model 〚33〛. This approach is very useful if an initial low-resolution structure is available, either based on a set of traditional NMR restraints or a model of a related molecule.
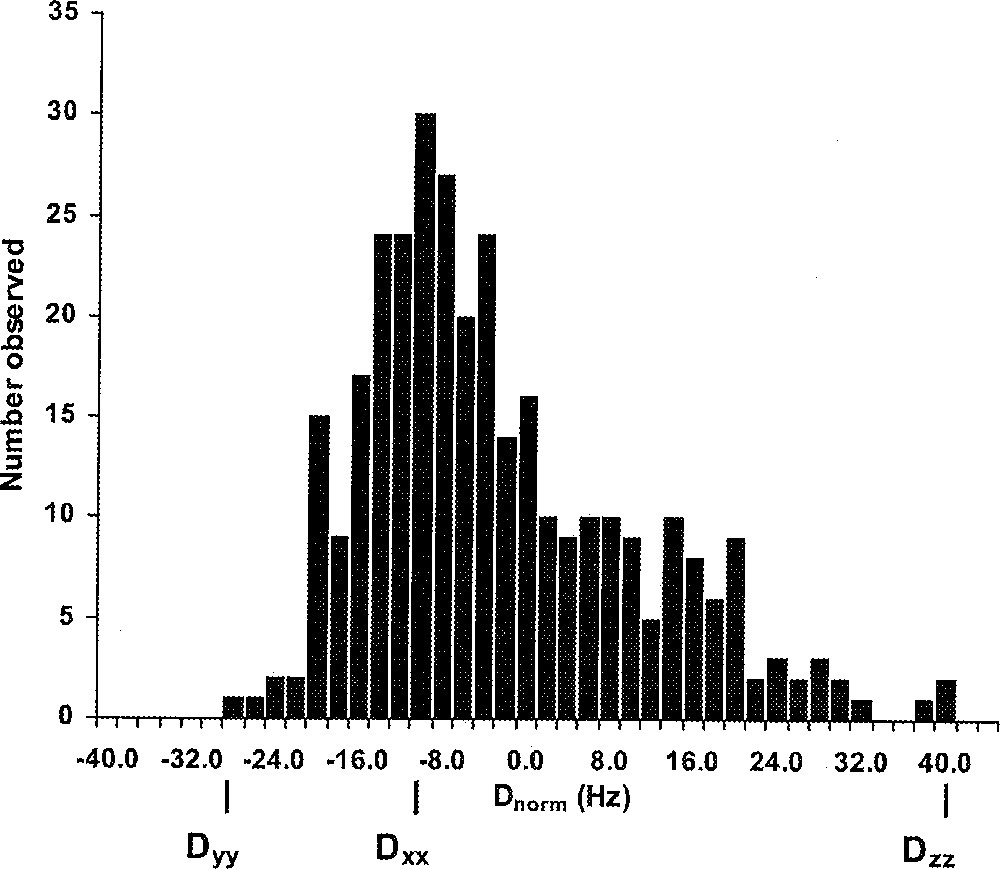
Histogram depicting the distribution of normalized residual dipolar couplings measured for the protein CV–N. The mode (Dxx) and extrema (Dyy and Dzz) of the distribution are indicated.
Refinement against dipolar couplings represents a difficult optimization problem, since each dipolar coupling is compatible with two orientations of the associated bond vectors, pointing in opposite directions. In addition, dipolar coupling constraints are of a very different qualitative nature, compared to NOE based distance constraints when used in a simulated annealing protocol. The success of the latter for structure determination based on distance constraints is based on the fact, that the experimentally determined inter-atomic distances are highly correlated. For example, if amino acid 20 is close to amino acid 90 within a folded protein structure, then the distance between atom X of amino acid 20 and atom Y of amino acid 90 constrains the distance between X and any other atom of amino acid 90, such as atom Z. Furthermore, it is highly probable, that any atom on residue 21 is also close to one or more atoms of residue 90. In this manner, the potential surface of the optimization in three-dimensional space resembles a funnel with a relatively smooth surface. For the case of dipolar couplings, no such straightforward correlation exists. On the contrary, dipolar couplings (or bond-vector orientations) tend to compete with each other and with the distances. If, for example, during the simulated annealing a NH bond becomes oriented such, that its dipolar coupling is satisfied, this does not necessarily lead to a better agreement for the dipolar coupling of the adjacent NC’ bond. Thus, each inter-atomic vector orientation represents an independent orientational parameter. As a result, calculational strategies based on dipolar couplings alone are fraught with difficulties. But even the simple addition of a constraint term for dipolar couplings to proven methodologies of NMR structure calculations can result in structures becoming trapped in deep local minima. As a consequence, the convergence properties of the procedure can be severely curtailed and careful adjustment of the protocols is necessary.
In order to avoid such trapping in the determination of a NMR structure, it is advantageous to employ a two stage simulated annealing protocol. In the first stage, all conventional experimental constraints, such as NOE and H-bond based distance constraints, dihedral angle constraints, coupling constants and carbon chemical shifts are employed to calculate an ensemble of models. During the second stage, each structure of the ensemble is refined against the dipolar couplings. These are added as constraints in an appropriate simulated annealing protocol, frequently employing both high temperature and low temperature slow cooling steps.
Inclusion of the additional dipolar constraints improves the precision of the structures considerably 〚35〛. In particular, significant increases in coordinate precision are observed, both for backbone atoms and for side chains atoms. As an example, families of structures calculated without and with inclusion of residual dipolar couplings are displayed in Fig. 2 for the potent HIV-inactivating protein cyanovirin-N, vividly demonstrating the improved precision.
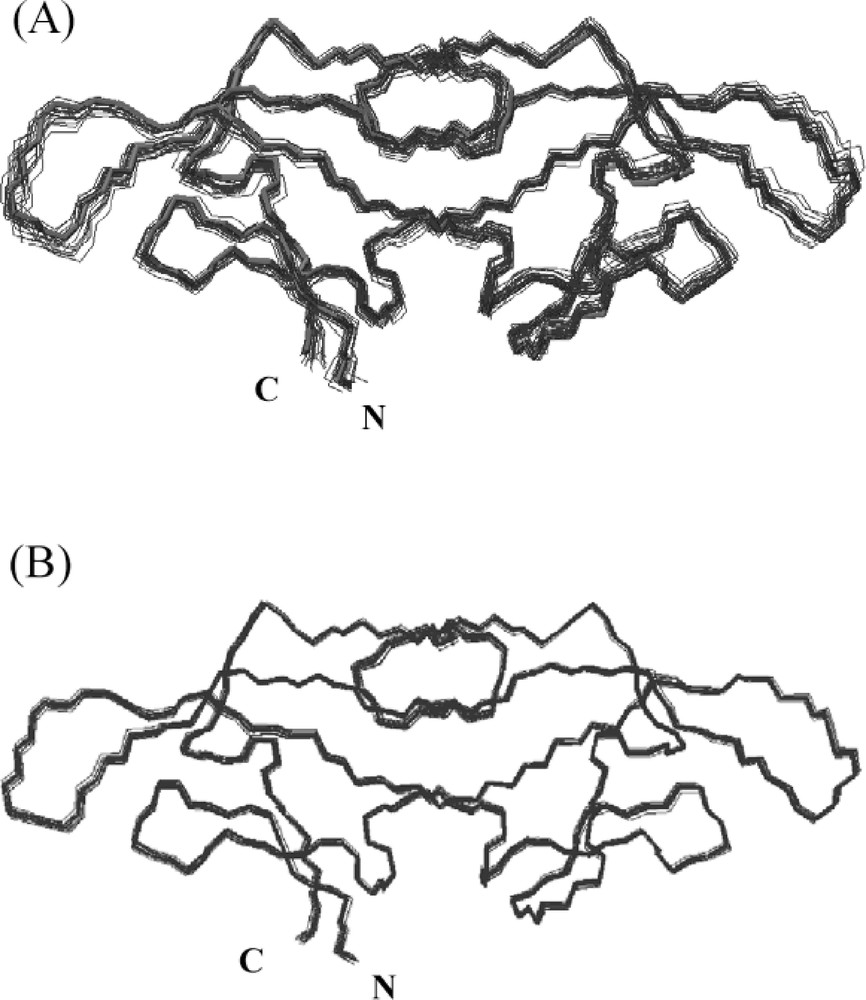
Best-fit superpositions of backbone (N, Cα, C’) atoms of 20 CV-N structures calculated without (A) and with residual dipolar coupling constraints (B). Individual conformers are shown in blue and the minimized average structure in red.
The most significant improvements in the quality of the structures upon inclusion of all residual dipolar couplings relate to the Ramachandran statistics. The percentage of residues found in the most allowed regions of the Ramachandran map generally increases from ca. 80% to over 90% and the number of bad contacts is reduced by more than 50%. Therefore, structures calculated with a one-bond residual dipolar couplings exhibit superior packing characteristics, even without the inclusion of a conformational database potential 〚36〛.
5 Validation of protein folds
The most powerful and attractive use of residual dipolar couplings lies in their application for validation of structural models. In this context, structures may be derived from modeling, either ab initio or homology, or from low-resolution experimental data. If the model structure is accurate, one will observe excellent agreement between the dipolar couplings calculated based on the structure and the experimentally measured ones. Exploitation of this fact may lead to the most direct and important contribution of NMR based methodology in ‘Structural Genomics’. Although large efforts are underway to determine as many protein structures by NMR in a high throughput manner, traditional methodology for NMR structure determination for proteins of intermediate size (∼30–50 kDa) is still relatively slow. For instance, to solve the structure of the 40 kDa complex between the N-terminal domain of enzyme I and HPr, the NMR measurement time alone extended over almost five months 〚37〛, with an additional time of at least 6 months for data interpretation and structure calculation. It therefore seems imperative to explore alternative avenues, if NMR methodology to become more powerful for structure determination in the post-genomic era. Exploitation of strategies based on residual dipolar couplings may overcome the traditional shortcomings indicated above, and could lead to new conceptual applications of NMR. Here again, the alignment tensor or Saupe matrix has to be determined and any of the methodologies outlined previously for refinement can be applied for this purpose. Naturally, it is important to include sufficient experimentally determined couplings. Given that there are five independent Saupe matrix elements, it generally is easy to include a much larger number of observables (measured residual dipolar couplings) than variables in the fitting procedure, rendering the SVD approach superior to the shape-based prediction of the alignment tensor.
A very promising direction combines structure prediction and experimental validation or selection. Based on our increasing understanding of the important factors that govern folding and stability of proteins, it has been proposed that only a finite set of protein folds exist 〚38〛. With improving force fields it may be conceivable, that for a particular amino acid sequence a set of several thousand different possible folds can be generated, one of which may be the correct one. The crucial question in this scenario then remains, how to identify this particular one. Using NMR may provide the answer. It is relatively fast and straightforward to obtain backbone resonance assignments for isotopically labeled proteins, and the measurement of residual dipolar couplings in alignment media can also be achieved with relative ease. It therefore is possible to experimentally obtain residual dipolar couplings for the backbone, which in turn can be compared with those calculated for the different theoretically predicted structures. This approach is illustrated with an example in Fig. 3. A homology model for a circular permuted variant of cyanovirin-N was constructed and, based on the coordinates of this model, residual dipolar couplings were calculated. Comparison of the calculated and experimentally measured couplings allowed verification of the modeled fold. Thus, NMR can be used as an experimental filter for any theoretically predicted structures. Initial results exploring such methodologies are clearly promising.
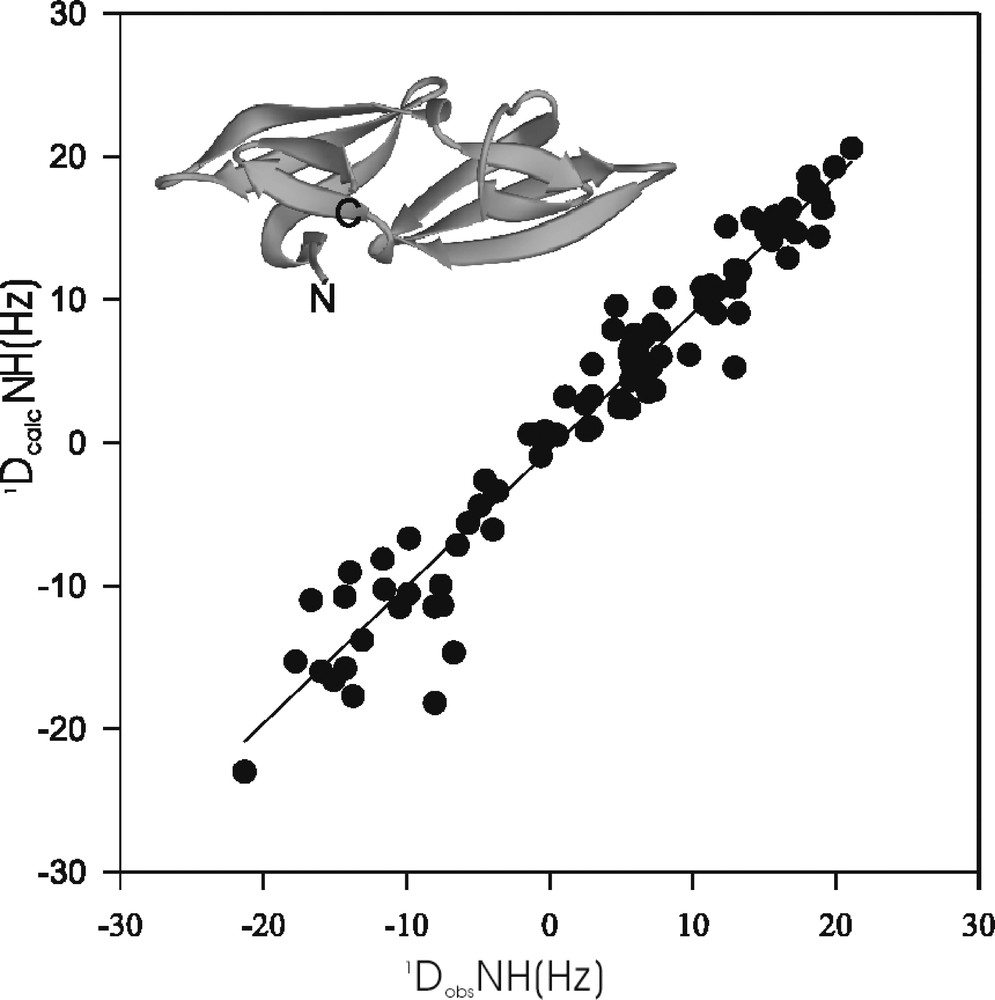
Correlation between experimentally measured dipolar couplings (1DobsNH) and those calculated (1DcalcNH) on the basis of the coordinates of a homology model (inset) for a circular permuted variant of the protein CV-N.
6 Concluding remarks
The accessibility of anisotropic NMR parameters in solution for biological macromolecules has opened the door for future imaginative exploitation of this diverse wealth of physical information. Applications with respect to improving the accuracy of protein structures 〚39〛 in defining the long-range orientation of the RNA in a protein–RNA complex 〚40〛 and domain orientation in multi-domain proteins 〚41–44〛, as well as recognition of protein folds 〚45, 46〛 have already been reported. No doubt further developments will occur. In the era of structural genomics, NMR is poised to make a major contribution. Since dipolar constraints can be readily measured on partially aligned proteins, they can be incorporated into powerful methodologies for validation of theoretical models. Not every protein structure of the completed human genome and those of model organisms will be solved experimentally by NMR or X-ray crystallography. However, all of them are amenable to structure prediction, and validation of these predicted structures and folds can be achieved rapidly by NMR using residual dipolar couplings.
Acknowledgements
I am indebted to all my collaborators and colleagues mentioned in the text and references who have developed the technologies described in this article and who have been a constant source of stimulating discussions. Particularly, Drs. Ad Bax, Nico Tjandra and Anatoliy Dobrodumov provided invaluable intellectual contributions. The work in the author’s laboratory was in part supported by the Intramural AIDS Targeted Antiviral Program of the Office of the Director of the National Institutes of Health.