

1 Introduction
All the genomes sequenced so far feature tandem gene arrays (TGAs), i.e. chromosomal organizations of tandem repeated paralogous gene copies. Each TGA derives from a common ancestor by successive events of gene duplication. Ample evidence has been accumulated showing the importance of gene duplication in evolution. Ohno postulated, in 1970, that gene duplication is one of the driving forces of evolution [1]. In the Ohno's model, one of the duplicate copies retains the function of the ancestral gene, while the other copy is free to accumulate mutations. In some cases, this leads the mutated copy to encode a new gene product. This evolutionary process, called neofunctionalization, explains the preservation of duplicate copies of genes. Other mechanisms, like subfunctionalization and concerted evolution, were proposed as responsible for the retention of duplicate genes in genomes [2,3]. Subfunctionalization is invoked when both duplicate copies accumulate mutations in different functional domains and, thus, share the ancestral function. In concerted evolution, there is no evolutionary innovation but a conservation of the original function to provide a quantitative advantage by increasing the level of gene product. In the case of some organisms, an increased gene dosage is quite frequent in connection with the acquisition of resistance to toxic compounds or pathogens [4,5]. The fate of genes after duplication does not always prove “advantageous” since mutated copies may gradually become non-functional copies of genes that are termed pseudogenes. This scenario of evolution by gene duplication refers to pseudogenization.
An original in silico method was used to predict these particular gene clusters that are TGAs in nine hemiascomycete yeast genomes [6]. The analysis of the set of TGAs detected by this method has given information about their frequency and structure (TGA size and relative orientation of tandem genes) in yeasts [6]. In the present work, to the data provided by this method, we added those resulting from the analysis of two additional species (Pichia sorbitophila and Arxula adeninivorans), representing new branches among the hemiascomycetes, in order to have a broader view of the structural properties of TGAs in yeasts. Results confirm with more precision those previously found by Despons et al. [6], which allow us to review general features of yeast TGAs. In this work, we also determined functional characteristics of tandem gene products on the basis of Gene Ontology (GO) annotations and orthology between genes of Saccharomyces cerevisiae and other yeast species. The evolutionary aspect of TGAs is discussed from study results of a S. cerevisiae model multigene family, the DUP family. It is the second largest family of the sequenced strain S288c and consists of two subfamilies named DUP240 and DUP380 [7]. In this reference strain, several DUP240 members are organized as two tandem direct repeats: five copies on chromosome I and two copies on chromosome VII. DUP380 genes are dispersed subtelomeric paralogs in S288c, but clustered as tandem repeats in other strains. Comparative genomic sequence analyses show that high polymorphism exists between these DUP repeats at intra- and inter-species levels. Moreover, using an in vivo approach, we demonstrated that the tandem DUP240 loci are target sites for chromosomal reshaping events in S. cerevisiae.
2 Results and discussion
2.1 Frequency, physical location and structural characteristics of TGAs
The in silico method of Despons et al. [6] applied to 11 hemiascomycete complete genomes provides a unique catalog of yeast genes that constitute tandem arrays (see the Supplementary Material). The analysis of this data set reveals that genes present in TGAs represent 2% of total genes (Table 1). The average percentage of genes in TGAs is lower in yeasts than in plants or vertebrates where it varies from 9 to 21% [8,9]. This result suggests that TGAs exploded in multicellular organisms, and thus that the TGA frequency may be related to the genome compaction and/or the cell structure complexity of an organism. More investigations in comparative genomics and experimental biology are needed to address this interesting point. Genes organized in tandem arrays are twice as frequent in Debaryomyces hansenii. The greater efficiency of tandem gene duplication in this marine yeast species may be a response to environmental constraints. Another explanation is that TGA formation may be equally frequent in all yeast species, but TGA stability may be higher in D. hansenii. The species Pichia sorbitophila was isolated from a 70% sorbitol solution. It is highly resistant to salt stress [10] as well as the most closely related species to D. hansenii. But, interestingly, P. sorbitophila does not display more TGAs than other yeasts (Table 1). This observation supports the second hypothesis regarding the high number of TGAs in D. hansenii. The frequency of yeast TGAs containing pseudogenes is not negligible since it averages 10% (Table 1). This parameter is unknown in multicellular eukaryotes, but its value may be high. In fact, our method applied to the chromosome 4 of the plant Arabidopsis thaliana reveals 22% of TGAs with pseudogenes (data not shown).
Statistics of TGAs in genomes of hemiascomycete yeasts.
| Species | Number of TGAs | Genome size | Average | Total number of genes | Number of genes in TGAs | Percentage of genes in TGAs | |
| Total | With pseudogenea | (Mb) | TGA density (kb per one TGA) | ||||
| S. cerevisiae | 52 | 3 | 12.071 | 232 | 5859 | 111 | 1.895 |
| C. glabrata | 44 | 1 | 12.318 | 280 | 5200 | 114 | 2.192 |
| Z. rouxii | 47 | 8 | 9.765 | 208 | 4991 | 97 | 1.943 |
| K. thermotolerans | 37 | 6 | 10.393 | 281 | 5092 | 77 | 1.512 |
| S. kluyveri | 51 | 4 | 11.346 | 222 | 5311 | 105 | 1.977 |
| K. lactis | 36 | 2 | 10.689 | 297 | 5075 | 72 | 1.419 |
| A. gossypii | 31 | 2 | 8.742 | 282 | 4718 | 70 | 1.484 |
| D. hansenii | 128 | 19 | 12.152 | 95 | 6264 | 273 | 4.358 |
| P. sorbitophila | 75 | 1 | 21.460 | 286 | 11,175 | 153 | 1.369 |
| A. adeninivorans | 78 | 9 | 11.622 | 149 | 6012 | 169 | 2.811 |
| Y. lipolytica | 43 | 7 | 20.503 | 477 | 6426 | 80 | 1.245 |
| All 11 species | 622 | 62 | 141.062 | 227 | 66,123 | 1321 | 1.998 |
a TGAs containing at least one annotated gene and one pseudogene.
The yeast TGA density is too low (on average, one TGA per 293 kb, Table 1) to see whether their distribution along the chromosomes is random or not. In human, mouse and rat, the TGA repartition is heterogeneous along chromosomes and some chromosomes appear enriched in “TGA forests” or “TGA deserts” [11]. Pericentromeric regions of human chromosomes tend to be preferred locations for TGAs. In contrast, TGA density is low in centromeric and pericentromeric areas of rice and A. thaliana chromosomes and is positively correlated with recombination rate [8].
The TGA size is variable, although the TGA distribution decreases when the number of tandem gene copies increases (Fig. 1). Eighty-six percent of TGAs are constituted of two repeated units (2 genes and “1 gene-1 pseudogene” classes of TGAs). This value falls to 10% with three units (3 genes and “2 genes-1 pseudogene” classes). The D. hansenii genome has the largest TGA with 16 paralogous gene copies of unknown function. The maximum size of a TGA in the S. cerevisiae S288c strain is five genes plus one highly degenerated pseudogene. These genes belong to the DUP240 family and their function is not known either [7]. The relative orientation of genes in the tandem arrays is mainly direct (78% of TGAs, Fig. 1). Nineteen percent of TGAs have genes in opposite orientation and only 3% have genes in both types of orientation (mixed TGAs). We hypothesize that several tandem gene duplication mechanisms with different degrees of efficiency may be the cause of the different orientations observed (“head-to-head”, “tail-to-tail” or “head-to-tail” orientation). The structural features of yeast TGAs are found in other eukaryotic species [8,9] suggesting that their formation mechanism(s) is universal.
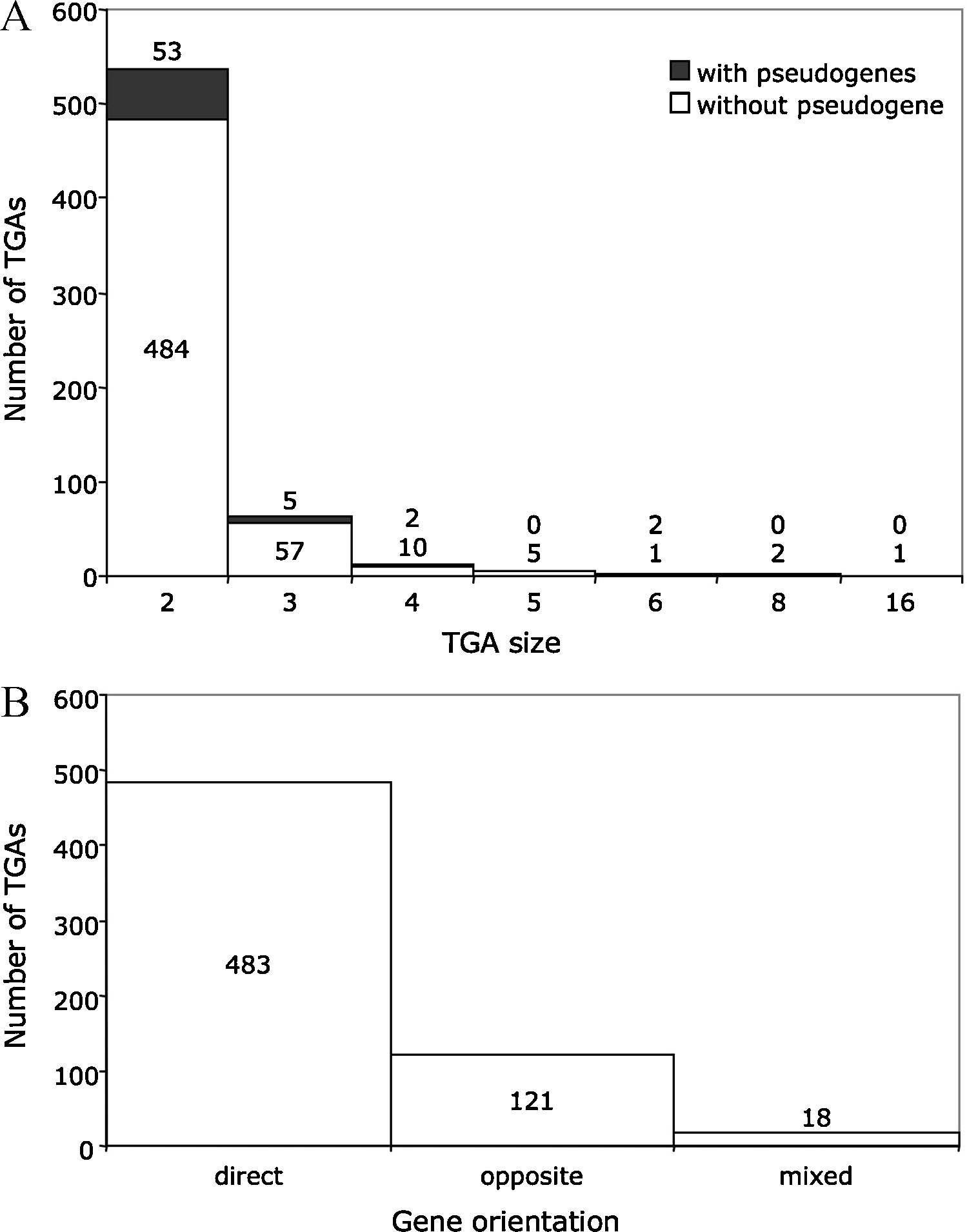
Distributions of the size and gene orientation of TGAs. Except for P. sorbitophila and A. adeninivorans, the data are taken from the study published by Despons et al. [6]. A. The distribution of the number of genes in a TGA. The TGAs constituted of genes and pseudogenes (with pseudogenes) are distinguished from those exclusively composed of genes (without pseudogene). B. The distribution of the orientation of genes in a TGA. The direct orientation refers to TGAs in which all genes share the same orientation (sense →→ or antisense ←←). When TGAs are composed of two genes located on different DNA strands (convergent →← or divergent ←→), their orientation is named “opposite”. Mixed TGAs contain at least one gene pair in direct orientation and one gene pair in opposite orientation. Masquer
Distributions of the size and gene orientation of TGAs. Except for P. sorbitophila and A. adeninivorans, the data are taken from the study published by Despons et al. [6]. A. The distribution of the number of genes ... Lire la suite
2.2 Functional roles of TGAs
The functional bias of yeast tandem arrayed genes was estimated on the basis of the Gene Ontology (GO) term annotation information [12]. Genes involved in TGAs are more frequently associated with the following GO terms than the other genes: cell wall, extracellular region and plasma membrane (Table 2). Therefore, the subcellular location of the TGA gene products would be preferentially at the cell surface or periphery. This hypothesis is consistent with the biological processes overrepresented among TGA genes: cell wall organization, transport and response to chemical stimulus. Furthermore, Table 2 shows an enrichment in transporter genes for TGAs (ratio of 2.9 in the column “molecular function”). A high number of tandem duplicate copies of these genes is not surprising because it often responds to a growing need for proteins with a transporter activity. Genomic amplifications of transporter genes were reported by Gresham et al. [13] who have observed the evolutionary dynamics of adaptation of S. cerevisiae populations in controlled nutrient-limited environments. The gene SUL1 encoding a high-affinity sulfate transporter is almost invariably amplified when yeasts adapt to sulfate limitation. Such an increase in gene dosage has been proposed by Ohno in his evolutionary model as one explanation for the preservation of duplicate genes [1]. On the other hand, in Table 2, the overall process of gene expression, including translation and protein folding, and processes that do not require extracellular contacts are underrepresented in the TGA gene class.
Frequencies of GO-Slim terms for TGA genes in comparison with all other genes.
| Biological process | Molecular function | Cellular component | |||
| GO-Slim termsa | Ratiob | GO-Slim termsa | Ratiob | GO-Slim termsa | Ratiob |
| Fungal-type cell wall organization | 5.490 | Transporter activity | 2.897 | Cell wall | 7.716 |
| Peroxisome organization | 2.442 | Protein kinase activity | 1.974 | Extracellular region | 6.894 |
| Cellular carbohydrate metabolic process | 2.223 | Oxidoreductase activity | 1.940 | Plasma membrane | 4.712 |
| Transport | 2.127 | Peptidase activity | 1.824 | Membrane fraction | 2.464 |
| Pseudohyphal growth | 1.963 | Peroxisome | 2.229 | ||
| Cellular lipid metabolic process | 1.800 | Cellular bud | 1.903 | ||
| Cell budding | 1.749 | Membrane | 1.674 | ||
| Response to chemical stimulus | 1.696 | ||||
| Sporulation resulting in formation of a cellular spore | 1.603 | ||||
| Cellular component morphogenesis | 0.299 | Protein binding | 0.262 | Ribosome | 0.257 |
| Nucleus organization | 0.262 | Isomerase activity | 0.093 | Chromosome | 0.226 |
| Cellular respiration | 0.255 | RNA binding | 0.074 | Cell cortex | 0.179 |
| Mitochondrion organization | 0.206 | Lipid binding | 0.072 | Nucleolus | 0.041 |
| Translation | 0.200 | Lyase activity | 0.000 | ||
| Heterocycle metabolic process | 0.178 | Motor activity | 0.000 | ||
| Ribosome biogenesis | 0.141 | Translation regulator activity | 0.000 | ||
| Protein folding | 0.000 | Nucleotidyltransferase activity | 0.000 | ||
| Phosphoprotein phosphatase activity | 0.000 |
a Only GO-Slim terms corresponding to a ratio ≥ 1.600 or ≤ 0.300 are mentioned.
b Ratio is the frequency of TGA genes associated to a given GO-Slim term divided by the frequency of all other genes associated with the same GO-Slim term.
Numerous examples of TGAs in S. cerevisiae or other well-studied yeasts show that TGA gene products interact with extracellular components or with other cells for the purpose of rapid adaptation to environmental stresses or changes in medium conditions. Tandem amplification of the metallothionein-coding CUP1 locus confers resistance to copper toxicity [14]. The number of CUP1 copies is directly correlated with the concentration of copper ions in the external medium. HXT4, HXT1 and HXT5 tandem loci produce hexose transporters. Each of these membrane transporters has a functional specificity: a different affinity for glucose [15,16]. This HXT array is an example where all members of a TGA have not exactly the same function. Neo- and/or subfunctionalization might have contributed to the evolution of some tandem duplicate genes. The distinction between the acquisition of a new function that is different from the ancestral one (neofunctionalization) and the partition of the ancestral function (subfunctionalization) is all the more difficult to make since a model combining these two evolutionary scenarios (subneofunctionalisation) was proposed by He and Zhang [17]. FLO genes encode proteins involved in the cell-cell adhesion process named yeast flocculation. Three of them are tandem clustered with paralogous pseudogenes in the S288c strain [18]. But, putative functional orthologs of S. cerevisiae FLO genes are tandem organized in A. gossypii (TGA no.320 in the Supplementary Material) and C. glabrata (TGAs no.15 and no.29 in the Supplementary Material) species. Finally, the tandem arrayed SAP1 and SAP4 genes express aspartic proteases that play a role in the virulence of Candida albicans clinical isolates [19].
Many TGAs in all species, especially the largest TGAs, are of unknown function. In the case of the DUP240 multigene family, five and two members are organized in two distinct TGAs in the S288c strain. Simultaneous deletion of all 10 DUP240 members is viable and the knock-out strain grows normally on several culture media [20]. Although the Dup240 proteins are supposed to be implicated in membrane trafficking [7], their function remains unknown. Such paralogous genes have probably subtle functional specificities and/or are sensitive to fine modifications of environmental conditions. Moreover, they could belong to the class of genes that define the properties of species because TGA genes are often specific to one species or a clade of few species.
2.3 Genetic evolution of TGAs
Tandem gene duplication produces new gene copies identical to the original gene. Duplicate copies are subsequently subject to two antagonistic processes: sequence divergence and sequence homogenization. High single nucleotide polymorphism (SNP) and numerous indels are quite frequently observed between genes of the same TGA in many eukaryotic organisms. In the S. cerevisiae strain S288c, nucleotide identity between tandem DUP240 genes varies from 54.3 to 98.0%. Multiple alignment of the amino acid sequences shows that only the protein domain architecture of Dup240p is very well conserved. These proteins share the following structure: C1-H1-H2-C2-C3 where C is a conserved domain and H a hydrophobic domain predicted as a transmembrane segment [20]. For other examples of SNP between tandem duplicate genes, we can cite the genes encoding human antigens [21], plant disease resistance factors [4], Arabidopsis ankyrins [22] and insect chemosensors [23]. For all these cases, it is tempting to say that a strong selection should have acted to preserve SNP between tandem repeated genes under the pressure of homogenization by gene conversion. An advanced evolutionary analysis performed on numerous TGAs across a large number and a great diversity of species must be done to demonstrate the selective forces that act upon the TGAs. On the contrary, gene conversion is the predominant mechanism ensuring the integrity of other TGAs such as ribosomal DNA arrays [24]. Therefore, tandem duplicate paralogs appear to undergo a dynamic balance between mutation and conversion controlled by the functional necessity (diversity or uniformity).
The copy number polymorphism is another type of polymorphism very common for tandem arrayed genes. The variation in gene copy number is well illustrated by the DUP TGAs at intraspecific [25] and interspecific levels (Tables 3 and 4). Interestingly, heterozygous state for the DUP240 copy number is observed in some natural diploid strains of S. cerevisiae (chromosome I of CLIB413 and chromosome VII of YIIc17, Table 3). Only the laboratory strain S288c has two tandem repeated DUP240 genes on chromosome VII, other strains have one or no DUP240 gene. Strain TL229 is devoid of these genes at both loci. The largest DUP240 TGAs contain seven gene copies and are located on chromosome I of the strains CLIB382 (both allelic loci) and CLIB413 (a single locus). Complete sequencing of the S. cerevisiae clinical strain YJM789 confirms that the DUP240 tandem locus of chromosome I is a highly polymorphic region [26]. A mixed TGA composed of one DUP240 and three DUP380 copies is present in the subtelomeric region of chromosome VIII in S288c (Table 4). DUP genes are found in four other hemiascomycete yeast species, Z. rouxii, K. thermotolerans, S. kluyveri and K. lactis, but tandem arrayed DUP genes are identified only in three species (none in K. lactis, Table 4). DUP TGAs are composed either exclusively of one subfamily of genes (DUP240 or DUP380), or a mixture of both subfamilies. Paralogous pseudogenes are frequent in these TGAs. The highest number of tandem DUP copies (46 distributed in 9 TGAs) and the largest TGA (9 copies) are in Z. rouxii. Such results (Tables 3 and 4) suggest that the copy number polymorphism observed between TGAs is obviously due to inter- or intrachromosomal rearrangements by allelic or ectopic recombination. On the basis of this hypothesis, we developed an experimental system for the selection of deletion events occurring in the DUP240 TGA of S288c chromosome I (Jauniaux et al., submitted). First, results revealed that the loss rate of genetic markers (URA3 and TRP1) is six times as high for the DUP240 TGA locus as for a control chromosomal region devoid of TGA. Second, we demonstrated that the selected deletion events are notably due to intra-chromosomal recombination between tandem repeated DUP240 and gene conversion with other DUP240 paralogs on chromosome VII. These experimental results prove that the studied tandem arrays are hotspots of chromosomal reshaping. Nevertheless, the existence of very large TGAs in some yeast strains shows that a strong selection must have worked sometimes to maintain numerous gene copies under the pressure of deletion by homologous recombination. Further experimental analyses would be needed to confirm this assumption. Another study performed on populations of C. glabrata shows two major types of intra-species variations: chromosomal translocations and copy number polymorphism in TGAs [27]. Many tandem arrayed genes concerned by this polymorphism encode putative or confirmed cell wall proteins. Duplications/deletions of these genes might serve adaptive purposes.
Copy number polymorphism of tandem DUP240 genes in S. cerevisiae strains.
| Strain | Chromosome I | Chromosome VII | ||
| DUP240 copy number | DUP240 copy number | |||
| Allele 1 | Allele 2 | Allele 1 | Allele 2 | |
| S288ca | 5 | – | 2 | – |
| YIIc17 | 5 | 5 | 1 | 0 |
| R12 | 5 | 5 | 0 | 0 |
| CLIB413 | 3 | 7 | 1 | 1 |
| CLIB410 | 6 | 6 | 1 | 1 |
| CLIB382 | 7 | 7 | 0 | 0 |
| CLIB219b | 6 + 1 | 6 + 1 | 1 | 1 |
| TL229 | 0 | 0 | 0 | 0 |
a Only the laboratory reference strain is haploid. Other natural strains are diploids.
b In addition to the chromosome I DUP240 copies there is one paralogous pseudogene.
Interspecific copy number polymorphism of tandem DUP genes.
| Species | DUP genesa | DUP TGAsb | |||
| DUP240 copy number | DUP380 copy number | Total number | Minimum size | Maximum size | |
| S. cerevisiaec | 8 | 3 | 3 | 2 | 5 |
| Z. rouxii | 21 | 25 | 9 | 2 | 9 |
| K. thermotolerans | 5 | 4 | 3 | 2 | 4 |
| S. kluyveri | 12 | 0 | 5 | 2 | 4 |
a Gene copies of both subfamilies which are tandem arrayed (isolated copies omitted).
b TGAs composed of DUP240 and/or DUP380 copies. TGA size is given in DUP copy number.
c In the strain S288c, TGAs are located on chromosome I (5 DUP240), chromosome VII (2 DUP240) and chromosome VIII (1 DUP240 and 3 DUP380).
Combining all the information about duplicate genes given above and in the literature (for reviews, see references [28,29]), Fig. 2 summarizes the genetic evolution of TGAs. Three major evolutionary paths open in front of TGAs:
- (i) concerted evolution;
- (ii) gene birth-and-death evolution;
- (iii) a combination of these two models.
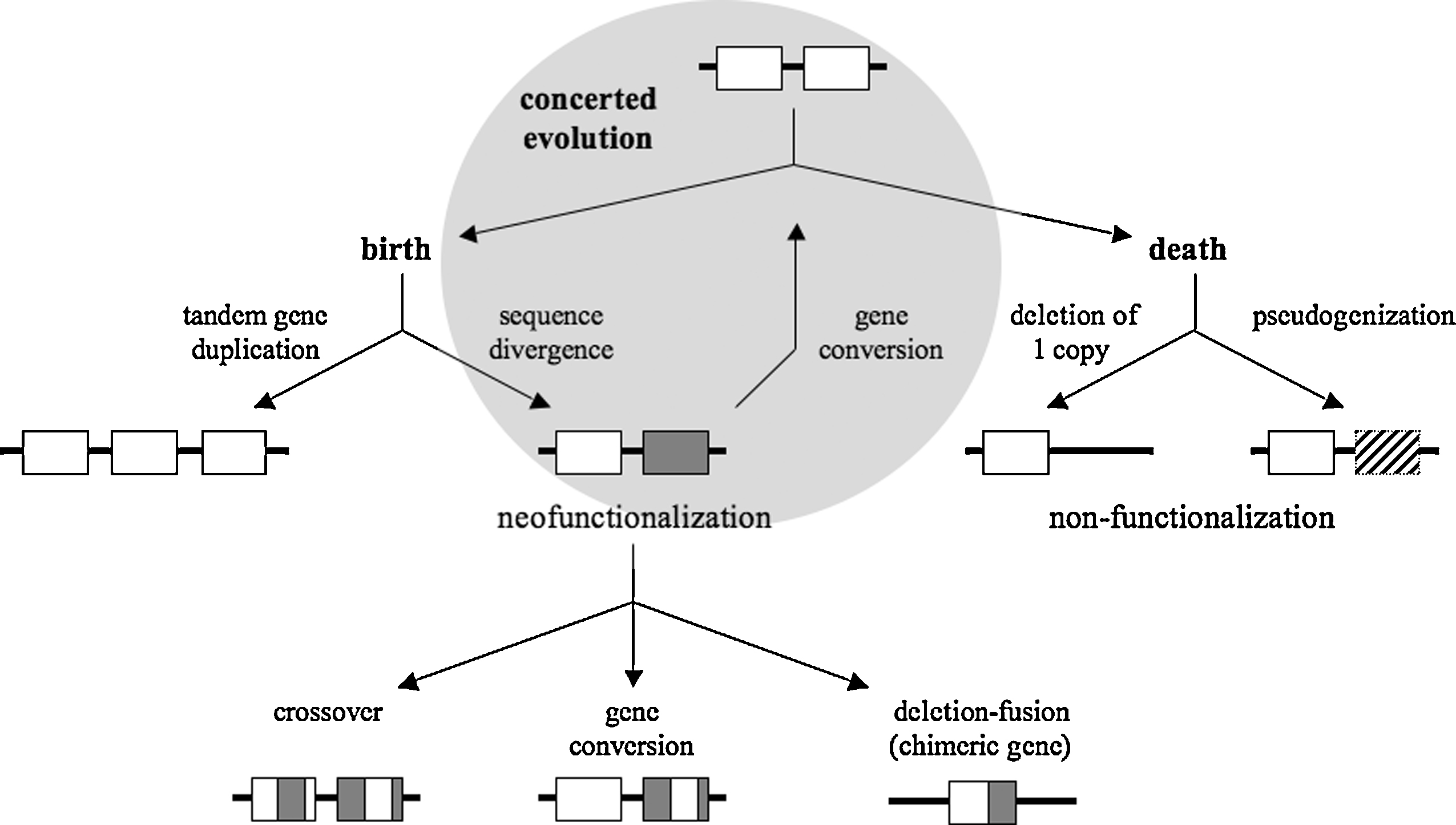
A model proposed for the evolution of TGAs. A repeat of two tandem duplicate genes may evolve along three major paths: the gene birth-and-death evolution, the concerted evolution (in the shaded circle) or a combination of these two evolutionary models. Genes are represented by open boxes and pseudogenes by hatched boxes. Shaded boxes are duplicate gene copies that have undergone mutational events and thus diverge from their ancestral gene.
In path (i), members of a TGA do not evolve independently of each other but rather evolve in a concerted way. Sequences of TGA members become homogenized by gene conversion events that preserve gene function. In path (ii), new genes are born by gene duplication and some of them stay in the genome and may diverge functionally, whereas others die by inactivation or deletion. The birth-and-death evolution under the influence of strong purifying selection is an alternative model to the model of concerted evolution for homogenization of gene sequences. Functional diversity is initially generated by mutational events in duplicate gene copies and then amplified by recombination events giving rise to chimeric gene formation (deletion-fusion) or gene domain exchanges (crossover if reciprocal exchange, if not gene conversion; Fig. 2). This neofunctionalization process is inseparable from the non-functionalization process within the framework of an evolution by birth and death of genes. In fact, sequence divergence within a gene may create a new function or lead to the formation of a pseudogene and recombination may give a new structure to the gene or delete the gene.
3 Concluding remarks
TGAs are particular chromosomal organizations frequently found in yeast genomes (2% of total genes). They encode proteins mainly located at the cell surface and then in contact with the extracellular environment. They are privileged sites of chromosomal rearrangements and copy number polymorphism because high sequence similarity between contiguous paralogs induces homologous recombination. Moreover, single nucleotide polymorphism is often detected between tandem repeated paralogs. Therefore, it seems that, in a general way, these very dynamic structures are sites of gene birth and death probably for purpose of rapid adaptation to environmental changes.
Similarly to TGAs, subtelomeres are unstable structures, being hotspots for chromosomal rearrangements. In yeast, subtelomeres are 20–30 kb long chromosomal regions located upstream of telomeric caps (reviewed in [30]). They have high AT percentage, low gene density and are subjected to epigenetic silencing. Paralogous gene copies are often located on different subtelomeric regions. Some of them are organized in tandem clusters, but the majority of them are isolated paralogs. Presence of multigene families repeated in subtelomeres favors recombination. Ricchetti et al. [31] have demonstrated that double-strand break repair is more efficient at chromosomal ends because gene conversion and break-induced replication occur between subtelomeric duplicate genes (FLO and COS = DUP380). Moreover, it was shown that subtelomeric gene families evolve faster than other families [32]. Subtelomeres have another feature in common with TGAs: they contain many families of species-specific genes [33]. This suggests that these genes may serve a unique molecular function in species or be adapted to a special environment. Taking these remarks together, we reach the conclusion that TGAs, like subtelomeres, are sources of genomic plasticity and genetic innovation.
4 Materials and methods
4.1 Tandem Gene Arrays (TGAs) detection
TGAs were predicted by the Despons et al. [6] in silico method. This original method allows the detection of functional or non-functional (pseudogenes) paralogous gene copies that are organized as tandem repeats in eukaryotic genomes. In most cases, no intervening spacer gene is present within a TGA, but sometimes one spacer is authorized and very rarely more than one spacer.
4.2 Genome databases
We applied our computational method on 11 genomes of hemiascomycete yeast species: Arxula adeninivorans, Candida glabrata, Debaryomyces hansenii, Eremothecium (Ashbya) gossypii, Kluyveromyces lactis, Kluyveromyces thermotolerans, Pichia sorbitophila, Saccharomyces cerevisiae, Saccharomyces kluyveri, Yarrowia lipolytica and Zygosaccharomyces rouxii. These genomes have been sequenced by the Génolevures Consortium (website http://www.genolevures.org/; [34]), except for S. kluyveri (collaboration with Mark Johnston, Washington University Department of Genetics), S. cerevisiae (database available on http://www.yeastgenome.org/; [35]) and A. gossypii (http://agd.vital-it.ch/Ashbya_gossypii/index.html; [36]).
4.3 Gene annotations and GO-Slim frequencies
Gene annotation information about the 11 yeast species analyzed here (Pichia sorbitophila and Arxula adeninivorans included) was extracted by Tiphaine Martin and Pascal Durrens from the genome databases mentioned above.
The mapping of all S. cerevisiae gene products to yeast-specific GO-Slim terms was downloaded from Saccharomyces Genome Database (SGD) website (http://www.yeastgenome.org/; [35]). GO-Slim terms were associated to yeast genes on the basis of orthology with S. cerevisiae gene products, considering that all GO-Slim terms of a S. cerevisiae gene are transferable to its ortholog. In order to define the functions over- or underrepresented in the subgroup of genes located in TGAs, the GO-Slim frequencies of the tandemly arrayed genes and of all other genes were calculated and compared.
Disclosure of interest
The authors declare that they have no conflicts of interest concerning this article.
Acknowledgements
We thank Fabien Pertuy for help in analyzing yeast genomic sequence data to search DUP genes. We are grateful to Claude Coulomb for the language corrections of the manuscript. This work was supported in part by funding from CNRS (GDR 2354, the Génolevures Consortium) and in part by ANR (ANR-05-BLAN-0331, GENARISE).