

1 Synteny, an old genetic concept with a new meaning in comparative genomics
1.1 Synteny in the “linkage” sense
The first use of the word synteny dates back to the early seventies (Fig. 1) when new methods for gene mapping based on somatic hybrid cell lines were developed. Synteny originally described the colocalization of several markers on the same chromosome. As human chromosomes were preferentially lost in man-rodent hybrid cells, two genes could be attributed to the same chromosome when simultaneously present or absent from a hybrid cell population whatever the genetic distance separating them. These physically linked, but not necessarily genetically linked, genes were called syntenic genes. Etymologically, the term synteny means “on the same ribbon” (from the Greek syn = together and taenia = ribbon). Although relatively limited in number until the 1990s, nearly all published scientific papers referring to synteny involved gene mapping studies based on hybrid somatic cells in human and also in many primate, cattle and rodent species [1–4]. These methods led to the development of high-density radiation hybrid maps during the 1990s [5]. In the last 20 years, the number of synteny-related papers published each year has linearly increased to reach more than 200 scientific reports in the year 2009. It is interesting to note that in yeast, the number of publications dealing with synteny has always been quantitatively negligible since this term was first invented (Fig. 1). However, several experimental studies based on electrophoretic karyotyping and later on, on comparative genomic hybridization, have allowed an exploration of the chromosome structures and their evolution in yeast [6–11].
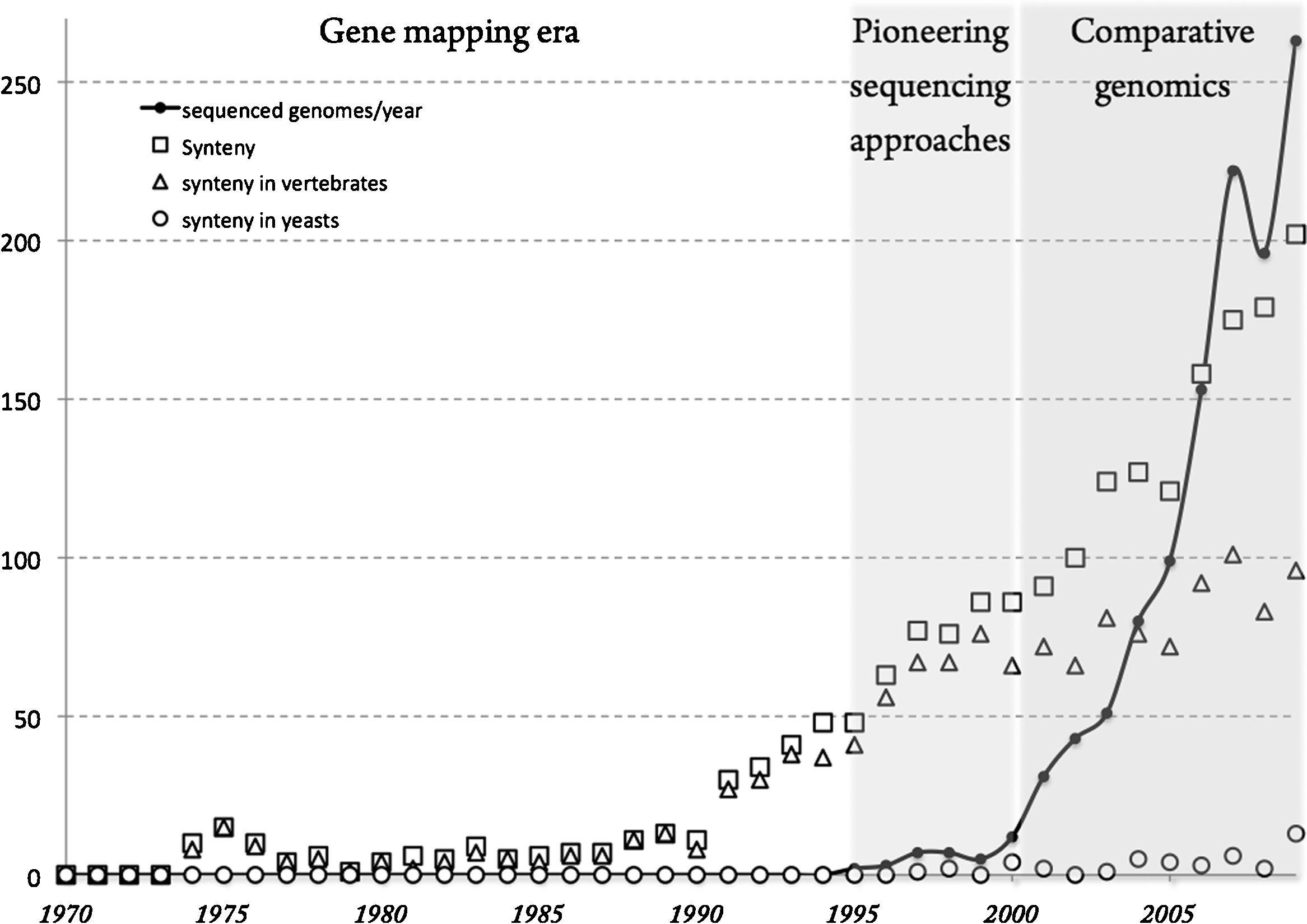
The use of the term synteny in the scientific literature. The ‘Synteny’ plot (open squares) corresponds to the total number of publications citing the word synteny in either the title or the abstract sections identified in PubMed between 1970 and 2009. The ‘synteny in vertebrates’ plot (open triangles) corresponds to the fraction of these publications that in addition comprises one of the following terms: mammal* or mouse or human or primate or fish or cattle or rodent or dog or rat or mouse or vertebrate*, in either the title or the abstract sections. The ‘synteny in yeasts’ plot (open circles) corresponds to the fraction of the total synteny publications that comprises one of the following terms: yeast* or Saccharomyces or Candida or Kluyveromyces, in either the title or the abstract sections, followed by manual curation to remove publications citing yeast for methodological reasons (such as YAC). The black curve represents the number of completely sequenced genomes (eukaryotes, bacteria and archaea) published and referenced in the Genome OnLine Database (http://www.genomesonline.org/). From the year 2000 where the number of completely sequenced genomes rapidly increased, the relative prominence of vertebrates (open triangles) in the synteny-related literature has partly declined probably to the profit of plant and bacteria studies while the total number of publications dealing with yeast in the field of synteny (open circles) has always remained anecdotic. Masquer
The use of the term synteny in the scientific literature. The ‘Synteny’ plot (open squares) corresponds to the total number of publications citing the word synteny in either the title or the abstract sections identified in PubMed between 1970 and ... Lire la suite
1.2 Synteny in the conserved gene order sense
Chromosomes do not remain collinear over evolutionary time because rearrangements such as translocations, inversions, duplications and deletions shuffle the order and orientation of large genomic segments between genomes. When genetic maps became available for several related species, researchers started to compare genomes in order to understand how chromosomes are evolving. In this context, the notion of shared-synteny (or synteny conservation) was increasingly used in the literature. However, this notion was employed with a meaning different from the original definition of synteny. Instead of describing the linkage of genes on chromosomes in different species, the concept of shared-synteny rather described the preservation of gene order between homologs along chromosome segments in different species. Some geneticists rejected this use of the term synteny and noticed that a majority of the scientific papers did not use the term synteny according to its original meaning [12]. It is probably because a term of reference was lacking to describe the conserved order of common markers in different species that the term “shared synteny” has been diverted from its original meaning. Subsequently, this term was gradually stripped of the word “shared” (or conserved) and in today's researcher's vocabulary, synteny, on its own, (abusively) means conserved gene order between different species rather than linkage of two or more markers on a chromosome per se.
In the last decade, sequencing technologies have taken over traditional methods of gene mapping. With the growing availability of genome sequences, the large prominence of vertebrates in the synteny-related literature has partly declined (Fig. 1) probably to the profit of plant and bacteria studies (Fig. 1). Concomitantly, synteny studies have moved from the experimental field to the bioinformatics field. Although the total number of publications dealing with yeast in the field of synteny has remained anecdotic (Fig. 1), pioneering genome-wide explorations of gene content and gene order based on sequencing data only were first developed between related yeast species [13–16]. These studies paved the road for the birth of a new field called comparative genomics aiming at understanding the mechanisms of genome evolution through the comparative analysis of chromosomes between related species. Comparative genomics was concomitantly developed in vertebrates, with the sequencing of a compact fish genome, Tetraodon nigroviridis [17], to help for the annotation of the human genome [18,19], as well as in yeast with the Génolevures program [20] which represented the first large exploratory sequencing project between related species aiming at deciphering the mechanisms of genome evolution. Among other things, the Génolevures 1 program sought for the mechanisms of chromosome map reorganization through the study of synteny conservation [21]. Since then, the study of synteny has been the tool of choice, both in yeasts and vertebrates, to unravel major conceptual advances in our understanding of genome evolution such as orthology/paralogy relationships and the relative contributions of segmental vs whole genome duplication (WGD) events. Synteny has also allowed the determination of the relative rates of chromosome rearrangements in individual lineages of yeast and vertebrate as well as the reconstruction of ancestral genomes. Finally, the study of the structure and the repartition of synteny breakpoints gives access the mechanisms of chromosome rearrangements and to the models of genome evolution. However, no study has so far put into perspective the relative levels and rates of chromosomal reorganization between yeast and vertebrates.
2 The evolution of synteny in yeasts and vertebrates
2.1 Major structural and functional differences between yeast and vertebrate genomes
Yeasts and vertebrates harbor very different genome characteristics in terms of size (a 200-fold difference on average, Table 1), number of genes, proportion and size of introns, number of transposable elements and repeat sequences, gene density and proportion of coding and noncoding DNA (see [22] and [23] for a review of yeast and vertebrate genome architectures, respectively). In addition, major functional properties that can have a profound impact onto genome dynamics also differ between yeasts and vertebrates. Firstly, outcrossing between germ lines is the only mode of propagation of vertebrates, implying that the chromosome rearrangements that can be transmitted to the next generation and eventually reach fixation in populations are restricted to the meiotic divisions and the subsequent mitotic amplification of the gamete cell lines. The life cycle of wild yeasts is more complex, including clonal reproduction, outcrossing, and inbreeding. Yeast reproduction is principally characterized by a rapid clonal expansion when the environmental conditions are favorable. The proportion of sexual reproduction varies between lineages. Many lineages seem to be completely asexual while for those that undergo meiosis, mating mainly occur between ascospores originating from the same tetrad (inbreeding), hence limiting the level of outcrossing. It was calculated that Saccharomyces species undergo one sexual cycle every 1000 asexual divisions and that the proportion of outcrossing would be limited to once in every 50,000 to 100,000 asexual generations [24,25]. The rates of meiotic recombination are also very different because 1 centimorgan corresponds to approximately 3 kb in yeast but to about 1 Mb in human [26]. This implies that the two organisms have similar genome sizes in centimorgans. Secondly, it is well known that mitotic mutation rates vary between organisms [27,28]. From recent sequencing data, the intergeneration substitution rate is estimated to 1.1 × 10−8 per base per human haploid genome [29] and about 3 × 10−10 per base per division in either diploid or haploid cells of Saccharomyces cerevisiae [30,31]. These figures correspond to a 36-fold difference in the per-base probability of mutation. This difference is probably due to the cell divisions that occur in the germ line between two generations in human, while in yeast, one cell division corresponds to one asexual generation. In human, the number of cell divisions in the germ line per generation is limited to 30 cell divisions in women because oogonia cease replication during fetal life but is close to 200 divisions in a 20 year old man where spermatogenesis takes place throughout life [32]. Finally, another major functional difference between yeasts and vertebrates is the generation time that could differ by several orders of magnitude (few hours in yeasts compared to few months or years in vertebrates). This implies that for a similar evolutionary time the number of generations would be much higher in yeasts than in vertebrates although the average generation time for yeast populations in natural environments must be much longer than a few hours because they would often have to face critical growth conditions (such as long periods of starvation, low temperatures, etc.).
List of the 18 yeast and 13 vertebrate species with completed genome sequences.
| Class | Species | Genome size (Mb) | Chromosome number | Scaffold number | Reference |
| Saccharomycetes | Candida albicans | 14.3 | 8 | 8 | [44] |
| Saccharomycetes | Candida dubliniensis | 14.6 | 8a | 8a | [45] |
| Saccharomycetes | Candida glabrata | 12.3 | 13 | 13 | [35] |
| Saccharomycetes | Candida tropicalis | 14.6 | 8 | 23 | [46] |
| Saccharomycetes | Clavispora lusitaniae | 12.1 | 8 | 9 | [46] |
| Saccharomycetes | Debaryomyces hansenii | 12.2 | 7 | 7 | [35] |
| Saccharomycetes | Eremothecium gossypii | 8.7 | 7 | 7 | [47] |
| Saccharomycetes | Kluyveromyces lactis | 10.7 | 6 | 6 | [35] |
| Saccharomycetes | Lachancea kluyveri | 11.3 | 8 | 8 | [48] |
| Saccharomycetes | Lachancea thermotolerans | 10.4 | 8 | 8 | [48] |
| Saccharomycetes | Lachancea waltii | 10.7 | 8 | 10 | [49] |
| Saccharomycetes | Lodderomyces elongisporus | 15.5 | 9 | 27 | [46] |
| Saccharomycetes | Pichia guilliermondii | 10.6 | 8 | 9 | [46] |
| Saccharomycetes | Pichia pastoris | 9.4 | 4 | 6 | [50,51] |
| Saccharomycetes | Pichia stipitis | 15.4 | 8 | 9 | [52] |
| Saccharomycetes | Saccharomyces cerevisiae | 12.1 | 16 | 16 | [53] |
| Saccharomycetes | Yarrowia lipolytica | 20.5 | 6 | 6 | [35] |
| Saccharomycetes | Zygosaccharomyces rouxii | 9.8 | 7 | 7 | [48] |
| Mammalia | Canis familiaris | 2400 | 39 | 39 | [54] |
| Actinopterygii | Danio rerio | 1700 | 25 | 25 | Unpublished |
| Mammalia | Equus caballus | 2689 | 32 | 32 | [55] |
| Aves | Gallus gallus | 1000 | 40b | 30 | [56] |
| Mammalia | Homo sapiens | 3080 | 23 | 23 | [18,19] |
| Mammalia | Macaca mulatta | 2871 | 22 | 21 | [57] |
| Mammalia | Mus musculus | 2644 | 20 | 20 | [58] |
| Marsupialia | Opos monodelphis | 3475 | 9 | 9 | [59] |
| Actinopterygii | Oryzias latipes | 800 | 24 | 24 | [60] |
| Mammalia | Pan troglodytes | 3100 | 24 | 22 | [61] |
| Mammalia | Ratus Norvegicus | 3000 | 21 | 21 | [62] |
| Aves | Taeniopygia guttata | 2644 | 28 | 29 | [63] |
| Actinopterygii | Tetraodon nigroviridis | 350 | 21 | 21 | [36] |
a Pseudochromosomes obtained by mapping onto C. albicans chromosomes [45].
b Including microchromosomes that were not assembled.
2.2 Chromosome evolution in yeasts and vertebrates
Because of these radically different structural and functional properties and also because important efforts to understanding genome evolution have been made so far separately in yeasts and vertebrates, it was interesting to compare the dynamics of chromosome map reshuffling between these two groups of eukaryotes. Large sequencing data sets are presently available for 51 vertebrates (http://www.ensembl.org/index.html) and 32 yeasts from the Saccharomycotina subphylum [33]. However, there is a great diversity in the completeness of genome sequences. Because fragmented genome assemblies would introduce a high number of artificial synteny breakpoints, we excluded species where the genome sequence is broken into too many small contigs and focused on the 13 vertebrate genomes and the 18 yeast genomes for which chromosomes are represented by a single or a limited number of sequencing scaffolds (Table 1).
To look for common or different evolutionary themes and to test whether there exists some sort of molecular clock for chromosome rearrangements, we computed the blocks of conserved synteny between all pairs of species applying exactly the same criteria (see legend of Fig. 2) to the 78 and 153 possible pairwise comparisons of species within the groups of vertebrates and yeasts, respectively. A unit to measure evolutionary time that would be common to both yeast and vertebrate is nevertheless needed in order to compare the evolution of the number and the size of synteny blocks in these two groups of species. Estimations of evolutionary time in Myr for yeast are weak due to the absence of reliable fossil records. In addition, generation times are very different between yeasts and vertebrates. Therefore, we decided to use the average protein divergence between orthologs as the common unit of evolutionary range. Previous analyses using the global level of divergence of orthologous proteins revealed that the evolutionary range covered by the Saccharomycotina yeasts exceeds that of vertebrates and is similar to the span covered by the entire phylum of Chordata [34–36].
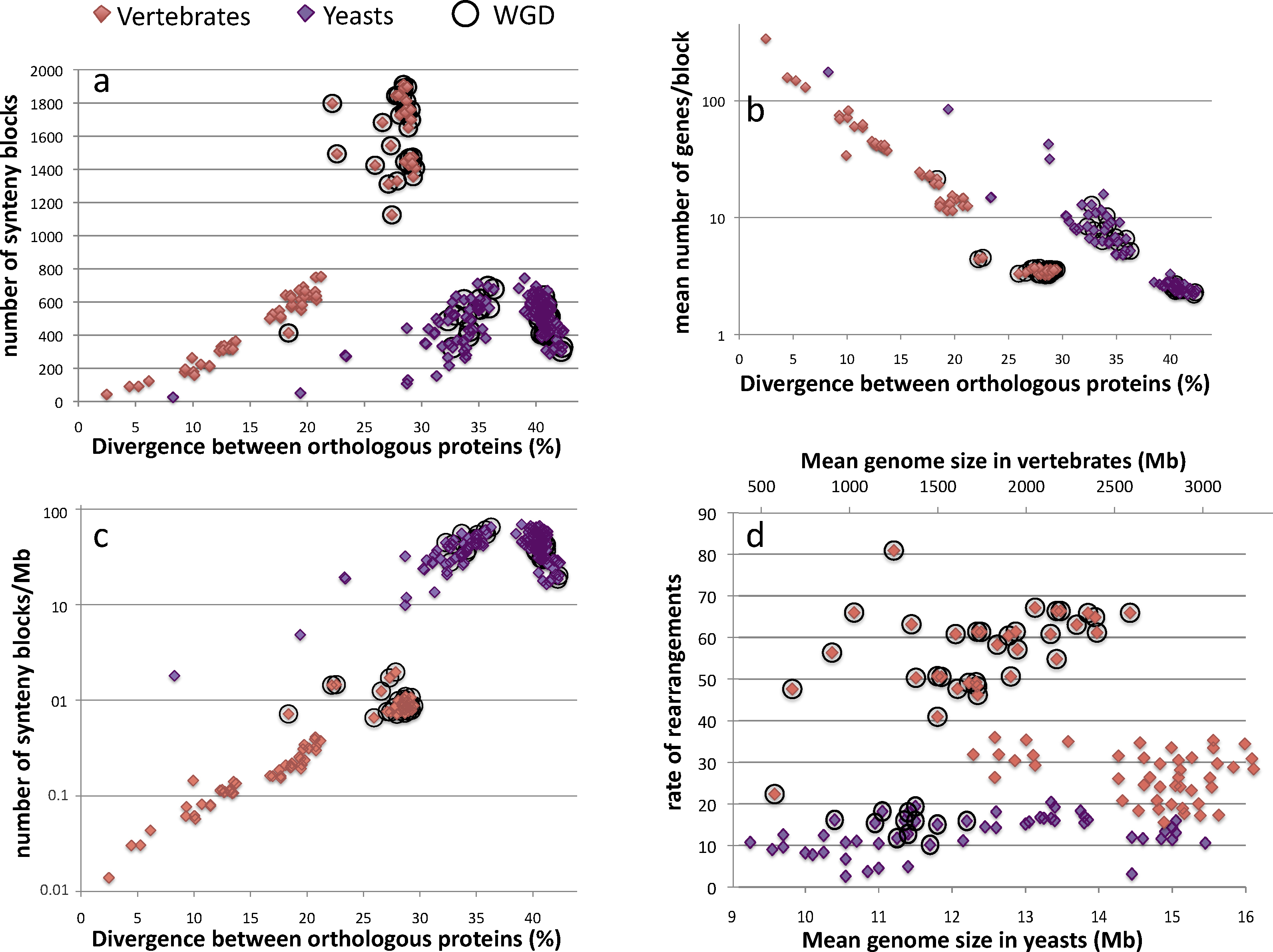
Comparative analysis of genome reorganization in 13 vertebrate and 18 yeast species (Table 1). Pairs of genes were considered as orthologs if their products were reciprocal best-hits with at least 40% similarity in sequence and their sequences were less than 30% different in length as previously described [39]. Synteny blocks were defined as series of neighboring pairs of orthologs separated by less than 5 nonneighboring reciprocal best-hits in the two compared genomes. Synteny blocks were constructed for the 78 and 153 possible pairwise comparisons between the 13 vertebrate (orange diamonds) and 18 yeast (purple diamonds) species, respectively. Black circles indicate pairwise comparisons involving at least one species that undergone a lineage-specific ancestral whole genome duplication (WGD) event (D. rerio, O. latipes and T. nigroviridis in vertebrates and S. cerevisiae and C. glabrata in yeasts). Protein divergence values correspond to the mean divergence between syntenic reciprocal best hits for each pair of compared genomes. a. Evolution of the number of synteny blocks as a function of protein divergence in vertebrates and yeasts. b. Evolution of the number of genes per block with increasing phylogenetic distances. c. The number of synteny blocks is normalized by the mean size of the 2 compared genomes and plotted as log-scale. d. The number of synteny blocks is used to approximate the number of rearrangements (comprising more than 5 genes) accumulated between 2 genomes for all comparisons involving a level of protein divergence lower than 36%. For higher level of divergence, the number of synteny blocks cannot be used to approximate the number of rearrangements because it decreases with increasing evolutionary distances (see a.). Rearrangement rates correspond to the number of rearrangements divided by mean ortholog divergence between the compared paired of species. All 78 possible pairwise comparisons were taken into account for vertebrates while only 55 out of the 153 pairwise comparisons were considered in yeast (below the threshold of 36% divergence). Masquer
Comparative analysis of genome reorganization in 13 vertebrate and 18 yeast species (Table 1). Pairs of genes were considered as orthologs if their products were reciprocal best-hits with at least 40% similarity in sequence and their sequences were less ... Lire la suite
In vertebrates, the number of synteny blocks increases exponentially with increasing divergence time, varying from a very small number of blocks, 43 between human and chimpanzee, to more than 1900 blocks between dog and zebrafish (Fig. 2a). The highest numbers of blocks are found for comparisons involving a fish genome (circled in black on Fig. 2). Such large numbers are in good accordance with the large phylogenetic distance that separates fish from tetrapodes. However, Actinopterygii species have undergone a lineage specific WGD event that was subsequently followed by a massive loss of gene duplicates. Some synteny blocks could result from these local deletion events rather than from large chromosomal rearrangements per se (see below). It is also possible that these large numbers could partly result from an increase of rearrangement rates after the WGD event. In yeasts, the number of synteny blocks is more restrained, varying from 26 between Candida albicans and C. dubliniensis up to 744 between Debaryomyces hansenii and Pichia pastoris. The number of blocks also exponentially increases along with protein divergence but only between 8 and 36% of divergence. At increasing phylogenetic distances, the number of synteny blocks decreases (Fig. 2a). This trend is most likely due to the repeated accumulation of breakpoints that lead to the reduction of the size of the synteny blocks below the minimal threshold of 2 neighboring genes (Fig. 2b) and also to a less efficient recognition of orthologous protein when divergence increases (not shown). Two yeast genomes (S. cerevisiae and Candida glabrata) have also undergone a WGD event followed by rediploidization (circled in black in Fig. 2). But, as opposed to vertebrates, all the comparisons that involve either of these 2 species are scattered throughout the plot because of their intermediate phylogenetic position relative to other yeast species.
For comparable evolutionary distances, where ranges of protein divergence overlap between yeast and vertebrate (i.e. between 8 and 30% of protein divergence), the number of synteny blocks between 2 vertebrate genomes is about 6 to 8-fold higher than between 2 yeast genomes (Fig. 2a). This shows that despite a lower evolutionary range, the raw level of chromosome map reorganization is much higher in vertebrate than in yeast. This result shows that, for comparable evolutionary distances, more chromosomal rearrangements occurred on average between 2 vertebrate genomes than between 2 yeast genomes. However, the genome sizes being on average 200 times larger in vertebrates, the physical density of synteny breakpoints along chromosomes (measured by the number of synteny blocks per Mb) is consistently higher in yeasts (between 5 and 65 blocks per Mb) than in vertebrates (between 0.01 and 2 blocks per Mb, Fig. 2c).
For both yeast and vertebrate, the average number of shared orthologs per synteny block decreases exponentially with increasing evolutionary distance until it asymptotically reaches the threshold of 2 genes below which it is impossible to recognize conserved synteny blocks (Fig. 2b). Surprisingly, in the overlapping evolutionary range (i.e. between 8 and 30% of divergence), the number of genes per block is higher in yeasts than in vertebrates (54 vs 21 on average, respectively). This higher number of genes per synteny block is best explained by the conjunction of a higher gene density in yeast (only 4 times as many genes in vertebrates than in yeasts while genome sizes are on average 200 times larger) and a higher number of rearrangements in vertebrates that is limited to only 6 to 8 times that of the yeast genomes.
Then, we estimated the rates of rearrangements by approximating the number of synteny blocks to the number of chromosomal rearrangements that occurred since two species diverged from their last common ancestor. Our analysis only accounts for rearrangements involving more than 5 orthologous genes because we tolerate up to 5 consecutive nonsyntenic homologs within a synteny block. For instance, small inversions involving less than 5 genes are not counted here. In yeast, approximating the number of rearrangements to the number of synteny blocks holds true only for pairwise comparisons involving average protein divergence below 36%. For higher levels of divergence, the superimposition of numerous rearrangements leads to the progressive destruction of recognizable synteny blocks and therefore to a strong underestimation of the number of rearrangements that actually occurred (see Fig. 2a and legend of Fig. 2d). The rates of rearrangements correspond to the number of rearrangements that occurred per unit of evolutionary time, which corresponds here to 1% of divergence between orthologous proteins (Fig. 2d). Mean rates of rearrangements are statistically different between the two groups (40 ± 4 vs 13 ± 1 rearrangements/%divergence in vertebrates and yeasts, respectively; T-test P-val = 5.4 × 10−23). On average, rearrangement rates are 3-fold higher in vertebrates than in yeasts.
In yeast, rearrangement rates do not convincingly correlate with genome sizes (R2 = 0.11, P-val = 0.02) while in vertebrate, rearrangement rates appear to be anti-correlated with genome sizes (R2 = −0.60, P-val = 5.8 × 10−9, Fig. 2d) because small genomes seem to be more rearranged. However, this anti-correlation uniquely relies on the presence of the small duplicated fish genomes (all 3 fish used in the analysis have the smallest vertebrate genomes) and vanishes when the corresponding data points (circled in black in Fig. 2d) are removed from the analysis (R2 = −0.23; p-value = 0.12). In fish genomes, rearrangement rates are confounded by the lineage specific rediploidisations subsequent to the WGD, which only involve local deletions, not gene-reordering rearrangements. In reality, these fish genomes are remarkably stable and show little rearrangements. For example, Medaka (Oryzias latipes) has been subjected to zero interchromosomal event since it splits from the pufferfish (Tetraodon nigroviridis) lineage more than 100 Myrago (Hugues Roest Crollius, pers. com.). Therefore approximating the number of rearrangements by the number of synteny blocks for these postduplicated genomes might lead to an overestimation of the rearrangement rates in vertebrates. When comparisons involving duplicated fish (O. latipes, D. rerio and T. nigroviridis) and yeast (S. cerevisiae and C. glabrata) genomes are excluded from the analysis, the mean rearrangement rate remains significantly 2-fold higher in vertebrates than in yeasts (27 ± 2 vs 13 ± 1 rearrangements/%divergence, respectively). It has been shown that both in yeasts and in vertebrates, rearrangement rates are variable between individual lineages [37–40]. For instance, rearrangement rates are smaller between S. cerevisiae and Lachancea waltii (12.7) than between S. cerevisiae and C. glabrata (15.9) and also smaller between human and dog (20.9) than between human and mouse (26.5), as previously reported [40,41]. Despite these lineage-specific variations, we show here that the global rates of rearrangements are higher in vertebrates than in yeasts, arguing against the hypothesis of a molecular clock for rearrangements. However, because of very large genome sizes in vertebrates, the average rearrangement rate per Mb is about 50-fold higher in yeasts than in vertebrates (1.04 vs 0.02 rearrangements/%divergence/Mb in yeasts and vertebrates, respectively).
Because vertebrates have emerged within the Chordata phylum approximately 450 Myr ago [42], the average rate of 40 ± 4 rearrangements/%divergence can be translated into time unit and would correspond to a rate of 2 rearrangements/Myr (918 blocks on average divided by 450), close to previous estimates on mammalian genome evolution (3.2 chromosomal rearrangements per million years on the mouse branch from the murid rodent ancestor; 3.5 chromosomal rearrangements per million years on the rat branch; and 1.6 chromosomal rearrangements per million years on the human branch [37]). A similar translation would be less reliable in yeast because estimated emergence time for the Saccharomycotina subphylum vary between 400 and 1000 Myr ago [43] and also because at large evolutionary distance (ortholog divergence greater than 36%) the number of synteny blocks cannot be used to approximate the number of rearrangements that actually happened.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We thank Hugues Roest Crollius for critical reading of the manuscript and for our regular scientific discussions that have contributed to the realization of this work. We are highly grateful to Jean-Luc Souciet, Bernard Dujon and Claude Gaillardin for having given rise to the Genolevures adventure and for allowing us to contribute.