

1 Introduction
In cerebral ischaemia, the reduction in the supply of glucose and oxygen to the brain leads to a complex cascade of cellular events, resulting in both acute and delayed neuronal death. Many aspects of ischaemic neurodegeneration have been demonstrated in animal models; however, these represent a complex environment in which it is not easy to dissect out the mechanisms involved in ischaemic neurodegeneration. Cell culture systems, while further from the in vivo situation, represent a more controlled microenvironment.
In organotypic slice cultures, cell stoichiometry and regional connectivity are substantially retained when compared to classical mixed neuronal and glial cultures 〚1, 2〛. In addition, hippocampal slice cultures allow both stimulation and recording on the same slice while retain a similar glutamate receptor population compared to that found in adult animals 〚3〛. This in vitro preparation is thus being regarded increasingly as a useful model for investigating anoxic/ischaemic neurodegeneration 〚4–8〛. These studies have demonstrated that fundamental aspects of ischaemic damage were replicated in organotypic slice cultures, including greater vulnerability of CA1 pyramidal cells, delayed nature of the damage and neuroprotection by glutamate-receptor antagonists 〚9〛. As glutamate-mediated mechanisms may play an integral role in the generation of ischaemic damage, extracellular glutamate concentrations will be monitored in organotypic hippocampal slice cultures to assess excessive glutamate-mediated excitation 〚10〛. In addition, evoked field potentials will be recorded as an index of slice viability following ischaemia. Finally, as ischaemia is known to be associated with neuronal and glial swelling, a non-transmitter compound, phosphoethanolamine, will be also monitored that may be an indicator of membrane disorders 〚11, 12〛.
The combination of this sensitive monitoring with insult-modelling on organotypic hippocampal cultures allowed approaching the following questions. (1) What are the temporal dynamics of extracellular glutamate and phosphoethanolamine? Microdialysates were sampled with short intervals (2 min) and their content was determined by capillary electrophoresis with laser-induced fluorescence detection (CE–LIFD). Ischaemia conditions were modelled by metabolic inhibition to induce a rapid and controllable ATP depletion: brief perfusion of potassium cyanide (KCN) in combination or not with 2-deoxyglucose (2-DG) perfusion and glucose removal to simulate anoxic, hypoglycaemic and ischaemic insults. (2) Do increases in glutamate efflux coincide with particular changes in electrical activity? Ischaemic cell death occurs after extracellular glutamate accumulation and tissue depolarisation 〚13, 14〛. (3) How do glutamate, phosphoethanolamine and field potentials insult-evoked changes respond to protective treatments with the NMDA-receptor antagonists MK-801 and D-AP5 (D-2-amino-5-phosphonovalerate)? Their effects on ischaemia-induced glutamate accumulation in hippocampus, which have been shown to be controversial in previous in vivo studies 〚15–19〛, could be well studied in our model, for a better understanding of the mechanism and pathophysiological role of enhanced glutamate levels following ischaemia. (4) Do the L-type calcium channel blocker diltiazem has protective effects? This experiment was designed to dissect out the MK-801 effects, since it has been shown that this NMDA-receptor antagonist may produce neuroprotection via additional mechanism(s), including the blockade of L-type calcium channels 〚20〛.
2 Materials and methods
Organotypic hippocampal slice cultures were prepared based on the method described by Stoppini and co-workers 〚2〛. Hippocampi were dissected out from neonatal rats (seven-day-old) and slices were placed on porous and transparent membranes, at the interface between atmosphere and the culture medium. Animals’ care and sacrifice for the preparation of cultures were in accordance with the NIH Guide for the Care and Use of Laboratory Animals and were approved by the ‘Swiss National Research Foundation’ and the ‘Office vétérinaire cantonal’. Ten- to 15-day old cultures were used for all subsequent studies. For each experiment, a visual control and electrophysiological recordings were used to assess slice culture viability: only slices having satisfactory evoked responses (amplitude greater than 0.5 mV) were selected and thus included in the results.
Combined microdialysis and electrophysiology approaches on organotypic hippocampal slice cultures were performed using a recent method described in details elsewhere 〚21〛: the multi-electrode array of a Physiocard® system 〚22〛 was used to record evoked extracellular field potentials 〚stimulation in CA3 area, recording in CA1 region, while a microdialysis probe was located between CA1 and CA3, in the apical dendritic field of CA1 to monitor extracellular levels of glutamate and phosphoethanolamine. The perfusion chamber of the Physiocard® was continuously perfused (100 μl min–1) with a modified Ringer’s solution of the following composition (in mM; referred to electrophysiological medium): NaCl 124, KCl 1.6, CaCl2 2.5, MgCl2 1.5, KH2PO4 1.2, NaHCO3 24, glucose 10, ascorbic acid 2 (pH 7.4), allowing a good survival of the culture. The microdialysis probe was perfused with artificial CSF (in mM; NaCl 145.0, KCl 2.7, MgCl2 1.0, CaCl2 2.5, NaH2PO4 0.45, Na2HPO4 1.55, pH 7.4) at a perfusion rate of 1 μl min–1. For electrophysiology, stimulation frequency was once every 15 s. Quantification of the amount of endogenous compounds in the dialysates was accomplished on a CE-LIFD system 〚23〛: glutamate and phosphoethanolamine were given fluorescent using a continuous flow derivatisation system for mixing fluorescent reagent tag (naphthalene-2,3-dicarboxaldehyde) and dialysate fractions 〚21〛. Derivatised dialysates were then automatically injected onto the CE-LIFD apparatus by on-line coupling. This automated analysis system allowed to determine glutamate and phosphoethanolamine concentrations in 2-min hippocampal dialysates.
According to the model described by Vornov and co-workers 〚8, 24〛, metabolic inhibition was used to model insults on hippocampal slice cultures: rapid inhibition of glycolysis was induced by 10-min perfusion of 5 mM 2-DG added to the glucose-deficient electrophysiological medium (hypoglycaemia), 10-min KCN perfusion (1 mM) was used to produce chemical anoxia, and simulated ischaemia (combined hypoglycaemia and anoxia) was induced using the same protocol as that described for chemical anoxia, except that the medium used during exposure to anoxia was devoid of glucose and contained 5 mM 2-DG. Cresyl violet staining 24 and 48 h after the ischaemic insult was performed to ensure that cell integrity was not affected.
Drug treatments with MK-801 (30 μM), D-AP5 (50 μM) and diltiazem (100 μM) were performed during (chemical anoxia) or throughout (20 min before, during and 20 min after) the insult paradigm (ischaemia) by infusion through the perfusion chamber of the Physiocard® 〚21, 22〛. Stock solutions were prepared in water, except for D-AP5, which was dissolved in 1 equiv sodium hydroxide. Final dilutions were performed in control- or insult-electrophysiological medium. Drug concentrations were chosen to produce high protection in hippocampal slices against anoxic/ischaemic insults 〚8, 20〛.
Time presented in the figures corresponds to real time of collection of the microdialysis fractions. Field potential amplitudes are presented as means of four consecutive recordings. Concentrations of glutamate and phosphoethanolamine as well as field potential amplitudes were expressed as percent of basal values (mean ± SEM), measured over a 20-min control period that started at least 60 min after implantations of the probe and electrodes. Statistical analysis was carried out by: (i) one-way repeated-measure analysis of variance within each group to determine the effect of insult paradigm and/or protective treatment over time, (ii) two-way repeated-measure analysis of variance to compare between treatment groups, and (iii) Student’s t test to test the existence of a relationship between the maximal glutamate or phosphoethanolamine increases and the amplitude of field potentials after 1 h. Statistical significance was set at 0.05 for all comparisons.
3 Results
3.1 Control
The average dialysate concentrations were 320 ± 90 nM for glutamate and 52 ± 8 nM for phosphoethanolamine (n = 71; not corrected for recovery), and the mean amplitude of field potentials was 0.91 ± 0.17 mV (n = 111).
3.2 Glucose deprivation
After perfusion of a 2-DG-containing (and glucose-free) medium for 10 min (hypoglycaemic model), no significant changes in glutamate and phosphoethanolamine were observed (Fig. 1A and B; P = 0.51 and P = 0.42, respectively). A small but significant increase in field potential amplitude (maximum: +23%; P < 0.01) was observed between +9 and +23 min after the beginning of 2-DG perfusion (Fig. 1C).
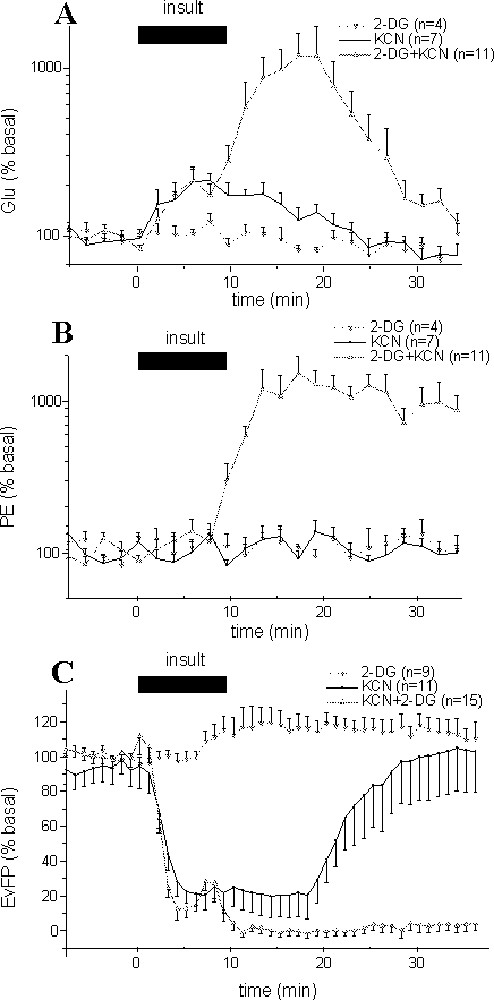
Effects of different insult paradigms on glutamate (Glu) (A), phosphoethanolamine (PE) concentrations (B) in hippocampus dialysates and on evoked field potential (EvFP) amplitudes (C). Ten-min perfusion of 2-DG (with no glucose), KCN or (2-DG, KCN and no glucose) solutions were performed, so as to simulate hypoglycaemic, anoxic and ischaemic insults. Each point represents mean ± SEM. For clarity, EvFP amplitudes are presented as means of four consecutive recordings, i.e., one point every minute.
3.3 Chemical anoxia
The perfusion of a KCN containing medium for 10 min (anoxic model) induced both a significant (P < 0.001) increase in glutamate concentrations for 16 min that peaked (+104.5%) at +6 min (Fig. 1A), and a strong decrease in field potential amplitude (P < 0.001) that was maximal from +2.5 to +16.5 min (–81%) before returning to baseline values within 25–30 min (Fig. 1C). No significant changes were observed for phosphoethanolamine (Fig. 1B; P = 0.70).
3.4 Simulated ischaemia
Combined 2-DG and KCN perfusion (and no glucose) for 10 min (ischaemic model) resulted in potentiated increases in glutamate and phosphoethanolamine concentrations, and in an irreversible decrease of evoked responses (Fig. 1). Note that despite these modifications, no cell damage was observed after 24 or 48 h, as indicated by Nissl staining (not shown).
The kinetic of glutamate increase was biphasic: during the initial phase, glutamate started to increase very early after the onset of ischaemia to reach at +2 min a steady state (+88%; P < 0.001) and stabilise for 6 min. After this short plateau, glutamate concentrations strongly rose to its maximum (× 11.9 at +18 min, P < 0.001; Fig. 1A) before returning to baseline values within 35 min. In the linear part of this large increase (from +8 to +18 min), glutamate increased at 0.43 ± 0.03 μM min–1. During the decrease (from +18 to +28 min), the rate was similar, with –0.38 ± 0.06 μM min–1. In contrast, phosphoethanolamine levels remained unchanged during the initial increase of extracellular glutamate concentrations, but started to increase from +8 min to reach a plateau at +12 min (1018% of basal values; Fig. 1B).
Various changes in field potentials were detected during the ischaemic insult (Fig. 1C). A primary decrease in the amplitude was recorded within 1–2 min after the onset of ischaemia. During this initial phase, the amplitude strongly decreased (P < 0.001) to stabilise at –86% between +1.5 and +4.75 min. Then, a transient increase was observed at ∼6 min to reach –67%, often followed by the appearance of a transient epileptiform activity (Fig. 2). Finally, a total and rapid disappearance of the field potentials occurred after 7.0 ± 0.3 min that lasted during at least one hour (Fig. 1B). It is interesting to note that the onset of the second massive glutamate increase and of the large increase in phosphoethanolamine concentration occurred just after the second field potential depression at +7 min.
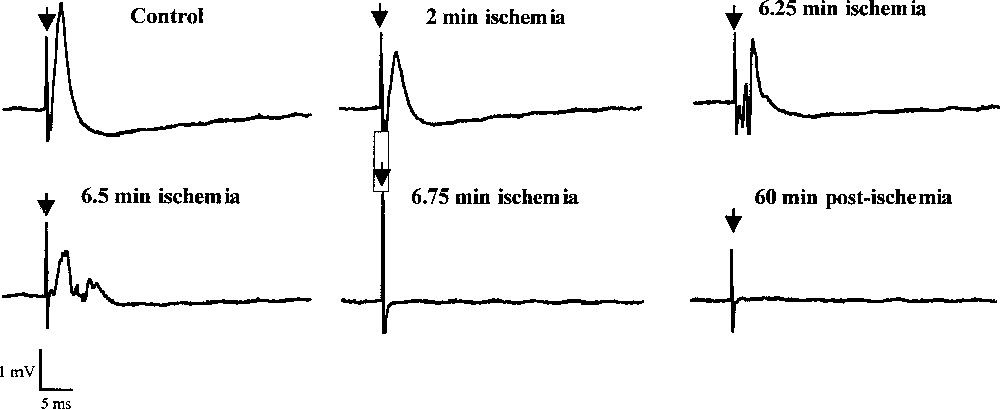
Effects of ischaemia on the CA1 evoked field potentials. Responses were evoked by stimulation in the CA3 area. Stimulation rate was once every 15 s and duration of ischaemia (induced by perfusion of KCN, 2DG and glucose-free medium) was 10 min.
3.5 Effects of drug treatments
The effects of MK-801, D-AP5 and diltiazem on extracellular glutamate and phosphoethanolamine levels as well as on field potential amplitude on their own when perfused for a 20-min period in absence of any insult are shown in Table 1: only diltiazem affected glutamate levels that were reduced to 59.4% of basal values (P < 0.01). Unlike glutamate levels, none of the drugs affected extracellular phosphoethanolamine concentrations and electrical activity on their own.
Effects of pharmacological treatments by MK-801, D-AP5 and diltiazem on the extracellular levels of glutamate (Glu) and phosphoethanolamine, and evoked field-potential amplitudes.
| Glu (% control) | PE (% control) | EvFP (% control) | |
| MK-801 30 μM | 95.9 ± 4.0 (n = 5) | 101.1 ± 13.4 (n = 5) | 100.7 ± 1.4 (n = 6) |
| D-AP5 50 μM | 81.8 ± 4.5 (n = 7) | 80.9 ± 7.6 (n = 7) | 117.9 ± 2.4 (n = 11) |
| Diltiazem 100 μM | 59.4 ± 4.8 (n = 5) | 102.4 ± 17.1 (n = 5) | 101.8 ± 1.0 (n = 8) |
3.5.1 Chemical anoxia
Administration of MK-801 and diltiazem during the 10 min perfusion of the 1 mM KCN solution totally blocked the anoxia-induced glutamate increase; in contrast, D-AP5 only partially blocked this increase (Fig. 3A). No significant changes in phosphoethanolamine concentrations were observed for any pharmacological treatment, although there were some variations between slices for D-AP5 (Fig. 3B). The recovery of field potential amplitude was similar to the control group for NMDA-receptor antagonists, but unfortunately lower after diltiazem administration, although the reason for such a difference remains unclear (Fig. 3C).
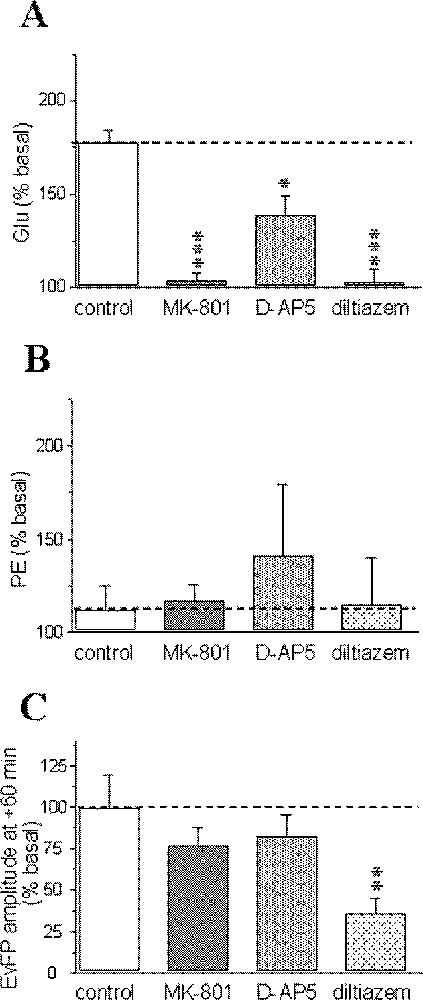
Pharmacological sensitivity of the anoxia-induced glutamate (Glu) (A), phosphoethanolamine (PE) accumulations (B) and recovery of evoked field potentials (EvFP) (C). Data correspond to maximal changes and are expressed as percentages (mean ± SEM) of pre-anoxic values for each group (n = 5–7 for microdialysis data and n = 5–12 for electrophysiology). *** P < 0.001, ** P < 0.01, * P < 0.05 vs corresponding levels in the control group (indicated by a horizontal dotted line).
3.5.2 Simulated ischaemia
The initial phase of the ischaemic-evoked glutamate increase from +2 to +8 min was highly reduced after diltiazem (+8.0%, P < 0.001), whereas no significant changes were observed after MK-801 (+69.5%, P = 0.21) or D-AP5 treatments (+116.2%, P = 0.55, Fig. 4A1). In contrast, the maximal glutamate increase occurring between +8 and +28 min was reduced by both MK-801 (+71.7% at +20 min, P < 0.001) and diltiazem (+118% at +14 min; P < 0.01), but not by D-AP5 (× 14.1 at +12 min, P = 0.74; Fig. 4A2).
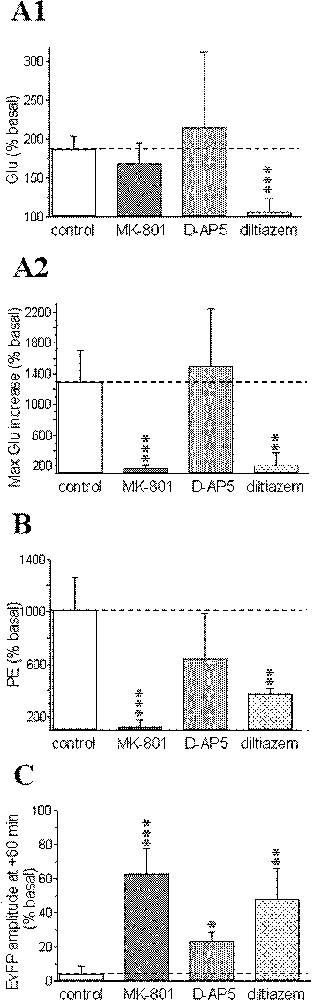
Pharmacological sensitivity of: the early (A1) and the second massive component (from +8 to +28 min; A2) of the biphasic ischaemia-induced glutamate (Glu) efflux; the phosphoethanolamine (PE) accumulation induced by ischaemia (B); the recovery of evoked field potentials (EvFP) (C). Steady-state values of early Glu accumulation (from +2 to +8 min) are presented in (A1); maximal Glu increases obtained during the second massive Glu efflux in (A2); maximal PE levels (at +30 min) in (B); and the amplitude of EvFP at +60 min in (C). Data correspond to maximal changes and are expressed as percentages (mean ± SEM) of pre-ischaemic values for each group (n = 5–11 for microdialysis data, n = 6–16 for electrophysiology). *** P < 0.001, ** P < 0.01, * P < 0.05 vs corresponding levels in the control group (indicated by a horizontal dotted line).
Phosphoethanolamine accumulation induced by the ischaemic insult displayed quite similar changes to those observed for the second phase of glutamate increase (Fig. 4B).
In parallel, field potentials first displayed similar changes to those previously reported in the control (ischaemic) group for any pharmacological treatments envisaged: primary decrease, transient increase with epileptiform activity and total disappearance of the responses. Unlike the control group however, significant recovery was observed from +25–30 min for MK-801 diltiazem D-AP5 (∼70, 47 and 25% of basal values at +60 min, respectively; Fig. 4C).
3.6 Relationships between extracellular levels of glutamate or phosphoethanolamine and electrical activity
Finally, we examined the hypothesis that the severity of the insult could be related to the extent of glutamate efflux elicited by ATP depletion in our models. For this purpose, we tested the existence of relationships between the maximal injury-evoked glutamate increase, and the amplitude of field potentials after 1 h for the anoxic and ischaemic insults. We also examined the existence of such relationships for phosphoethanolamine instead of glutamate increases. For the anoxic model, no significant correlation was found between glutamate or phosphoethanolamine increases and the recovery of electrical activity (r = 0.310, P = 0.14 for glutamate and r = 0.246, P = 0.25 for phosphoethanolamine; n = 23; Fig. 5A). In contrast, comparison between glutamate or phosphoethanolamine increases and field potential recovery for the ischaemic model yielded significant correlations (exponential decays): r = 0.488, P = 0.025 and r= 0.552, P = 0.002, respectively (n = 28; Fig. 5B).
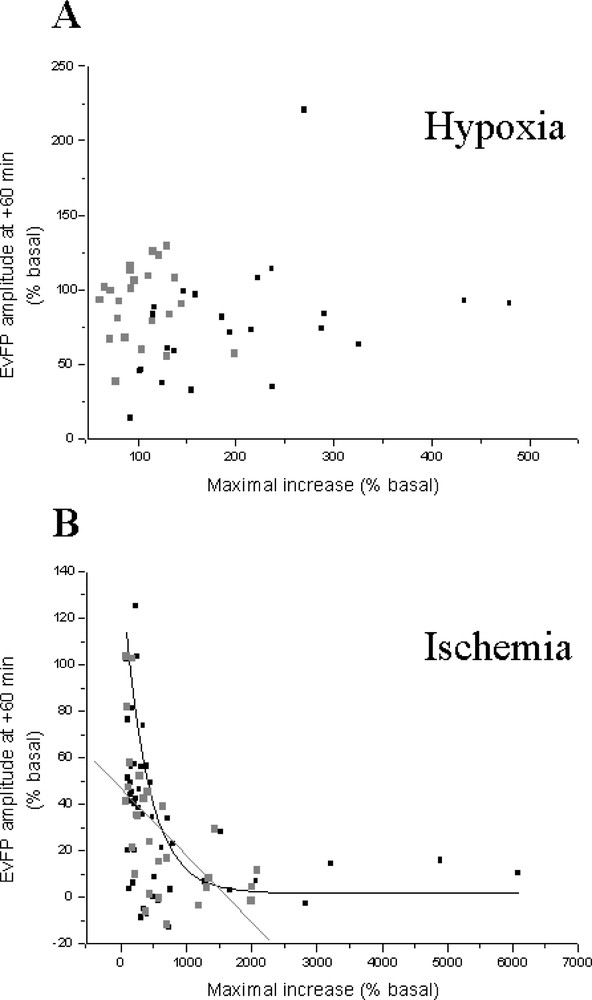
Relationship between the maximal insult-induced glutamate (Glu) or phosphoethanolamine (PE) increases and the field potential (EvFP) amplitude one hour after chemical anoxia (A) and ischaemia (B). All experiments presented in the previous figures are included.
4 Discussion
Organotypic cultures of brain slices offer a promising compromise between complex animal models and more simplified dissociated neuronal cell cultures or acutely prepared brain slices 〚1〛. By combining microdialysis monitoring with high time resolution and precise electrophysiological recording 〚21〛, we are able to detect the relative occurrence of changes in extracellular glutamate and phosphoethanolamine levels and neuronal activity in organotypic hippocampal slice cultures.
From the different ways known to induce in vitro cerebral ischaemia, metabolic blockade was preferred in the present study over oxygen, or oxygen and glucose deprivation. Substrate deprivation is difficult to achieve in tissue culture by simple removal of oxygen and/or glucose: organotypic hippocampal cultures require at least 30–60 min of incubation in a medium lacking glucose and oxygen to cause substantial neuronal injury 〚5, 7, 9〛. The present study on the changes in neuronal functioning occurring in early stages of injury required rapid effect through brief exposure and controlled conditions. Thus, we have used inhibition of oxidative metabolism and/or glycolysis induced by brief perfusion of cyanide in combination (or not) with 2-DG perfusion and glucose removal 〚8, 25〛. This model has been shown to reproduce elements of the time course, regional vulnerability, and pharmacological sensitivities of in vivo ischaemic hippocampal injury 〚8〛.
Three experimental paradigms of metabolic inhibition were tested to model cerebral ischaemia: 2-DG perfusion with glucose removal (hypoglycaemia), KCN perfusion (chemical anoxia), and a combination of both procedures (simulated ischaemia). Paradoxically, 2-DG perfusion with glucose removal induced a small and transient increase in evoked response amplitude. This increase could be related to the long-term potentiation phenomenon observed after application of 2-DG on hippocampal slices 〚26〛. The smaller effect observed in the present study could be explained by the lower deprivation time as well as by the use of culture versus acute hippocampal slices.
Such a biphasic increase in glutamate concentrations was to our knowledge never reported in hippocampus, either in vitro or in living animals, although this has been previously reported in cortex or striatum of living animals after ischaemia. Moreover, we have confirmed in our in vitro culture model characteristics of glutamate efflux occurring during cerebral hypoxia or ischaemia, including a dependence of its amplitude on the severity of the insult 〚27, 28〛, a very early increase at the onset of ischaemia 〚28〛 and a moderate accumulation in hippocampal slices after cyanide or hypoxia 〚29, 30〛. In parallel, the present observations also confirm previous findings according to which ischaemia resulted in elevated extracellular levels of phosphoethanolamine 〚11, 31〛.
As assessed by field potential recordings, changes in electrical activity observed after cerebral ischaemia are in agreement with previous findings or concepts: (1) ischaemia induced a rapid depression in field potentials and excitatory postsynaptic potentials (EPSP) within 1 min 〚32, 33〛; (2) after this initial decrease, transient epileptiform activity was observed, followed by a steep-shape abolition 〚34〛; (3) time course of changes in field potentials observed after ischaemia is in agreement with the sequence of events occurring during anoxic depolarisation; in particular, the sudden abolition of evoked responses could correspond to the rapid depolarisation of the membrane occurring after ∼7 min of combined glucose and oxygen deprivation 〚13, 35〛, and (4) the amplitude of the field potential abolition was related to the severity of ischaemia, with almost complete recovery after chemical anoxia and very poor or no recovery after ischaemia 〚27〛. It has been suggested that the extent of neuronal damage may be correlated to the time period during which the brain tissue is in a depolarised state 〚36〛: in our case, it could correspond to the time period during which field potentials were abolished. As the extracellularly-recorded anoxic depolarisation in vivo 〚37〛 and the intracellularly-recorded rapid depolarisation of membrane in acute hippocampal slices 〚35〛, the massive and sudden abolition of field potentials we observed at +7 min could be considered as a critical event of the pathogenesis of ischaemia. Moreover, we found that this event was coincident with the beginning of phosphoethanolamine accumulation and of the second massive glutamate increase at the end of ischaemia. These suggest that glutamate increase may not be the cause of anoxic depolarisation.
We observed that the NMDA-antagonists MK-801 and D-AP5 had no effect on their own on glutamate output and field potentials 〚38, 39〛. In contrast, the lack of effect of D-AP5 we observed is at variance with the increase reported by Liu and Moghaddam 〚40〛. The different experimental conditions (whole brain vs slice) might explain this discrepancy because of the lack of extrinsic inputs in slice cultures. We also found that diltiazem pre-perfusion induced a decrease in glutamate concentrations. Even if calcium influx through voltage-sensitive channels lead to glutamate exocytosis, N- and mainly P-type rather than L-type channels have been shown to play a predominant role in glutamatergic synaptic transmission in the hippocampus 〚41〛. The high concentration used in the present study, which corresponds to a neuroprotective dose 〚18〛, could explain the observed effect through non-calcium-channel-mediated mechanisms 〚42〛.
The non-competitive antagonist of the NMDA receptor MK-801 induced an important recovery of field potentials after ischaemia, which is in agreement with its highly neuroprotective properties 〚18〛. In contrast, blockade of NMDA-receptors with the competitive antagonist D-AP5, even at relatively high concentrations, was less effective in preventing the ischaemia-induced field potential depression in the hippocampus. This is in agreement with a recent study on organotypic hippocampal cultures showing that D-AP5 had weaker neuroprotective potency than MK-801 〚6〛. Finally, since it has been suggested that MK-801 may possess L-type calcium blocking properties 〚20〛, we evaluated the effects of diltiazem in protecting the organotypic slice culture preparation against metabolic ischaemia. Calcium channel blockers such as ω-conotoxins and L-type antagonists (including diltiazem) show contrasted protection against ischaemia-induced damages 〚5, 20, 34, 43〛. Our results, showing that diltiazem induced a significant recovery of field potentials after ischaemia, suggest however that this L-type calcium channel blocker may effectively possess some neuroprotective properties against ischaemic neuronal damage.
The efflux of glutamate evoked by chemical anoxia was mainly dependent on the activation of calcium channels sensitive to diltiazem and MK-801, i.e., L-type calcium channels 〚20〛. The two components of the biphasic ischaemia-evoked glutamate efflux did not show similar pharmacological sensitivities: the early component was found to be highly sensitive to L-type calcium channel blockade, but not to both NMDA-receptor antagonists tested. These results agree with an implication of calcium at a presynaptic site for glutamate release 〚44〛, as well as with the existence of an interplay between voltage-sensitive calcium channel function and rise in glutamate concentration in the ischaemic setting 〚45〛. The second massive glutamate overflow was found to be highly sensitive to both MK-801 and diltiazem, but not to D-AP5. These tend to indicate that several mechanisms, pre- and post-synaptic, may contribute to this massive extracellular glutamate efflux following ischaemia, which cannot be discriminated. Our findings on similar rates for massive glutamate increase and subsequent decrease may suggest that a single active mechanism could be mainly involved in this overflow. This might be glutamate uptake reversal followed by normal active re-uptake, since it has been shown that severe ATP depletion (which is likely to occur in our model of ischaemia) reversed neuronal uptake of glutamate in hippocampal slices 〚30〛. In addition, our data on simultaneous increases in extracellular glutamate and phosphoethanolamine levels following ischaemia may also suggest the implication of other non-active mechanisms like leakage across destabilised membranes. Phosphoethanolamine levels remained elevated later than those of glutamate, probably reflecting the absence of rapid reuptake mechanisms for this non-transmitter compound. Our observation of a pharmacological sensitivity of the massive phosphoethanolamine and glutamate increases after ischaemia to MK-801 and diltiazem suggests that these changes in membrane permeability may be the consequence of NMDA-receptor and calcium-channel-mediated events.
Equivocal data have been reported on the sensitivity of hippocampal ischaemia-induced glutamate accumulation to NMDA-receptor antagonists 〚10, 15–18, 46〛. We observe opposed effects of MK-801 and D-AP5: this may be related to the weaker neuroprotective potency of D-AP5 displayed in acute or organotypic slices when compared to MK-801 〚6, 20〛.
A significant relationship between the maximal glutamate increase and the amplitude of the field potentials 1 h after the insult was observed in the ischaemic, but not in the anoxic model. In ischaemia, the recovery of evoked responses was lesser when glutamate accumulation was high. In contrast to ischaemia where very high extracellular glutamate levels were reached, the lower increase elicited by chemical anoxia could explain the lack of correlation. Such a discrepancy between the insult-evoked glutamate efflux and the subsequent recovery of the field potential amplitude in the two insults for control- and treated-groups raises the possibility that anoxia and ischaemia induce glutamate (and phosphoethanolamine in ischaemia) accumulation in the extracellular space via different mechanisms 〚47〛. The existence of a correlation after ischaemia suggests that high extracellular glutamate accumulation could be considered as an index of neuronal damage in this in vitro model of ischaemia on organotypic hippocampal slice cultures. As recently hypothesised, neurotransmitter efflux could be a generalised consequence of ischaemia, increasing with the severity of the insult 〚37, 48〛.
In conclusion, our results show new elements on ischaemia-evoked glutamate accumulation sensitivity to some neuroprotective drugs and previously described concepts in in vitro or in vivo models of ischaemia. Multiple mechanisms, including calcium-dependent, exocytotic release, reversal of sodium-dependent membrane transporters, and changes in membrane permeability, seem to be involved in excitatory amino acid release during anoxia/ischaemia. Our in vitro anoxic/ischaemic models represent a complementary approach to complex in vivo studies because of more controlled microenvironment, more easy application of drugs, no functional vascular component and improved access and resolution. Such a sensitive in vitro detection of injury could allow new insights for screening tests of putative therapeutic molecules in cerebral ischaemia. Finally, even if such systems could be considered as simplified because slice cultures are deafferented, we could envisage to add back extrinsic projections using co-culture techniques to test their role in mediating injury, so as to dissect out in further experiments the relative importance of such elements in ischaemia.
Acknowledgements
This work was supported by the Swiss National Research Foundation (FNRS 31-49799.96 to L.S.), Chemodyne SA (Geneva), the French ‘Institut national de la santé et de la recherche médicale’ (INSERM; post-doctoral fellowship to F.R.), the Sandoz Foundation (post-doctoral fellowship to F.R.) and the ‘Région Rhône-Alpes’ (doctoral fellowship to L.B.). Special thanks to Dr Philippe Vallet (Bel-Idée Hospital, Geneva) for enlightening discussions, to Lorena Parisi for the preparation of organotypic cultures and to Agostino Maseiro for technical assistance.