

1 Introduction
The taxonomy of fungal species has been debated for a long time. Among fungi, the taxonomy of hemiascomycetes provides an extra challenge. Although it has been proposed that hemiascomycetous yeasts have evolved for as long as the chordates [1], this taxon is morphologically very homogenous. Therefore, characteristics such as morphology that are still valuable in establishing a fungal taxonomy are less used, if used at all, in yeasts.
The most comprehensive description of yeast species, The Yeasts: a taxonomic study, is now 13 years old and describes over 750 species [2]. A new edition of this series is now published [3]. It lists 1500 yeast species [3]. The major work by Suh and collaborators on the discovery of new taxa in beetles [4] led Boekhout [5] to estimate the number of yeast species to be discovered by 2010 to be close to 3000. The discrepancy between the number of described species and the predicted number of species to be described suggests that a number of ecological niches have not been investigated yet and that cryptic species may have not received enough attention. These figures are somewhat low compared to the 1.5 million predicted extant fungal species [6]. Yeast species are distinguished according to the following characteristics: cellular morphology, type of conidiogenesis, comparative physiology, type of coenzyme Q and G + C content. Since these characteristics are prone to intra-species variability, the DNA/DNA reassociation technique was retained as the method of choice to distinguish species. Although it is still the recognized method used in bacterial taxonomy, in yeast taxonomy, this method may be affected by the large amount of highly conserved and highly repetitive sequences of ribosomal DNA and by the occurrence of hybrids. Furthermore, this method is time-consuming, inapplicable to a large number of strains and it does not provide a consistent phylogeny [7].
Molecular systematics has revolutionized taxonomy and our view of yeast evolution. It is interesting to look back and analyze how the choices of molecular markers have developed. The favorite marker for molecular taxonomy is rDNA, since it is slow evolving and therefore well conserved, thus allowing easy sequence comparison and facilitating of PCR amplification [8]. The first comprehensive analysis based on the entire small subunit 18S rDNA gene was published in 1993 [9]. This approach also proved time-consuming, since this part of the rDNA unit is around 1800 bp long. Nevertheless, the data allowed a clear separation of the hemiascomycetes from the filamentous euascomycetes.
Works by several authors on various fungi belonging to basidiomycetous and hemiascomycetous yeasts as well as euascomycetes led to the choice of the around 600 bp long D1/D2 variable region at the 5′ end of the 26S rDNA [10–13]. Most authors concentrated on this region and a comprehensive database of D1/D2 sequences for over 500 species became available in 1998 [14,15]. It provided the D1/D2 rDNA barcode for identification of hemiascomycetous yeasts to the species level for the ensuing decade, contributing thereby to a more straightforward phylogeny. Since 1998, numerous parts of the rDNA unit have been used for this purpose, including various studies using Restriction Fragment Length Polymorphism (RFLP) of different parts of the rDNA unit for rapid identification and species delineation, mainly the Internal Transcribed Spacers (ITS) [16] and the Non-Transcribed Spacers (NTS) [17]. Considering the properties of D1/D2 (short size, ease of amplification, and ubiquity), the use of other parts of rDNA was abandoned. However, in the meantime, work on fungi started using protein coding genes, which yielded better species delineation and led to evidence for sexuality in some fungi at the end of the 1990s [18,19]. First, the single-copy genes RPB2 [20] and ACT1 [21,22] were used in addition to D1/D2, but the taxonomy and the phylogeny of hemiascomycetes really developed concomitantly with the availability of genome sequences [1,23–34].
2 Multigene analysis
The sequence of the complete genome of Saccharomyces cerevisiae and the first Genolevures project opened up new horizons for yeast taxonomy [23,24]. The classification of the so-called “Saccharomyces complex” clade was brought in a pioneer work [35] based on the concatenation of various sequences: rDNA repeat (18S, 26S), single-copy nuclear genes (translation elongation factor 1, actin, RNA polymerase II) and mitochondrially encoded genes (rDNA small-subunit, COXII). It provided a new standard for the delineation of genera, based on the exclusion of polyphyly [36]. In particular, it made the relationship clear between the genus Saccharomyces, in the current sense (so-called sensu stricto) and now reduced to six species, and its closest neighbors, the Saccharomyces sensu lato species that are now classified into various genera such as Kazachstania, Lachancea and Naumovia. (Fig. 1).
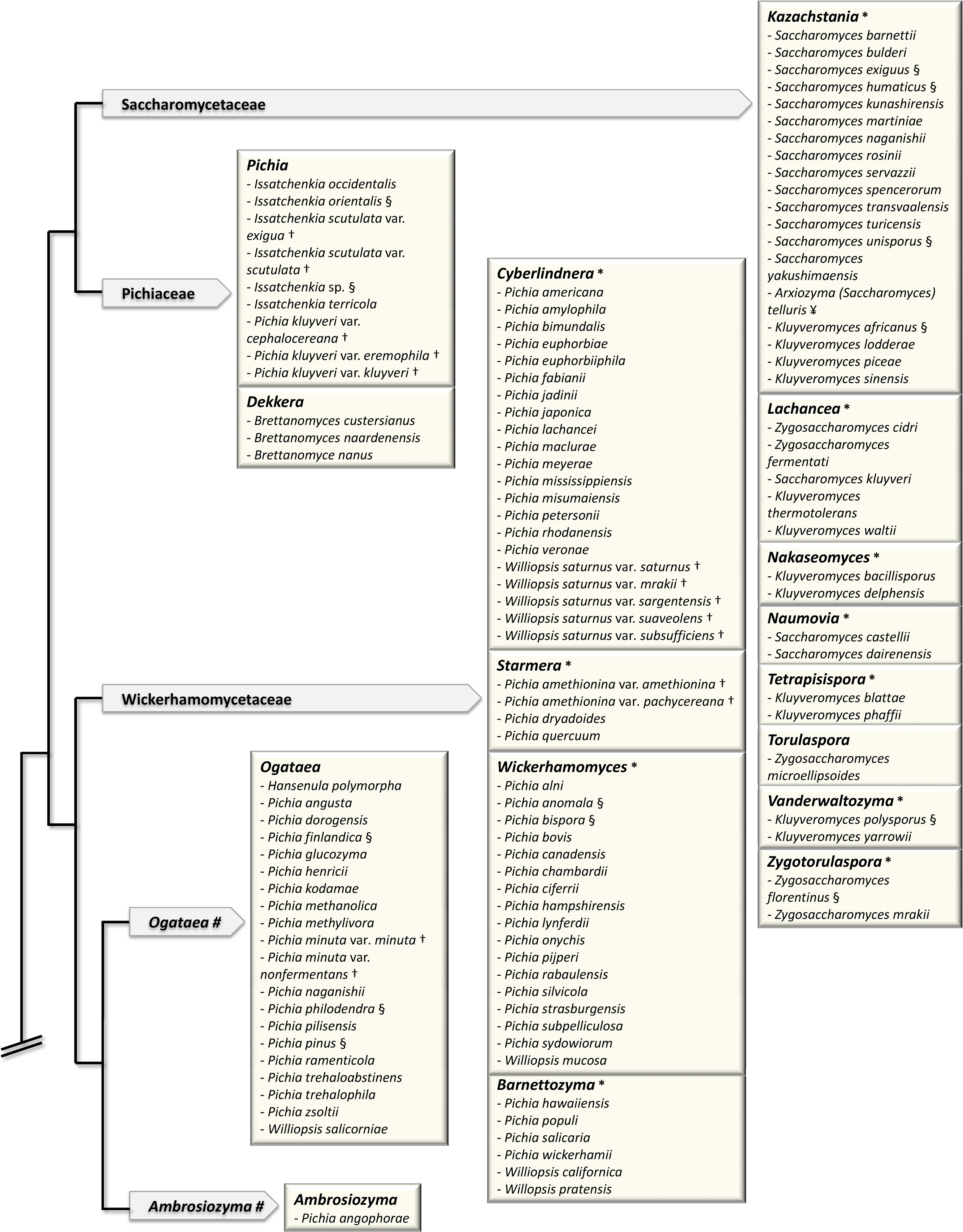
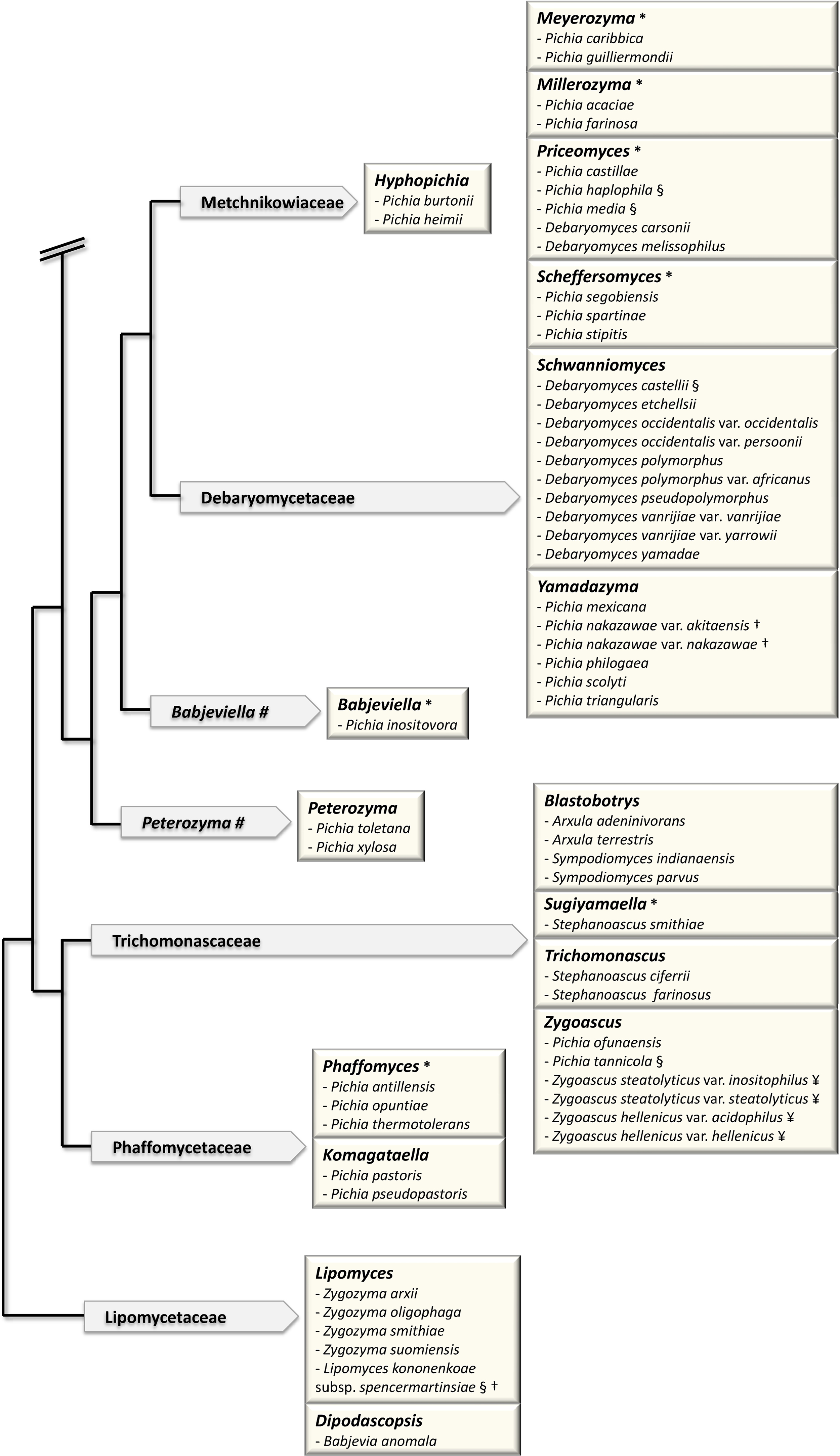
Major changes in the classification of hemiascomycetes since 1998. The schematic tree of the families is adapted from [43]. Arrows contain names of families. Boxes correspond to genera in bold letters. (#) indicates genera, which are not yet assigned to a family. (*) indicates newly defined genera. Only the species, which have been reassigned to a different genus (that defined by the box) are shown. The previous name of the species, i.e. the genus to which each species belonged previously, is shown. For example, the species Saccharomyces servazzii is found in the box defined by the new genus Kazachstania. Saccharomyces servazzii is now Kazachstania servazzii. (§) indicates the species, which have also changed names: Saccharomyces exiguus = Kazachstania exigua; Saccharomyces humaticus = Kazachstania humatica; Saccharomyces unisporus = Kazachstania unispora; Kluyveromyces africanus = Kazachstania africana; Kluyveromyces polysporus = Vanderwaltozyma polyspora; Zygosaccharomyces florentinus = Zygotorulaspora florentina; Issatchenka orientalis = Pichia kudriavzevii; Pichia finlandica = Ogataea wickerhamii; Pichia philodendra = Ogataea philodendri; Pichia pinus = Ogataea pini; Pichia anomala = Wickerhamomyces anomalus; Pichia bispora = Wickerhamomyces bisporus; Pichia haplophila = Priceomyces haplophilus; Pichia media = Priceomyces medius; Debaryomyces castellii = Schwanniomyces capriottii. The two subspecies of Lipomyces kononenkoae var. kononenkoae and var. spencermartinsiae had their name changed to L. kononenkoae and L. spencermartinsiae, respectively. (†) indicates the species, whose name was previously a variety; for example Pichia nakazawae var. akitaensis is now Yamadazyma akitaensis. (¥) indicates the species, which were subdived into several species. Saccharomyces (Arxiozyma) telluris species is now subdivided into Kazachstania telluris and Kazachstania bovina. Zygoascus stearolyticus and Zygoascus hellenicus varieties are now replaced by Z. hellenicus and Z. meyerae.
Note that the Candida species that were shown to have a teleomorph were not included in the figure.
Kurtzman and his collaborators have applied this method to a large number of clades. This led to the circumscription of many genera [36–42]. Most of the transfers of existing species to new genera established by Kurtzman and his collaborators are shown in Fig. 1. A more detailed account of Kurtzman's work is also described in his recent review [43]. It is noteworthy that in this analysis of 83 species, Kurtzman used only three markers, two being rDNA markers, which have been questioned for the bias they can introduce into phylogenetic analysis [44–46]. For instance, rDNA analysis incorrectly groups Zygosaccharomyces rouxii with Nakaseomyces delphensis/Candida glabrata [1,15], the latter having undergone Whole Genome Duplication (WGD) [1] like Saccharomyces cerevisiae [47]. The branching of Z. rouxii in rDNA phylogenies is therefore inexact, since Z. rouxii has diverged from the ancestor of the clade before WGD occurred [48]. The need for genomic markers, other than repetitive sequences such as rDNA, that are informative from the phylogenetic point of view is therefore crucial. Genome sequence data have provided the potential for solving these problems.
3 Phylogenomics
With the increased availability of complete genome sequences, multigene analysis can extend to a large number of genes as long as real orthologs can be compared. Duplicated genes cannot be used for phylogeny, since the two copies generally evolve independently. It has been demonstrated that roughly 40% of yeasts genes have paralogues [1], which exclude them for phylogeny. With the availability of large datasets, an old question reappeared: which of the number of genes or taxa available are the most important in phylogeny reconstruction? An early study analyzing 14 yeast genomes and 106 genes [49] proposed that robustness of phylogenies was linked to the number of genes used, whereas the number of species had hardly any effect on the phylogenies. This study may seem audacious, since the species, which were analyzed, were widely diverging. Later on, studies that were more cautious included all available fungal genomes to yield the beginning of a tree of life for this part of the eukarya. A total of 531 genes derived from the euKaryotic Orthologous Groups (KOGs) of 25 species led to a well supported unique tree [45]; however, by simply reducing the number of genes used by 1/3, these authors showed that the phylogeny of some parts of the tree could not be resolved. Fitzpatrick et al. [44] found that, using 153 universally distributed orthologs of 42 genomes, robust relationships could not be established between some species like C. glabrata or Saccharomyces castellii and the WGD clade. The conflicting data obtained using various methods suggested that more taxa were needed to resolve this node. A similar result was obtained in a study in which no prior selection of genes or sites was performed [50]. Further work by Kuramae et al. [51] on 33 genomes helped to solve this problem. These studies therefore revealed that the number of species analyzed was crucial when inferring phylogenies by these approaches.
One of the most interesting conclusions of these studies was that the number of taxa to introduce in phylogenomic analysis depended on the genetic distance which separates the taxa. Indeed, the aforementioned studies analyzed species that span the whole fungal tree and that evolved over 1 billion years. Considering these large genetic distances, it was not clear whether this approach would be successful with closely related species. The difficulty at inferring robust relationships between closely related species was confirmed in a systematic comparison of all the models used for elaborating phylogenies (superalignments, supertrees, distance and gene content…) from complete genomes [52]. In response to Rokas and Carroll [49], it was even suggested that the inclusion of more genes in phylogenetic reconstructions could decrease accuracy, especially in the case of bias sampling [53]. Conversely, the reduction of those biases by the addition of extra taxa may result in the use of fewer genes for the phylogenetic analysis. A similar conclusion was reached by Aguileta et al. [54]. The direct consequence of this proposition is that a limited number of genes may be sufficient in order to establish robust phylogenies. This may be good news because we have to consider ten to hundred times more species in phylogenetic studies in the near future.
4 Hybrids, hybrids, hybrids…
The most famous example of yeast hybrids are the ones involved in beer making (reviewed by Kielland-Brandt et al. [55]). Since the discovery of the complexity of the brewing yeast genome that contained material DNA from at least two contributors, many hybrids between Saccharomyces species were evidenced (this is described in the Fermentative Saccharomyces chapter of this volume). The high occurrence of hybrids in this genus may be attributed to the fact that these yeasts have been used for millennia in biotechnology and that their genome was shaped by human activities. However, a few hybrids were found in other clades not belonging to the Saccharomyces genus. This is the case in several genera and species, e.g. Candida, Kazachstania, Metschnikowia, Zygosaccharomyces and Debaryomyces [56–59]. Hybrids are not specific to hemiascomycetes; they have been shown to exist in basidiomycetous yeasts [60].
Interspecific hybridization may yield stable haploid strains, which may or may not mate after the hybridization event, or after the resolution of the first hybridization events [61]. Examples can be found in a number of S. paradoxus strains in which Liti et al. [62] found an introgression of a large fraction of chromosome III of S. cerevisiae. More complex situations were found: for instance, the presence of several sub-telomeric Y’ sequences, a family of repeated DNA sequences of S. cerevisiae, were detected in some strains of Saccharomyces bayanus var. bayanus [17]. It is not known whether several Y’ sequences were transferred from S. cerevisiae or if a single Y’ sequence originated from S. cerevisiae that was subsequently duplicated in S. bayanus var. bayanus. The high variability of chromosomal organization in many hybrid strains of Saccharomyces pastorianus is also the result of many rearrangements including chromosome duplication, fusions, etc. subsequent to the original hybridization event(s) [17,63]. It was further shown that some S. pastorianus hybrids were the result of hybridizations involving a third species in addition to S. cerevisiae and S. bayanus var. bayanus; the third contributor to these hybrids remains to be isolated [64].
Recent work has shown that genetic diversity might be generated differently according to the yeast clade considered. Whereas classical sexuality maybe the rule in the species of the Saccharomyces complex, it may be otherwise in the clades like the CTG clade [65], a monophyletic group of yeast species, which share a deviation of the universal genetic code in which the CUG codon is read as Serine instead of Leucine. The much studied Candida albicans, which is diploid heterozygote, was recently shown to undergo loss of heterozygosity (LOH), leaving large regions of chromosomes or even entire chromosomes homozygote (see [66] and references therein). By studying crosses in Candida lusitaniae, recombinant and aneuploid progeny was obtained that may expand genetic diversity [65]. By applying a “gene genealogies” approach, which is used to evidence sexuality among cryptic fungal species [67] and by analyzing informative genomic markers, it was shown that Debaryomyces hansenii, the biotechnological species of the CTG clade, was in fact a complex made of cryptic species. Some of these species were partly made of diploid heterozygotes, which like C. albicans, undergo LOH [59,68]. The presence of cryptic species that form hybrids was observed in another species Millerozyma (Pichia) farinosa (Mallet et al., in preparation). One of the species belonging to the M. farinosa complex, the well known Pichia sorbitophila, was shown to be a diploid heterozygote that also underwent LOH (The Genolévures consortium, personal communication), indicating that this may be common to many, if not all, CTG clade species. The combined existence of hybrids and associated LOH can explain some of the difficulties encountered when reconstructing phylogenies in this part of the yeast tree. In most cases, the ploidy and heterozygote status of the appraised strains was not taking into account in previous phylogenetic studies [69,70], which led to discrepancies between the resulting trees. Indeed, in our experience, diploid strains were shown to contain markers belonging to different species that were redistributed following LOH (Mallet et al., in preparation). As a result, a phylogenetic analysis with multi-species markers led inevitably to erroneous trees.
Overall, the combination of numerous diploid heterozygotes, LOH and clonality will need to be considered in the future phylogenetic studies in specific clades like that of the CTG. The mating process does not seem well conserved for many heterothallic species, thus leading to mating between closely related species. Some of the progeny from these matings survive leading to an abundance of interspecific forms.
5 Bar coding with a unique molecular marker: the Graal in taxonomy
Whatever the type of organism studied, a unique universal marker is desirable. Indeed, although very efficient and reliable, identification and classification of yeast strains through the amplification and sequencing of several markers is burdensome. The attempts to adapt techniques devised for bacteria, Fourier-Transform Infrared Microspectroscopy (see [71] for review) and MALDI-TOF mass spectrometry [72], to yeasts and fungi show promise, and may solve the problem of rapid identification to diagnose infections due to fungi. Nevertheless, these methods cannot cater for (1) new taxa analysis, since by definition, the new species which is about to be described cannot be represented in databases and (2) phylogeny. One of the goals of modern taxonomy is to find a single easily PCR-amplifiable marker that is relatively short, to allow a single run of Sanger sequencing or pyrosequencing, and informative, to provide a clear distinction between all species. The preferred yeast rDNA D1/D2 marker cannot fulfill this role, since its reduced variability does not allow for differentiation of a number of taxa (see above). Attention was given to a similar type of moderately repeated marker in eukaryotes, the mitochondrial COX1 gene, in order to barcode biodiversity [73]. Like other markers, it proved to be useful [74], although problems associated with (1) the nature of mtDNA itself considering its peculiar inheritance and its mode of evolution, and (2) interspecific hybridizations, were observed (for review, see [75,76]). More practical considerations arose with the use of the COX1 gene, such as the variable location of introns within the gene of interest [77].
Fungal taxonomists turned towards the Internal Transcribed Sequence 1 and 2 separated by the slow evolving 5.8S gene (ITS), which by its nature is much more discriminating than the D1D2 part of rDNA. This proved to be useful in hemiascomycetes, although some exceptions were observed in basidiomycetous yeasts [78]. Indeed, the lack of strong selection pressure on the two non-coding regions is such that, although sufficiently variable to allow for barcoding, it is subjected to many indels leading to extraneous size variability, making it unsuitable for phylogeny in hemiascomycetes. Attempts have been made to use this marker as a barcode (G. Verkeij, personal communication). Our experience is that one could take advantage of its important size variation to strengthen species delineation (Weiss et al., in preparation). Nevertheless, in Debaryomyces, the ITS region is unable to differentiate between the cryptic species related to D. hansenii (our unpublished data), whereas spliceosomal introns of various housekeeping genes or coding sequence for actin can do this.
Again, comparative genomics could help in this matter and a bioinformatics search for genes that could perform as well as the large numbers of markers used to construct species phylogenies was undertaken. A first study based on 33 genomes and the comparison of distance matrixes of each KOG and that of the concatenated KOGs led to a number of single gene candidates [51]. A similar study based on the exhaustive comparison of the topologies of the phylogenies between orthologs from 30 genomes and single phylogeny topologies led to the selection of over 200 candidates [54]. Interestingly, these genes were shown to perform well, i.e. the phylogeny of these genes is very similar or identical to the phylogeny of the species established with 246 genes, independently of the set of species to be analyzed. The commonly used markers such as ACT1, RPB2, etc. did not perform well in this study. The first attempts at using the best of these selected markers, TSR1 and MCM7, are promising [79], but more genomes are needed to ascertain these candidates as “high phylogenic performers”. A major drawback of this approach is that the amplification of many of these genes is highly problematic because they are not well conserved. One may imagine that the constant accumulation of sequence data on these genes from many species can permit the design of a number of nucleotide primers, which could be efficient in PCR amplification when used as a mixture.
6 The future
A number of studies have highlighted the two key problems in reconstructing phylogeny: (1) the difficulty to find common markers that are informative enough for species distinction, (2) the difficulty of assessing whether a marker or a combination of markers can reflect the evolution of hemiascomycetous yeasts. Additional questions like the minimal data set necessary for molecular definition of a species are also relevant, since most of the newly described species have only “passed” the “D1D2 test”. It is also clear that the relevance of the use of ribosomal DNA as the source of unique markers is questionable. Finally, future work will attempt at harmonizing the combination of used markers (compare [69,70]).
What could change and/or improve taxonomy of hemiascomycetous yeasts in the future?
- 1. Population genomics is clearly the most informative approach to determine phylogenetic relationship between species as shown by Liti et al. [80] and Schacherer et al. [81]. Although the price of sequencing will continue to go down and this approach will be without doubt applied to major pathogens like Candida and the basidiomycetes Cryptoccocus, and to important biotechnological isolates, it is very unlikely that many clades will be analyzed in such a fashion.
- 2. Already started in bacteria, a large project aiming at sequencing all the existing type strains will certainly be undertaken for yeasts. “Dikaryome”, an international effort aimed at sequencing a large number of hemiascomycetous and basidiomycetous yeasts has recently been initiated. Such large-scale genome sequencing will solve most of the problems of taxonomy by providing a wide phylogeny of yeasts and reference genomes.
- 3. Yeast taxonomy has always been hampered by a certain self-consciousness at defining new species and by the lack of curiosity of exploring entire taxa, in contrast to only comparing type strains. Our recent work [59,68] has shown that at least in the CTG clade, the analysis of a large number of strains within a species could reveal cryptic species that were previously ignored, as well as unexpectedly larger biodiversity due to genetic exchanges between divergent strains and species at high frequency.
- 4. The need for integrated up-to-date databases is important, since the search through the large generalist sequence databases such as NCBI and EBI may prove disappointing; in these databases individual strains and ecological samples are over represented, largely diluting the type strains or representative strains. The trend toward unified taxonomy has led to many initiatives that better facilitate non-specialist needs such as Mycobank (http://www.mycobank.org/). Straininfo (http://www.straininfo.net) is one of the most innovative tools created recently. Such integrated databases may allow, through an ingenious updating system, the search of 13 international collections. In our view, an integrated database would gather databases on (1) taxonomical nomenclature like Mycobank, (2) genome sequence resources as that provided by Genolevures (http://www.genolevures.org) or the Candida database (http://www.broadinstitute.org), and (3) taxonomical marker sequence resources associated to easy-to-use tools; this taxonomy-dedicated marker database remains to be built. This would overcome the need to search for scattered information in independent, not always updated, databases. It must be stressed that more manual annotation (or less automatized annotation) is crucial for its success. Such a database would also associate with Biological Resource Centers constituted in networks like the one that the ongoing European program EMbaRC (http://www.embarc.eu) is currently building.
High-throughput sequencing has changed many aspects of biology, taxonomy not the least. It needs to be applied more systematically to new species as well as to previously discovered species. High-throughput sequencing of genomes and of specific markers may not have provided an immediate solution to taxonomical problems, but it surely has raised more questions, like the minimal data set necessary to characterize a species. No doubt, new generation sequencing will bring a number of surprises regarding the evolution of yeasts, and it can be foreseen that a robust taxonomy will be generated in the near future.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
This work has received funding from the European Community's Seventh Framework Programme (FP7, 2007–2013), Research Infrastructures action, under the grant agreement No. FP7-228310 (EMbaRC project). S.W. is a post-doctoral fellow in the EMbaRC project. G.M. is a PhD student supported by the CNIEL. This work was financially supported by INRA. The authors would like to thank the anonymous referees for their help in improving the manuscript. The authors are grateful to Vidya Rajan for reading the manuscript.