

1 Introduction
Transfusion is a life-sustaining therapy for sickle cell disease (SCD) patients. However, transfusion has side effects in SCD patients, mainly iron overload and alloimmunization. The main cause of alloimmunization against red blood cells in SCD is major blood group differences between donor and recipients. In this review, we will describe the characteristics of some blood groups in SCD, the consequences of alloimmunization in these patients and finally the life-threatening reaction known as the delayed haemolytic transfusion reaction (DHTR).
2 Blood groups and alloimmunization in sickle cell disease patients
Within the general population of transfused patients, SCD patients are among those most frequently alloimmunized [1]. The substantial polymorphism of immunogenic antigens between donors of European descent and these patients mostly of African descent is clearly the major factor contributing to the high prevalence of RBC alloimmunization. In Table 1, blood group differences between donors and recipients in countries with a European descent are shown. Indeed, the incidence of alloimmunization is lower when donors and patients have similar ethnic origins [2,3].
Blood group differences between donors of Caucasian descent and patients of African descent.
| % in Caucasian donors | % in Black recipients | |
| Common antigens | ||
| ABO groups | ||
| A | 43 | 27 |
| B | 9 | 20 |
| O | 44 | 49 |
| AB | 4 | 4 |
| RH | ||
| D | 85 | 92 |
| C | 68 | 27 |
| E | 29 | 20 |
| c | 80 | 96 |
| e | 98 | 98 |
| KEL | ||
| K | 9 | 2 |
| FY | ||
| Fya | 66 | 10 |
| Fyb | 83 | 23 |
| JK | ||
| Jka | 77 | 92 |
| Jkb | 74 | 49 |
| MNS | ||
| S | 51 | 31 |
| s | 89 | 93 |
| Partial RH antigens | ||
| Partial D among D+ | 1 | 7 |
| Partial C among C+ | 0 | 30 |
| Partial e among e+ | 0 | 2 |
| Low incidence antigens | ||
| VS (RH20) | 0.01 | 26–40 |
| Jsa (KEL6) | 0.01 | 20 |
| Rare blood groups | ||
| U neg- (MNS:-5) | 0 | 1 |
| HrS neg- (RH:-18) | 0 | 0.1 |
| HrB neg (RH:-34) | 0 | 0.1 |
| RN (RH:-46) | 0 | 0.1 |
| Jsb neg (KEL:-7) | 0 | 1 |
The antibodies produced by SCD patients are determined by both the differences between donors and recipients, and the relative immunogenicity of the foreign antigens, closely associated with the degree of histocompatibility promiscuity. The antibodies most frequently observed in transfused SCD patients are against C, E, Fya, Jkb and S [4,5]. Some less frequent antibodies are also found when patients carry a partial antigen or have a rare blood group. The antibodies linked to these situations are more difficult to manage, because the partial status is not always recognizable, and also because supply of rare blood types may not be sufficient.
The approach to match for E, C and Kell reduces the rate of alloimmunization, but does not take into account the numerous Rhesus (RH) variants that are encountered in these patients of Afro-Caribbean origin, representing additional potential risk of alloimmunization and DHTR. They are encoded by altered alleles at the RH locus. The RH variants have been widely described. Within the five main antigens (D, C, E, c, e), schematically, two types of variants are described, encoded by point mutations, multiple missense mutations or hybrid alleles. The first type is called partial, because they lack some immunogenic epitopes. Patients with partial variants were identified because they made alloantibodies against the missing epitopes when exposed to the complete antigen through transfusion or pregnancy. Antibodies proned by partial antigens can be potentially clinically significant and therefore patients with partial antigens should receive RBCs which do not express the Rh antigen to prevent alloimmunization. The second type of RH variants are called “weak” antigens. Since no epitopes of the antigens are lacking, individuals carrying weak antigens do not get immunized when exposed to the normal antigen. In Afro-Caribbean patients, partial antigens are frequent (Table 1). Within D variants, the DAR antigen, as well as the DIIIa, DIVa and some DAU types, are prone to immunization. But only DAR has been shown to be involved in delayed hemolytic transfusion reaction [6]. However, not all D variants transfused with D-positive units will develop anti-D mostly because the risk is not similar to the situation of D-negative individuals who lack the full Rh antigen receiving D-positive red cells. It should also be noted that not all patients are high responders. The question whether these patients should receive D-negative units as a way to prevent anti D immunization is currently under debate mostly due to shortage of D-negative RBCs, already extensively used for SCD patients to prevent anti-C and anti-E immunization. In the Paris area, 26% of the units transfused to the D+C-E-c+e patients are D-C-E-c+e+ [5]. Another potential negative consequence of transfusing D-negative units to patients with Rh variants is that it will increase their exposure to the common antigens (Fya, Jkb, S) expressed by the D-negative RBCs that are mainly from Caucasian donors. These antigens pose a higher risk of inducing alloimmunization and DHTR compared to the risk encountered by the RHD variant status of the patient. The same concerns arise in discussing variants produced by the RHCE gene. Some of these variants are well known to induce alloimmunization such as partial C encoded by (C)ceS and RN and the associated antibodies cause DHTR [7]. Substitutions on the Rh polypetides can encode antigens with missing epitopes as described in the partial Rh variants, but can also create new epitopes. These new epitopes can elicit alloantibodies in the recipient lacking the epitopes. These epitopes are called “low incidence antigens” relative to their low incidence in the Caucasian reference population. In the Black population, some of them are frequently expressed, such as the RH20 (VS) antigen, encoded by the RHCE*ceS allele Considering that this antigen is expressed in about 40% of individuals of Afro-Caribbean origin, a VS-negative SCD patient receiving blood from a donor with the same ethnic background can be exposed and potentially get immunized to VS. It should be more frequently the case in the Caribbean's Islands. Many other “low incidence” antigens are described in the Black population.
Serological tools are not effective to type variants, but molecular tools have been developed. However, given the high cost of the tests, variants whose associated antibodies are not clinically significant need not be typed. Ongoing studies to determine clinical significance of antibodies against variants are critical for transfusion management of patients.
Another difficult situation to handle in SCD patients is the occurrence of rare blood groups characterized by the absence of expression of a high incidence antigen. This situation makes the provision of RBC units almost impossible, as all common RBCs will express the high incidence antigen lacking in the patient. The most frequent is the U-negative phenotype (MNS:-5), recognizable by the absence of expression of the common S and s antigens at the serological typing. Another type is the Jsb-negative phenotype (KEL:-7), which needs molecular tools to be characterized, as no commercial serologic reagents are available to type RBCs. Finally, in the RH blood group, the RH:-18, RH:-46 and RH:-34 are the most frequent “rare phenotypes”. In all of these cases, alternative treatments should be discussed very early in order to avoid alloimmunization and definitive shortage of compatible RBCs.
On a wider point of view, understanding the mechanisms and risk factors associated with alloimmunization will aid in developing strategies to prevent and inhibit production of antibodies in transfused patients [8]. Specific characteristics of the underlying disease are likely to play an important role in favouring alloimmunization in certain patient populations.
Inflammation, which is a specific feature of SCD, could play a role. In mouse models, inflammation has been shown to play a central role in inducing alloimmunization [9]. However, the extrapolation of mice data to the situation in SCD patients is still difficult to demonstrate because of the wide diversity of the clinical presentation of the disease, which involves genetic or acquired patient-related factors that can interfere with the process of alloimmunization.
It is clear that alloimmunization leads to transfusion becoming both more complicated and more dangerous for the patient. The first consequence is the delay before transfusion due to the difficulty of finding blood units that match the antibodies produced. The results of screening tests can be also difficult to resolve when many antibodies are present, or when antibodies are produced in a patient with a partial or a rare blood group. The most serious consequence of alloimmunization in SCD patients, however, is the risk of a delayed haemolytic transfusion reaction with hyper haemolysis (DHTR). This side-effect of transfusion has consequences for the outcome of the disease and can be life-threatening. Alloimmunization is also a risk factor for the production of autoantibodies against RBCs [10]. Autoantibodies can enhance the haemolytic reaction, participating in the so-called bystander haemolysis and explaining in some cases the destruction of both the patient's own and transfused RBCs. Autoantibodies also complicate screening tests, as specific and time-consuming techniques are required to detect relevant alloantibodies that can be masked by autoantibodies.
3 The haemolytic transfusion reaction in SCD: the most harmful effect of transfusion
3.1 Classic situations of haemolytic transfusion reactions
Haemolytic transfusion reactions is a well-known complication in multiply transfused patients. This reaction generally results from a conflict between antigens expressed on donor RBCs and antibodies produced by the recipient. For ABO blood group incompatibilities, the antibodies are naturally occurring rather than being the result of previous exposure to the antigens. In such situations, applying ABO compatibility rules is sufficient to prevent transfusion reactions. In cases of error, the haemolytic accident is immediate, and frequently dramatic, involving intravascular haemolysis through complement activation (Membrane Attack Complex). More frequently, transfusion reactions are delayed. Such delayed reactions result from a secondary immune response in a previously transfused or pregnant patient in whom an alloantibody has developed. Screening tests are intended to detect these antibodies, which can then be taken into account by giving RBCs free of the particular antigen. In some cases, however, the alloantibody titre may be below detectable levels at the time of pre-transfusion testing and the antibody is therefore ignored. Upon repeat transfusion, alloantibodies are restimulated, with increased cytotoxic capacities, and a switch to IgG1 and IgG3 subclasses; the titre, affinity, and capacity to bind complement all increase. The haemolytic transfusion reaction develops within 4 to 6 days. Screening tests of patient plasma and direct antiglobulin tests (which characterize the in vivo sensitization of donor RBCs by the recipient antibodies) will then give positive results. Donor RBCs are mostly destroyed by macrophages expressing immunoglobulin (Ig)Fc receptors in the spleen and in the liver. Complement opsonization may synergise IgG-mediated haemolysis through a C3b receptor on macrophages. Prevention of accidents of this type is currently based on taking any previously detected antibodies into account, and avoiding alloimmunization against the major immunogenic antigens carried by RBCs. Initially, these antigens are the RH and KEL antigens; when patients become immunized, they also include FY, MNS and JK antigens.
3.2 Specific features of the delayed haemolytic transfusion reaction in SCD patients
The characteristic features of DHTR in SCD are a delayed accident (more than 6 days after transfusion), a dramatic drop in post-transfusion haemoglobin (Hb) to below pre-transfusion levels caused by destruction of both donor and recipient RBCs, the presence of SCD-related manifestations, and exacerbation haemolysis on further transfusion. Profound reticulopenia is frequent and may contribute to the drop in the Hb concentration.
Various serological features may be associated with this post-transfusion hyperhaemolysis syndrome. The antigen differences between donor and recipient can lead to patients developing reactions related to alloimmunization due to production of clinically significant antibodies to donor RBC antigens. A specific feature in SCD patients in this standard situation is the involvement of antibodies which are generally not considered to be clinically significant (anti-M, autoantibodies) [11,12]. Also, rare blood groups or variant antigens carried only by SCD individuals of African descent are associated with the production of rare antibodies which can cause DHTR. Some cases are more enigmatic, and no relevant antibody can be detected. Many case reports or series of case reports have been published illustrating that in many cases of DHTR, the hyperhaemolysis syndrome is not due to a conflict between donor RBC antigens and the antibodies in recipients. For example, in a prospective study of 49 transfused SCD patients, two patients presented with DHTR without detectable antibodies [13]. This complication is thus unpredictable: additional transfusions exacerbate existing haemolysis, and induce severe complications of the disease.
Table 2 lists most of the publications since 1984 reporting cases of DHTR. All these publications, except our previous report, describe retrospective studies and do not indicate the incidence of DHTR in SCD. Nevertheless, they illustrate the substantial variability of the immuno-hematological presentation of DHTR. They include 70 cases, 70% involving antibodies, and in half of these cases, previous alloimmunization was documented. In many cases, the antibodies had been detected previously, but were not taken into account because they were not detected in the patient serum at the time of the transfusion. Also, in half of the cases, more than one antibody was detected. The main specificities were, as expected, RH, FY, JK, and Ss (68%). Antibodies against low-prevalence antigens were also found as well as autoantibodies alone. Antibodies that are not classically clinically significant (Lea, M, weak reactivity) were found in 18% of these cases. In almost one third of these case reports, no antibodies were detected.
Case reports of DHTR since 1984. The antibodies reported in the table are those found after DHTR has occurred.
| References | Cases with no antibody | Cases with antibodies (Ab): specificities |
| de Montalembert et al., 2011 [20] | 4 | 4: M+ weak Ab; D + S; D + S + C+ e + Fy3; weak Ab |
| Win et al., 2010 [21] | 1: S + Fy3 + Jkb + auto-Ab | |
| Chadebech et al., 2009 [13] | 2 | 1: not characterized |
| Islam et al., 2010 [22] | 1: E + M + Fya + Fy3 | |
| Elenga et al., 2008 [23] | 1 | 1: S |
| Win et al., 2008 [24] | 1 | |
| Noizat-Pirenne et al., 2007 [12] | 1: auto-Ab | |
| Proudfit et al., 2007 [25] | 1: E + Fya + C + c + Jka + I + auto-Ab | |
| McGlennan and Grundy, 2005 [26] | 1: S + Jkb | |
| Talano et al., 2003 [27] | 3 | 4: E + Lea, E; E; Fyb |
| Aygun et al., 2002 [11] | 1 | 4: auto-Ab; E + I; E + Jkb + S + C; K + E + auto-Ab |
| Noizat-Pirenne et al., 2002 [6] | 1: D + HrS | |
| Win et al., 2001 [28] | 1 (or HLA) | 1: auto-Ab |
| Fabron et al., 1999 [29] | 1: c | |
| Kalayanaraman et al., 1999 [30] | 1: s | |
| Anderson et al., 1997 [31] | 1: Jsa | |
| King et al., 1997 [32] | 2 | 3: E + S; E + kpa; Fya |
| Larson et al., 1996 [33] | 1: Goa | |
| Syed et al., 1996 [34] | 4: Jkb + S + M, allo Ab; Allo-Ab; allo or auto-Ab | |
| Thornton and Sams, 1993 [35] | 1: C + Fya + E | |
| Bowen et al., 1988 [36] | 1: Fya + Fy5 | |
| Cox et al., 1988 [37] | 3: RH18; C + E; RH46 | |
| Rao et al., 1989 [38] | 1 | 2: E + C + M; Jka + E |
| Milner et al., 1985[39] | 4 | 6: allo and/or auto Ab |
| Squires et al., 1985 [40] | 1: Cob | |
| Brumfield et al., 1984 [41] | 1: E + K + Jkb + Fya + Cw + HI | |
| Total cases | 20 | 50 |
In the following sections, we consider the appearance and clinical significance of autoantibodies in DHTR, and various possible reasons both for the severity of DHTR in SCD related to antibodies, and for DHTR without detectable antibodies.
3.3 Impact of autoantibodies related to alloimmunization in SCD
Autoantibodies frequently develop with erythrocyte alloimmunization [10]. The incidence of autoantibodies among transfused SCD patients is about 6 to 10%. A similar phenomenon is also observed in cases of thalassemia, as a result of alloimmunization.
Studies in mice give some insight into the mechanism of autoantibody development, and T regulators have been implicated [14]. A frequent problem with autoantibodies is that it can be difficult to characterize relevant alloantibodies when performing antibody screening given that it is complex to cross match blood units with the patient plasma. Therefore, to ensure compatibility, time-consuming techniques are required to characterize alloantibodies resulting in delays before safe transfusion can be provided.
In addition to this technical problem, autoantibodies may shorten red cell survival and contribute to the hyperhaemolysis observed in patients presenting with DHTR. Castellino et al. documented haemolysis due to autoantibodies in four of 14 patients with autoantibodies. In all cases, direct antiglobulin tests (DAT) detected IgG and C3 on the RBC surface [15]. Cold-reactive autoantibodies of the IgM isotype are less frequent, and do not seem to be harmful. In some cases, autoantibodies were the major factor in the development of DHTR, inducing typical autoimmune haemolytic anaemia (AIHA) [12]. In many of these life-threatening cases of post-transfusion AHAI, the antibodies involved are panagglutinins. Panagglutination makes the choice of blood and identification of associated alloantibodies very difficult.
When autoantibodies are found in a SCD patient, the extent to which they are likely to affect the outcome of transfusion is unclear. Nevertheless, their presence may indicate that a process of autoimmunization is under way, and therefore, that close clinical and laboratory observation is required after any subsequent transfusion.
An important issue, in addition to the possible development of AHAI, is the difficulty in some settings to distinguish between autoantibodies and alloantibodies associated with a variant antigen carried by the patient. This diagnosis of partial antigen can be considered in patients producing an antibody against an expressed RH antigen, as variants are very frequent in SCD patients. The most frequent types are anti-D in D+, anti-e in e+ and anti-C in C+. In vitro adsorption techniques for patients who have not recently been transfused and/or DNA testing are required to distinguish between a partial antigen and a normal antigen in the carrier. An additional problem with autoantibodies in SCD is extended phenotyping, which is sometimes needed to identify the antigens against which the patient can produce antibodies. The implementation of molecular analysis in laboratories may allow this particular difficulty to be overcome.
3.4 Harmfulness of antibodies and severity of the haemolytic reaction in SCD patients
Whether an antibody against blood group antigens is clinically significant is a major issue for the management of transfusion. This issue arises when the pre-transfusion screening test is positive, but also when post-transfusion haemolysis occurs with new antibodies and further transfusions are required. The antibody titre, isotype, optimal temperature, affinity for Fc gamma receptor and C1q binding, and also the density of the target antigen on RBCs all affect the likelihood of haemolysis and therefore contribute to whether or not an antibody is harmful in a recipient. The antibodies found by investigations of post-transfusion haemolysis in SCD frequently present the characteristics of clinically significant antibodies. Nevertheless, in many cases of DHTR in SCD, some antibodies are found that are not expected from these characteristics to cause haemolysis of transfused RBCs; also, antibody-dependent haemolysis reactions in SCD patients is frequently very severe. Therefore, patient status and host factors are presumably important. The inflammatory state of SCD patients can result in effector cells being activated. Hyper-reactive macrophages may be involved, and cytokines induce the expression of FcR on effector cells, such as macrophages and NK cells [16]. Another feature that is likely to contribute to dangerous incompatibility between host antibodies and transfused RBCs is the integrity of membrane of the transfused RBCs. The host environment may exert strong oxidative stress and this may have a deleterious effect on stored RBCs, facilitating their binding to antibodies and complement, and also the destruction of sensitized RBCs by macrophages. Phosphatidylserine (PS)-exposure in donor RBCs is significantly higher after incubation with pre transfusion plasma samples from patients with acute complications of SCD, than with samples from patients in steady-state [3]. Therefore, host effects on RBCs, probably due to the inflammatory status, may potentialize the “antibody effect”.
3.5 DHTR without detectable antibodies
The biological presentation of DHTR falls schematically into two classes: the classical conflict between antigens on transfused RBCs and antibodies in the patient plasma against transfused RBCs; and the more enigmatic situation without detectable antibodies. Hyperhaemolysis without detectable antibodies is poorly understood, and consequently difficult to prevent and to treat. This syndrome has been attributed to macrophage activation, bystander haemolysis, reactive haemolysis, and the possible continuation of the painful episodic haemolysis observed for autologous RBCs. RBCs incubated in pre transfusion plasma from patients suffering DHTR without detectable antibodies show significant eryptosis and this has led to the suggestion of accelerated senescence of transfused RBCs in these cases. PS-exposure of RBCs from concentrates may account for this destruction in the SCD environment, as PS increases RBC adhesion, facilitating engulfment in the slow blood flow of the spleen, and recognition by macrophages. Various factors in plasma may also contribute to donor PS-RBC destruction, one example being PLA2, produced in SCD with acute chest syndrome (ACS) [17]. Transfused RBCs may be particularly sensitive to the patient environment because they already suffer from storage lesions. However, this syndrome is observed only in SCD patients. Another consistent feature of post transfusion haemolysis in SCD is the recurrence of vaso-occlusive crisis (VOC) as the main symptom. In mice, post transfusion haemolysis can induce VOC, and this effect is inhibited by inhibitors of inflammatory cytokine receptors [18]. A study in humans showed that PS-exposure of autologous RBCs increased during post-transfusion haemolysis [13]. PS-exposed sickle RBCs have increased adherence capacity [19] and this may be an additional explanation of the recurrence of VOC.
4 How to prevent alloimmunization and DHTR in SCD
In routine practice, prevention of alloimmunization has to take into account the shortage of compatible RBCs from donors of the same ethnic background (Fig. 1). Transfusion with RBCs matched for all immunogenic antigens is not realistic, but transfusion with leucoreduced RH/KEL-matched RBCs should be the minimum standard care, RH and KEL being the most immunogenic antigens. However, extended phenotyping of the patients should be also required to identify potentially difficult situations, and to determine in advance the antigens the patient may be immunized against. This would save time in the management of acute situations, and help when the screening test is complex to interpret because of multiple allo and autoantibodies. It would also identify cases for which appropriate blood is scarce, and allow early decisions concerning possible alternative treatments. For similar reasons, antigen variants of clinical relevance should be studied. DNA typing for common and variant antigens will became more affordable as techniques are developed. Donor typing will also improve and rare donors will be able to be found without the need for rare serological reagents. However, prevention of alloimmunization and DHTR will be always limited, whatever the tools used, if donation by individuals of African origin does not increase. Consequently, the promotion of donation in the relevant communities is a very important objective.
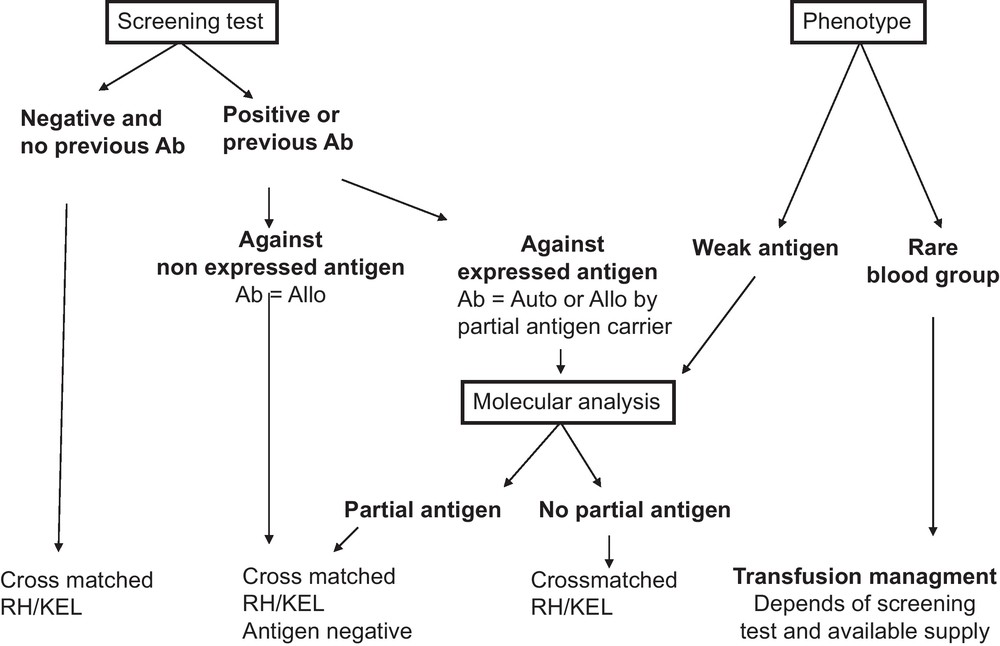
Recommendations for transfusion management in SCD patients. All recommendations are based on the results of the serum antibody screening test results and the extended RBC phenotype of the patient. For patients with no detectable antibodies, no known previous antibodies, and no abnormal RBC phenotype, transfusion of cross-matched RH/KEL compatible RBC units is recommended. When antibodies are identified at the screening test or known by history, two scenarios can be encountered: (i) the corresponding antigen is not expressed on patient RBCs and therefore the antibody is an alloantibody (Ab = Allo), and hence antigen-negative RBCs should be transfused; (ii) the corresponding antigen is expressed on patient RBCs and serological and/or molecular studies are needed to determine whether the antibody is an alloantibody produced in a partial antigen carrier or an autoantibody (Ab = Auto). If an alloantibody, and there are available donor units, compatibility should extend to other common non-expressed antigens. If an autoantibody, matching for the corresponding antigen is unnecessary. For the phenotype of the patient, two situations should be considered: (i) presence of a weak antigen, where molecular analysis will be needed to determine whether it is a partial antigen. If the patient has no detectable antibodies, transfusion with an antigen negative unit is preferred although antigen positive RBCs can also be issued, since the risk of alloimmunization in patients with weak antigens has not yet been determined (see text). However, close monitoring of possible immunization is needed if antigen-positive units are transfused; (ii) presence of a rare phenotype, where availability of high frequency antigen negative units is the determining factor for the transfusion strategy. If the patient is already immunized against the high frequency antigen, antigen-negative units are recommended for transfusion. If no rare units are available, the benefit/risk ratio of transfusion has to be considered. For patients who are not yet immunized against high incidence antigens, transfusion of antigen negative units are also recommended if such units are not in short supply. Otherwise, the standard RBCs for SCD can be issued, and the patients should be monitored closely. Given that antigen negative units for patients with rare phenotype are by definition in short supply (see text), transfusion management of these patients can become extremely challenging and we recommend that the risk/benefit ratio of alternative treatments, such as bone marrow or cord blood transplantation, is discussed with such patients.
To minimize the risk of antibody-mediated DHTR at each transfusion, all antibodies identified at any time in the patient's history, and that could therefore be re-stimulated, have to be taken into account. Consequently, well-maintained patient files are essential, as is the detection of all antibodies produced after transfusions (these antibodies may become undetectable subsequently, just before a new transfusion is indicated). A screening test should be performed between 2 to 4 weeks following each transfusion. The screening test should include the possibility of detecting antibodies against low-frequency antigens (RBC tests covering these antigens should be used), and/or systematically cross match patient plasma and the RBC of the units.
5 Conclusion
Blood groups are clinically significant in SCD, because they favour alloimmunisation, which is a critical issue in the management of transfusion for SCD and the main cause of DHTR. DHTR in cases of SCD has characteristic features that are probably related to the clinical conditions of the patients. Preventing alloimmunization by providing blood that is completely cross-matched for extended phenotypes is not realistic because of the shortage of donors of same ethnic background. Also, DHTR can occur without detectable antibodies. Therefore, improved transfusion safety in SCD patients requires both promoting donation by donors of African ancestry and continuing research into alloimmunization and post-transfusion haemolysis.
Disclosure of interest
The author declares that she has no conflicts of interest concerning this article.