

1 Introduction
We have been accumulating information on the protein-coding sequences of unidentified human transcripts for the last nine years [1,2]. Our cDNA analysis is unique in the fact that we have focused our sequencing efforts on large cDNA clones (>4 kb) encoding large proteins (>50 kDa). This approach is taken because (a) large cDNAs have not been extensively analyzed yet, (b) large proteins are often encoded by large cDNAs, and (c) large proteins are frequently involved in various mammalian-specific functions [3]. For this purpose, we constructed a set of strictly size-selected cDNA libraries which enabled us to isolate clones of the size of interest on a random sampling basis, and further selected them according to novelties of their end sequences and their protein-coding potentialities before entire sequencing [3]. The cDNA sequencing was done by a shotgun method with 5- to 10-fold sequence redundancy. The number of cDNA sequences thus identified, which are mainly derived from human brain and designated by a systematic gene code containing KIAA plus a 4-digit number, has reached 2000 to date (the number of cDNA sequences deposited to DDBJ/EMBL/GenBank is 2031 at the end of July 2003). The average size of KIAA cDNAs reported is approximately 5 kb and thus the total number of nucleotide residues sequenced is over 10 Mb. Since the number of genes encoding large proteins (>100 kDa) is expected to be less than 10% of the total number of human genes from genome analyses of other organisms, the number of KIAA genes must be significant as a set of genes producing large proteins in the brain.
As the human genome sequencing enters the final phase in which the draft sequences are converted to the finished ones, it becomes more evident that cDNA sequence data would serve as a complement to interpretation of the human genome. Furthermore, the cDNA data can offer many lines of information regarding post-transcriptional events, such as alternative splicing and RNA editing, which at present cannot be predicted in silico from the genome sequence alone. On the other hand, the genome sequence allows us to revise some errors in cDNA sequence data, if any, most of which originate from the fact that cDNAs are artificially synthesized molecules from mRNAs. Therefore, integration of our cDNA sequence data with the publicly available genomic sequence data would be an urgent and critical task for us, especially for moving beyond the identification of transcribed sequences. We describe here the current status of the Kazusa cDNA project and the results of comparative analyses of the KIAA cDNAs with the corresponding genomic sequence data. The results indicate that the integration of cDNA sequence data with the genomic sequence data will greatly help interpretation of cDNA structures, and vice versa.
2 The current status of Kazusa cDNA project
As described above, a distinct point of the Kazusa cDNA project from others is that our sequencing efforts have been focused on long cDNAs (>4 kb). However, we have taken various methods to select cDNA clones for entire sequencing. First, we used cytoplasmic RNA of a cultured myeloid cell line, KG1, as a source of cDNA templates and selected cDNA clones taking coincidence of the cDNA insert size and the corresponding mRNA size as a criterion. This criterion was expected to prevent us from contaminating our sequence data with seriously truncated cDNA. However, this preliminary experiment unexpectedly revealed that cDNA clones randomly selected in this manner did not always contain open reading frames (ORFs) which could be convincingly considered to encode functional proteins. Since we took this approach to select cDNA clones coding for unknown human proteins, the results of the preliminary experiment seriously concerned us. To solve this problem, we thus changed the primary clone selection criterion from the integrity of cDNAs to their protein coding ability. For this purpose, we took two different approaches, one based on experimental evaluation of cDNA clones in an in vitro transcription–translation system [3] while the other relied on in silico analysis of terminal sequences of large cDNA clones [4,5]. After genomic sequence data became publicly available, we could select cDNA clones in silico, which might have relatively large protein predicted in the genomic sequences lying between both of the terminal cDNA sequences [6]. As far as we examined, the experimental evaluation of cDNA clones in the in vitro transcription–translation system is the most reliable method to estimate the size of ORF in cDNA clones before sequencing. On the other hand, the in silico analysis of terminal sequences of cDNA clones could sometimes rescue cDNA clones overlooked by the experimental screening and enabled us to select cDNA clones encoding proteins with a particular domain [4,5]. The sources and clone selection methods for the cDNA clones analyzed to date are listed in Table 1. Besides these clones, we also analyzed approximately 360 human large cDNAs from spleen as a part of NEDO cDNA project (http://www.nedo.go.jp/bio/) [7,8].
Current status of Kazusa cDNA project∗
| RNA source | RNA fraction | Clone selection method | Number of analyzed cDNAs |
| Immature myeloid cell line (KG-1) | cytoplasmic | coincidence of cDNA size with the size of the corresponding mRNA | 268 |
| Brain | total cellular | coincidence of cDNA size with the size of the corresponding mRNA | 25 |
| Brain | total cellular | in vitro transcription–translation assay | 1416 |
| Brain | total cellular | in silico analysis of terminal cDNA sequences | 125 |
| Brain | total cellular | chromosomal location | 96 |
| Brain | total cellular | prediction of CDS from the genomic sequence lying between terminal cDNA sequences | 95 |
| aortic endothelial cell | total cellular | prediction of CDS from the genomic sequence lying between terminal cDNA sequences | 6 |
| total | 2031 |
∗ KIAA cDNA sequences which were deposited to DDBJ/GenBank/EMBL databases by July, 2003.
3 An overview of the results of the Kazusa cDNA project
Table 2 shows a summary of cDNA sequence data accessible in public databases as of April 2003. A consequence of clone selection based on the protein coding capacity of cDNA clones is that the mean size of proteins encoded by KIAA cDNA clones is approximately 900 amino acid residues. If we take it into consideration that about a half of KIAA cDNA clones are truncated, the mean size of KIAA gene products could be considerably larger than 900 amino acid residues. As shown in Table 2, the average number of exons in the KIAA genes is 3 times larger than that of genes on chromosome 22 while their average exon sizes are similar [9]. The characteristics of the predicted gene products of KIAA cDNAs have been analyzed, and the data are accessible through the HUGE database at our Web site (http://www.kazusa.or.jp/huge) [10]. The statistics of the occurrence rates of various protein domains in the KIAA gene products clarified that the clone selection method we took enabled us to efficiently collect genes relating to cellular communication and signaling, or nucleic acid management, or cell structure and motility. The detailed analysis data on the protein domain issue will be reported elsewhere.
Statistics of KIAA cDNA sequences1
| Average size | 5.1 kb |
| Average ORF size | 936 |
| Average size of the genomic region2 | 83 kb (19 kb)3 |
| Average number of exons2 | 16.7 (5.4)3 |
| Average size of exons2 | 383 bp (266 bp)3 |
1 The total number of KIAA cDNAs analyzed here is 2031.
2 These averages were obtained from 1522 KIAA cDNAs encoding proteins longer than 400 amino acid residues in the cDNAs which had the corresponding genomic sequence entries in a human genome database at National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/) as of April 14th, 2003.
3 The values in parentheses are those reported for genes on chromosome 22 [9].
As described below in detail, the comparison of KIAA cDNA sequences with the corresponding genomic sequences offers us a wealth of information in various aspects of transcript structures. In Table 2, the mean size of the genomic regions covered by KIAA cDNAs and the mean number of their exons are shown. According to Table 2, the average size ratio of the genomic region over the KIAA cDNA is 16.2. If this average value is valid for all the KIAA cDNAs we analyzed, 10 Mb of KIAA cDNA sequence data cover more than 160 Mb of transcribed regions on the human genome.
Since cDNA is nothing but an artificial copy of mRNA, it cannot always be free from artificial errors. In addition, because our cDNAs were synthesized from total cellular RNA as a template, some cDNAs were generated not from mature transcripts but from immature ones. In conventional cDNA cloning, these problems are circumvented by analyzing multiple clones for a single gene. However, we analyzed KIAA cDNAs on a single-clone-for-single-gene basis as in the case of conventional comprehensive cDNA analysis, mainly due to the limitation of sequencing capacity and cost. It should be kept in mind that any cDNA cloning method currently available has a risk of introduction of artifacts in cDNA sequences. In particular, if these artifacts cause nonsense mutations or frame-shift mutations, they seriously ruin the prediction of gene products from cDNA sequences. To solve this problem, we have developed an alert system for spurious interruption of protein coding regions (CDS) in cDNA [11]. When a cDNA sequence triggers this alert, a region suspected of causing CDS interruption was amplified by the polymerase chain reaction coupled with reverse transcription (RT-PCR), and directly sequenced. The direct sequencing result of the suspected region was used for revising the cloned sequence because it is expected to represent the most predominant sequence of the region in a tissue of interest. Among 2031 KIAA cDNAs shown in Table 2, 179 KIAA cDNA sequences were revised in this way. Through our cDNA project, we have learned that this evaluation step of cDNA sequences is highly critical for obtaining biologically significant data in comprehensive cDNA analysis. Moreover, based on the evaluation results of the integrity of CDSs in KIAA cDNAs, we are making efforts to tailor the cloned cDNAs to be full-length, to have longer CDSs, and/or to remove spurious CDS interruption by combination of currently available methods [12].
4 Comparison of the KIAA cDNA sequences with the corresponding genomic sequences
As described above, how to check the integrity and authenticity of cDNA sequences is a serious and general concern in a comprehensive cDNA project. In this respect, another promising solution arises from use of the human genome sequence data; the comparison of cDNA sequences with the predicted transcript sequences from the genome allows us to evaluate the integrity and authenticity of the cDNAs. Because a huge amount of the human genome sequence data is publicly available, this becomes practicable now. For this comparison, we used only the finished data of the human genome because the draft sequences contained a significant amount of ambiguity in sequence.
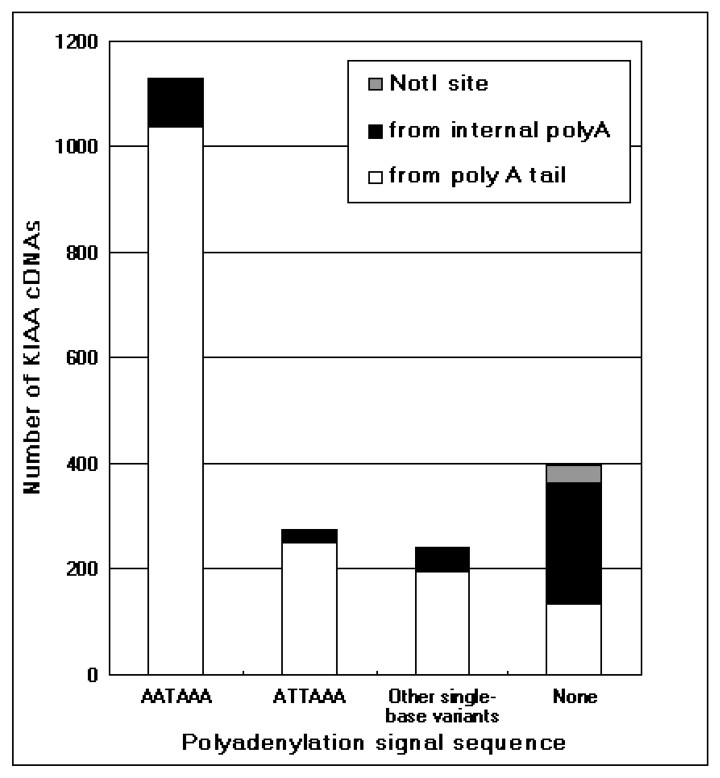
The integrity of 3′-end sequences of the KIAA cDNAs was first examined as described by Beaudoing et al. [13]. As shown in Fig. 1, 20.7% of KIAA cDNAs were found to miss 3′-end sequences. The comparison of the cDNA sequences with their corresponding genomic sequences showed that these cDNAs were synthesized with the oligo-dT primer annealing to an internal A-stretch, frequently in a repetitive sequence like Alu sequence. Coincidentally, these cDNAs were devoid of polyadenylatoin signal sequence at their 3′-ends as already described by Beaudoing et al. [13].
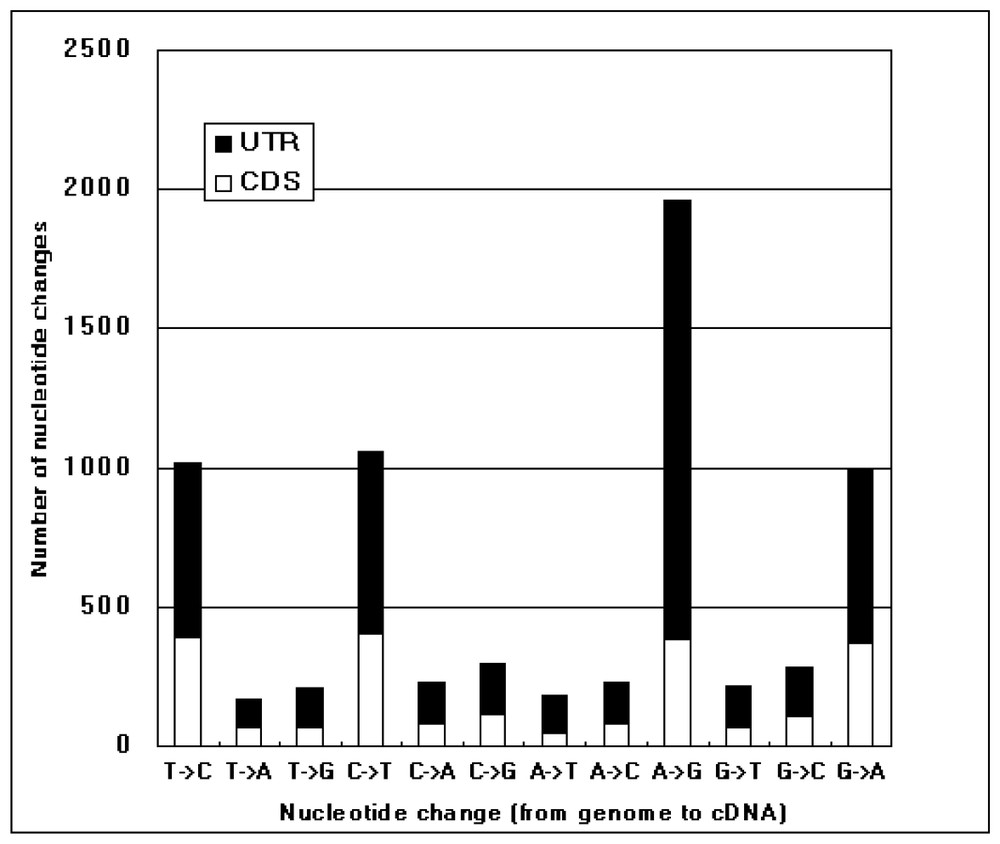
Comparison of the KIAA cDNA sequences with the corresponding genomic sequences. KIAA cDNA sequences were compared with their corresponding genomic sequences. In this figure, only single nucleotide differences between them are accounted. Types of nucleotide differences are shown along the horizontal line as (the residue in the genomic entry) → (the residue in the KIAA cDNA sequence). The nucleotide differences that occurred in CDS (assigned by GeneMark program [11]) and untranslated region (UTR) are displayed as open and filled bars.
By carefully comparing KIAA cDNA sequences with the genome sequence data, we found a considerable number of discrepancies between their nucleotide sequences. Fig. 2 shows the statistics of the nucleotide changes between KIAA cDNA sequences and the corresponding genomic ones. The observed nucleotide differences between the cDNA and genome occur at a rate of one every 1.4 kb, and are expected to be explained by one of the following causes: reverse transcription error, genomic polymorphism, RNA editing, and sequencing error. As far as we examined raw sequence data, no sequencing error was detected in these nucleotide positions. On the other hand, according to previous studies, the error rate of reverse transcriptase (MMLV reverse transcriptase) is approximately one every 40 kb [14]. Thus, these two causes are unlikely to account for most of the observed nucleotide differences. Because the occurrence rate of single nucleotide polymorphism (SNP) is known to be approximately one every 1 kb [15], SNP is the most likely cause of the observed sequence differences. In addition, the predominance of transitions over transversions is well explained by assuming that these nucleotide differences resulted from SNP. However, the predominance of A-to-G changes (from genome to cDNA) is difficult to explain by SNP (Fig. 2). In this respect, this reminded us that brain, from which most of KIAA cDNAs were derived, was reported to be rich in adenosine deaminase activity specific to double-stranded RNA (ADAR) [16]. ADAR is known to convert adenosine to inosine if adenosine is present in double-stranded RNA and the resultant inosine residue is reverse-transcribed to G in cDNA [17]. Thus, the action of ADAR could be a plausible explanation of the predominance of A-to-G changes in KIAA cDNAs. When we closely examined the observed A-to-G changes, it was noticed that the A-to-G changes were frequently clustered in a relatively short region in particular KIAA cDNAs (Table 3). If the A-to-G changes listed in Table 3 are excluded from counting the nucleotide differences, the occurrence rate of the remaining A-to-G changes becomes very close to those of other transitional changes observed. Interestingly, most of the regions listed in Table 3 consist of Alu elements. In particular, two regions including many A-to-G changes in single KIAA cDNAs listed interestingly correspond to two Alu elements inversely oriented. Such a structure is most likely to induce a stable double-stranded form of mRNA in vivo and thus explained why these regions were heavily modified by ADAR. It is well known that RNA editing has significant biological consequences in some cases, in which it results in critical changes of CDS. As all the observed A-to-G changes shown in Table 3 were found in untranslated regions in cDNA, the biological implication of the RNA editing in Alu elements has yet to be determined. Interestingly, an independent study of the identification of inosine-containing mRNA in human also reveals that most A-to-I modifications in human mRNA occur in untranslated regions [18]. Comprehensive cDNA analysis might help in uncovering the general biological meaning of RNA editing in the future.
Integrity of the 3′-end of the KIAA cDNAs. The integrity of the 3′-end of each of the KIAA cDNAs, which have the corresponding genomic sequence entries in a human genome database of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov/), was assessed according to the previous report [13]. In brief, the KIAA cDNA sequences were examined in terms of canonical polyadenylation signals (AATAAA, ATTAAA, and other single-base variants of AATAAA) usually observed within 30-nucleotides upstream of the 3′-end of the complete cDNA. In addition, the appearance of a poly(A) stretch within 10 nucleotides just downstream of the 3′-end of the cDNA on the genome was also checked to ascertain whether the cDNAs synthesized by internal priming were primed from an internal poly(A) stretch or actual poly(A) tail. Some cDNAs without any polyadenylation signal sequence have a Not I site at the 3′-end. These 3′-end Not I sites most likely originate from internal Not I site of cDNAs. The number of these clones is indicated as ‘Not I site’ in this figure.
KIAA cDNAs containing multiple A-to-G changes in a short region
| KIAA | Region | Number of A-to-G | Region |
| No. | (nucleotide residues) | changes | size (bp) |
| 0090 | 5264–5464 | 6 | 201 |
| 0134 | 4181–4380 | 5 | 200 |
| 0345∗ | 5152–5463 | 9 | 312 |
| 0345∗ | 6137–6468 | 14 | 332 |
| 0359 | 4475–4774 | 12 | 300 |
| 0412 | 3498–3781 | 6 | 284 |
| 0435 | 687–1014 | 14 | 328 |
| 0446 | 1275–1498 | 5 | 224 |
| 0485 | 3839–4186 | 11 | 348 |
| 0502 | 5254–5632 | 12 | 379 |
| 0503 | 2263–2593 | 19 | 331 |
| 0504 | 6232–6436 | 8 | 205 |
| 0621∗ | 5066–5367 | 11 | 302 |
| 0621∗ | 6119–6416 | 10 | 298 |
| 0628 | 5236–5514 | 14 | 279 |
| 0706 | 4596–4798 | 6 | 203 |
| 0739 | 3965–4164 | 5 | 200 |
| 0752 | 3110–3309 | 5 | 200 |
| 0754 | 4325–4721 | 13 | 397 |
| 0818 | 4172–4371 | 5 | 200 |
| 0825 | 5480–5900 | 26 | 421 |
| 0831∗ | 2820–3144 | 13 | 325 |
| 0831∗ | 3605–4008 | 16 | 404 |
| 0837 | 4612–4817 | 7 | 206 |
| 0884 | 3584–3783 | 5 | 200 |
| 0889∗ | 1689–1995 | 9 | 307 |
| 0889∗ | 2495–2730 | 6 | 236 |
| 0983∗ | 3067–3272 | 6 | 206 |
| 0983∗ | 3438–3679 | 7 | 242 |
| 0983∗ | 3895–4246 | 13 | 352 |
| 0983∗ | 4467–4733 | 10 | 267 |
| 1001 | 4050–4372 | 8 | 323 |
| 1048 | 3236–3753 | 17 | 518 |
| 1142∗ | 3419–3744 | 13 | 326 |
| 1142∗ | 4017–4302 | 7 | 286 |
| 1143 | 3615–3985 | 12 | 371 |
| 1262 | 5433–5729 | 16 | 297 |
| 1271∗ | 2939–3269 | 8 | 331 |
| 1271∗ | 3925–4256 | 19 | 332 |
| 1314∗ | 27–325 | 8 | 299 |
| 1314∗ | 865–1190 | 14 | 326 |
| 1314∗ | 1428–1663 | 7 | 236 |
| 1324∗ | 2404–2702 | 6 | 299 |
| 1324∗ | 2772–3081 | 10 | 310 |
| 1324∗ | 4944–5143 | 5 | 200 |
| 1353 | 382–736 | 10 | 355 |
| 1398 | 1580–1892 | 6 | 313 |
| 1497 | 22–493 | 16 | 472 |
| 1559 | 3768–4020 | 7 | 253 |
| 1615 | 2097–2296 | 5 | 200 |
| 1647 | 472–671 | 5 | 200 |
| 1649 | 3964–4163 | 5 | 200 |
| 1658 | 4324–4630 | 9 | 307 |
| 1659∗ | 3649–3897 | 6 | 249 |
| 1659∗ | 4022–4221 | 5 | 200 |
| 1661 | 5647–5986 | 11 | 340 |
| 1672 | 4816–5221 | 12 | 406 |
| 1791 | 292–554 | 9 | 263 |
| 1792 | 594–927 | 9 | 334 |
| 1829∗ | 3110–3475 | 12 | 366 |
| 1829∗ | 4210–4527 | 17 | 318 |
| 1868 | 3480–3704 | 5 | 225 |
| 1881 | 2442–2642 | 5 | 201 |
| 1919∗ | 3326–3594 | 8 | 269 |
| 1919∗ | 4499–4698 | 5 | 200 |
| 1948 | 32–234 | 6 | 203 |
| 1955 | 3075–3339 | 7 | 265 |
| 2003∗ | 3128–3343 | 7 | 216 |
| 2003∗ | 3722–4027 | 8 | 306 |
| 2016 | 3577–3923 | 12 | 347 |
∗ These KIAA cDNAs contained multiple regions rich in A-to-G changes.
Besides these interesting observations described above, integration of the cDNA and genomic sequence data allows us to evaluate the integrity of the 5′-end of KIAA cDNAs on a basis of predicted gene structure by a computer program. When the computer program predicts the presence of a further upstream region to the cDNA, we have tried to check it experimentally as far as possible. Moreover, the size of CDS in a cDNA can be assessed on the basis of the results of in silico gene prediction before sequencing of entire cDNA inserts, only if only terminal sequences of cDNAs can be mapped on the genome. This enables us to select cDNA clones having a long ORF in silico prior to sequencing of their entire region [6].
5 Concluding remarks
Our ultimate goal is to bridge between the human genome and the proteome through transcriptomic analysis. Because the complete human genome sequence will become available in the very near future, we consider it time to move beyond the identification of transcribed sequences in human. The Kazusa cDNA project aims to provide research communities in the fields of medical science, pharmaceutics, and basic biology with important resources of human long transcripts. The resources include not only the sequence information but also plasmid cDNAs as indispensable reagents for functional genomics. Furthermore, we are planning to prepare a set of antibodies raised against KIAA gene products. Because other cDNA analysis groups are also planning to offer similar resources derived mainly from relatively small cDNAs to the scientific community worldwide, the integration of our cDNA collection with theirs would greatly contribute to progress in comprehensive understanding of human transcriptome and eventually of human proteome. Toward this end, however, there still remain many technical problems in cDNA analysis. For example, we do not have any method to enable us always to isolate perfect copies of transcripts in terms of 5′-end, 3′-end, and their internal sequence, and any convincing way to systematically discriminate copies of non-functional transcripts from those of functional ones. Although various methods have been reported to solve these problems, none of them has turned out to be satisfactory in a practical sense. However, when we intend to use these cDNA clones for functional analysis of genes, it is highly critical to establish whether or not the produced proteins have authentic primary structures. If they do not have their authentic structures, the obtained data in functional experiments would be ruined and become less useful. Therefore, we are carefully evaluating the integrity and authenticity of KIAA cDNAs, verifying them experimentally, and, if necessary, revising them [12]. As we enter the end game of discovery of unknown human genes, more efforts have been made in this process than before. We believe this is a very basic process to move beyond the identification of human long cDNAs.