

1 Introduction
Pharmacological induction of fetal hemoglobin (HbF) as a potential therapeutic strategy for sickle cell anemia (SCA) arises from two key observations regarding the benefits of elevated HbF levels: firstly, persons who co-inherit hereditary persistence of fetal hemoglobin with SCA have minimal or no clinical manifestations, and secondly, infants with SCA have few clinical manifestations in the first 3–6 months of life. The HbF level in SCA is protective but declines rapidly during the first year of life and eventually reaches a nadir around age 5 years [1,2]; this latter value, often referred to as the “baseline” HbF value for individual patients, is strongly influenced by genetic factors [3,4] but still is not completely understood.
The biophysical basis for protection by HbF against sickling is primarily on the basis of reducing the rate of HbS polymerization within the erythrocytes. HbF tetramers dilute the intracellular HbS concentration and do not participate in the polymerization process. Based on these observations, efforts were made to identify therapeutic agents that could induce HbF, and hydroxyurea emerged as one of several effective compounds. Due to its oral route of administration and known toxicity profile, as well as its potent laboratory and clinical effects, hydroxyurea has been the primary focus of HbF induction strategies for SCA to date.
In the 1980, fairly simple but elegant “proof-of-principle” studies demonstrated the potential benefits of hydroxyurea (or hydroxycarbamide) therapy for adults with SCA [5]. Following short-term high-dose oral pulses of hydroxyurea, reticulocytes with increased fetal hemoglobin (HbF) were observed in the peripheral circulation, followed by an increased overall hemoglobin concentration and increased %HbF, documenting the ability of hydroxyurea to induce HbF production. Now almost 30 years later, we have accumulated a large amount of research data and clinical experience regarding the use of hydroxyurea in this patient population [6]. Hydroxyurea is currently recognized as a potent pharmacological agent for HbF induction, with proven laboratory and clinical efficacy for infants, children, adolescents, teens, and adults with SCA. HbF induction is the primary therapeutic effect of hydroxyurea in SCA. However, additional treatment benefits are derived including mild myelosuppression, macrocytosis, reduced cellular adhesion, improved rheology, and potentially local nitric oxide release [6].
2 Treatment indications
Clinical recommendations for when to consider hydroxyurea therapy are still evolving and consensus guidelines have not yet been established. Through a series of Phase I/II and Phase III clinical trials involving adults [7], school-age children [8], and even infants with SCA [9], hydroxyurea has documented laboratory efficacy and also significantly reduces the number of painful episodes (crises), acute chest syndrome (ACS) episodes, transfusions, and hospitalizations.
Based on efficacy data from these prospective clinical trials, frequent or severe painful vaso-occlusive events and ACS are the most common reasons to consider hydroxyurea (Box 1). Other clinical indications that often lead to hydroxyurea therapy include dactylitis, severe anemia, elevated transcranial Doppler (TCD) velocities, poor growth, and frequent hospitalizations. Additional potential indications include preservation of organ function or even reversal of organ dysfunction; hence patients with hypoxemia, proteinuria, or cerebrovascular disease may initiate hydroxyurea as well. Some clinicians feel that a laboratory profile reflecting disease severity (e.g., elevated white blood cell count, low %HbF, high LDH) also warrants consideration to initiate hydroxyurea. Ongoing and future research studies should clarify the role of hydroxyurea for these various indications. More recently, families are asking for hydroxyurea treatment, usually after one affected sibling is receiving therapy. In these settings, clinicians should consider the benefits of hydroxyurea for the otherwise “unselected” patient who has yet to develop clinical severity.
There is no ideal age to initiate therapy, but the safety profile of hydroxyurea has been documented in patients as young as 6–9 months of age [9,10]. Younger patients tend to have better treatment responses and better medication adherence than older patients, hence a decision to begin therapy should be consider early in life, ideally before 5–10 years of age. However, clinical efficacy can be demonstrated at any time; as noted above, even adults with severe symptoms had significant reduction in pain, ACS, transfusion, and hospitalization when started on hydroxyurea therapy [7].
3 Initiation and dose escalation
Hydroxyurea can be administered orally as capsules or by an extemporaneously prepared liquid formulation, given once a day [11]. The starting dose in the United States is typically 15-20 mg/kg/day, which is tolerated by virtually all patients with SCA. Most investigators then monitor with monthly blood counts and escalate by 5 mg/kg/d every 8 weeks, to reach the maximum tolerated dose (MTD), typically defined as mild myelosuppression such as ANC of 2.0–4.0 × 109/L (2000–4000 neutrophils/μL). However, myelosuppression to lower ANC values, e.g., 1.0–2.0 × 109/L appears to be safe, and no episodes of bacteremia or serious infection have been reported due to drug-induced neutropenia. Reticulocytes are also affected by hydroxyurea, and up to 25% of patients will develop reticulocytopenia as the main dose-limiting toxicity.
Most young patients reach MTD at 25–30 mg/kg/d, and at that dose will achieve key laboratory thresholds (hemoglobin concentration ≥ 9 g/dL and HbF ≥ 20%) without excessive myelosuppression. In Europe, clinicians often prefer a lower dose that can be termed the minimally effective dose (MED), typically 15–20 mg/kg/d, defined operationally as the dose where patients improve clinically and feel better. Compared to the MTD, patients who receive the MED tend to have lower treatment responses based on hemoglobin and HbF values [12].
Patients with renal dysfunction, low marrow reserve, or unusual body habitus (i.e., high body mass index) will not tolerate standard dose escalation and may have a low hydroxyurea MTD. In all patients, monthly monitoring is required initially to reach a safe and stable dose, whether MTD or MED, after which time the frequency of visits can be modified. Medication adherence is an important part of hydroxyurea therapy, and efforts should be undertaken to ensure that patients and families understand the importance of daily dosing with good compliance. Strategies to improve medication adherence have been described [11].
3.1 Treatment responses
Patients and families should understand that the effects of hydroxyurea are not immediate. Laboratory changes occur over the first few weeks, typically with a rise in MCV and a concomitant fall in the ANC and absolute reticulocyte count (ARC). As the hydroxyurea dose is escalated toward MTD, changes also become evident in the peripheral blood smear: macrocytosis without polychromasia, fewer sickled forms, more target cells, and lower numbers of circulating neutrophil and reticulocytes. Fig. 1 illustrates typical changes in the laboratory profile and peripheral blood smear of a young patient with SCA who initiates hydroxyurea with dose escalation to MTD. Beneficial effects on %HbF are not necessarily evident early in treatment, reaching maximum effects only after 6–12 months of treatment [8].
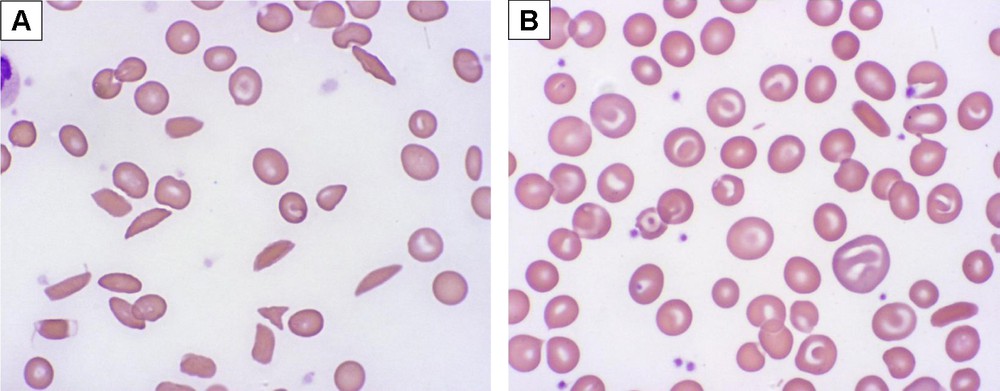
Peripheral blood smear morphology in sickle cell anemia and response to hydroxyurea therapy. Panel A illustrates a representative pre-treatment blood smear, with evidence of anemia and marked poikilocytosis, including numerous elongated sickled forms. Panel B illustrates a representative on-treatment blood smear at hydroxyurea MTD, with less anemia and reduced poikilocytosis; most cells are macrocytes (without polychromasia) and target cells rather than sickled forms.
At each visit, compliance and especially medication adherence must be emphasized. Each missed daily dose allows erythropoiesis to occur without the “influence” of hydroxyurea, so represents a missed opportunity to ameliorate the disease. Responses are sustained as long as adherence is maintained. Downward trends in MCV and HbF, or upward trends in ANC and ARC, often reveal problems with adherence. Periodic review of the peripheral blood smear with the patient and family can be helpful in promoting adherence [11].
3.2 Drug-related toxicities
When administered orally once a day, hydroxyurea treatment is very well tolerated with little short-term toxicity. Mild cytopenias, especially of granulocytes and reticulocytes, are expected and actually should be considered therapeutic goals of treatment. If cytopenias are ever excessive, counts recover quickly after a temporary treatment hold, typically one week. Patients typically require a complete blood count with reticulocytes every month during initial dose escalation, but monitoring can be reduced to every 2–3 months once a stable MTD is established. Mild gastrointestinal symptoms can occur but usually abate following adjustment of daily dosing, e.g., giving the dose before bedtime. Dermatological changes including melanonychia and hyperpigmentation do occur but are cosmetic only. Other toxicities such as hair loss, rash, skin ulcers, and hepatic or renal dysfunction are exceedingly rare, if they occur at all in this patient population.
3.3 Previous research
Hydroxyurea research in SCA is now approaching 30 years of experience (Fig. 2). In 1984 the first proof-of-principle study was reported [5], followed by prospective Phase I/II studies in adults [13], then children [8] and finally infants [10]. The landmark Phase III Multicenter Study of Hydroxyurea trial was published in 1995; this randomized placebo-controlled trial proved the clinical efficacy of hydroxyurea for the prevention of pain, ACS, transfusion, and hospitalization of adults with SCA [7].

Timeline of hydroxyurea research for sickle cell anemia.
Two major NIH-sponsored clinical trials involving hydroxyurea have been recently completed. In the Phase III infant hydroxyurea trial (BABY HUG, ClinicalTrials.gov NCT00006400), infants with SCA between 9–18 months old were recruited without regard to clinical severity, and randomized either to daily hydroxyurea (20 mg/kg/d) or placebo for 24 months of treatment. Although the primary study endpoints (prevention of organ dysfunction) were not met, secondary study endpoints of reduced pain, dactylitis, acute chest syndrome, transfusions, and hospitalizations were all significantly lower in the hydroxyurea treatment arm [9]. Based on these findings, the authors concluded that hydroxyurea can now be considered for all young patients with SCA.
A second major NIH-funded study was Stroke With Transfusions Changing to Hydroxyurea (SWiTCH, NCT00122980), which treated children with documented stroke and iron overload either with standard treatment (transfusions and chelation) or alternative treatment (hydroxyurea and phlebotomy) to prevent secondary stroke and better manage iron overload. This study was stopped early due to a failure of the alternative treatment arm to improve iron overloading between phlebotomy and chelation; at the time of study closure there was a stroke imbalance of 10% on hydroxyurea versus 0% on transfusions [14]. Additional lessons learned from the SWiTCH trial include the severe vasculopathy and clinical fragility present among most children with SCA and previous stroke, and the difficulty of a composite primary study endpoint in a multicenter randomized clinical trial.
3.4 Current research
Several NIH-funded clinical trials involving hydroxyurea for patients with SCA are currently underway. The first is TCD With Transfusions Changing to Hydroxyurea (TWiTCH, NCT01425307), where children with abnormal Transcranial Doppler velocities who receive chronic transfusions will be randomized either to standard treatment (continued transfusions) or alternative treatment (hydroxyurea). The primary endpoint is change in TCD velocity and this study is currently enrolling patients in the US and Canada. The second study is Sparing Conversion to Abnormal TCD Elevation (SCATE, NCT01531387), where children with conditional TCD velocities will be randomized either to standard treatment (observation) or alternative treatment (hydroxyurea) to prevent increases (conversion) to the abnormal threshold of greater or equal to 200 cm/s. This study includes clinical sites in the US, Jamaica, and Brazil and patient enrollment is ongoing. The Hydroxyurea Study of Long-Term Effects (HUSTLE, NCT00305175) continues to gather information on the pharmacokinetics and pharmacogenetics of hydroxyurea treatment for children with SCA, as well as the long-term effects of hydroxyurea on organ function. Finally, the BABY HUG cohort is now being following prospectively in a follow-up study (NCT00890396), to help determine the long-term risks and benefits of hydroxyurea treatment when initiated at an early age.
3.5 Long-term follow-up
Potential long-term toxicities continue to be of great concern and should be monitored in all patients with SCA who receive hydroxyurea therapy. To date, no increase in stroke, myelodysplasia, or carcinogenicity has been detected in SCA patient cohorts, now reaching 15–20 years of exposure for some treated adults and 10–15 years for treated children. Regarding the rare case reports of malignancy developing in patients on hydroxyurea, the background cancer rate in SCA must be considered [15] to prevent undue concern or ill-advised associations. Deleterious effects of hydroxyurea on fertility are suggested but not proven, in particular the effects on sperm production. However, clinical experience suggests no effect on fertility of either men or women with SCA. Similarly, teratogenicity is a theoretical risk has not been observed clinically, but a recent report from an adult cohort is reassuring with respect to pregnancy outcomes [16]. While most clinicians still recommend avoidance of pregnancy or conception while receiving hydroxyurea therapy, limited data indicate no adverse effects. In the setting of a pregnant woman with SCA on hydroxyurea, the risks of continuing hydroxyurea must be weighed carefully against the risks of discontinuing an effective treatment in a high-risk pregnancy.
From the standpoint of long-term benefits, the profile of hydroxyurea therapy is encouraging. Numerous studies document sustained laboratory and clinical benefits for patients who remain adherent, with improved growth/development in young patients. Two recent long-term adult studies (USA and Greece) have documented improved survival of patients with SCA and hydroxyurea exposure [17,18]; improved mortality on hydroxyurea provides an even more compelling reason to consider treatment for all affected patients.
3.6 Developing countries
Despite the accumulated evidence for benefits of hydroxyurea in children with SCA of all ages, there are scant data regarding its use in developing countries where the burden is greatest [19]. With over 300,000 babies born with sickle cell disease worldwide each year, the opportunity to provide the only disease-modifying treatment should be a high priority. However, the safety and feasibility of hydroxyurea treatment in the setting of co-morbidities such as general malnutrition, vitamin and micronutrient deficiencies, and infections (malaria, helminthes) have not been assessed.
While it is tempting to consider hydroxyurea suitable for current use in developing countries, especially in Africa, a careful assessment of its risks and benefits is warranted, ideally through prospective studies such as the proposed Realizing Effectiveness Across Continents with Hydroxyurea (REACH) pilot trial. As countries in Africa develop newborn screening programs to identify SCA, the widespread use of hydroxyurea may prove to be a useful treatment to help ameliorate the disease in resource-limited settings.
3.7 Knowledge gaps
An important area of future investigation is to explain the observed phenotypic variability of the hydroxyurea response. The ability to tolerate different doses of hydroxyurea is variable among patients, as is the HbF response. Recent investigation has suggested that pharmacokinetic differences exist as well as pharmacogenetic influences [20].
Another critical knowledge gap exists because almost all of the available data relate to patients with SCA (i.e., HbSS or HbS/β0-thalassemia); experience with HbSC and HbS/β+-thalassemia is limited to anecdotes and small series, and extrapolations to these patient populations should not be assumed. Prospective studies involving HbSC are badly needed, since clinicians are increasingly using hydroxyurea in this patient population. Although HbF does not have a clear benefit for patients with HbSC, the additional salutary effects such as lower WBC and reduced cellular adhesion may provide therapeutic benefits. Measurements of blood viscosity may prove useful in clinical trials involving HbSC patients, since increases in the hemoglobin concentration from hydroxyurea are common and could potentially be deleterious.
4 Summary
With almost 30 years of accumulated experience since the initial studies, we have ample documentation of the short-term safety and efficacy of hydroxyurea for patients with SCA. Long-term data are continuously accumulating and are to date equally reassuring. Taken together, the available evidence suggests that hydroxyurea represents an inexpensive and effective treatment option that should be offered to most, if not all, patients with SCA [21]. Current and future research will help define its role in the preservation of organ function. As the only currently available disease-modifying therapy for SCA, hydroxyurea may prove to be a safe and effective treatment for patients with SCA in both developed and developing countries.
Disclosure of interest
The author declares that he has no conflicts of interest concerning this article.