

1 Introduction
Suberin is a natural aliphatic-aromatic cross-linked polymer located in plant tissues where it favours protection from environmental damage. Suberin is, indeed, located in various parts of plants: roots, leaves, periderms from tubers, and in the outer bark of many higher plants. Although several models have been proposed [1,2], suberin structure remains unknown since in situ polymer analysis is hard to perform and available techniques such as 13C solid NMR provide only holistic data about the structure [3]. Furthermore the association of suberin with other plant cell-wall polymers such as lignin or cellulose has not yet been clearly established. However, suberin depolymerisation has allowed one to identify several aliphatic and aromatic monomers. Thus, many studies [4–7] based on various extraction protocols have described some of these aliphatic units: fatty alcohols, fatty acids, long chain ω-hydroxyacids, -diacids. These monomers may also have unsaturations as well as epoxyde or diol groups. The aromatic compounds isolated from suberin are close to those observed in lignin (coniferyl, synapilic and coumarylic alcohols) but acidic forms are mainly found in suberin [2,8].
Here we used different methods to depolymerise suberin from cork cropped from Quercus suber L. cultivated in the Maures (Var, France). The cork industry in Mediterranean regions is mainly developed in Portugal, which produces 50% of the world production. In the South of France, the industrial transformation of cork is linked to the production of furniture, isolating materials, local craftwork products. Recently, molecules extracted from cork have aroused interest in the cosmetic industry: in this context we have produced cosmetic-active extracts which have a smoothing effect on skin [9].
Here we tested various methods of pre-extraction and extraction in order to select and to optimise a methodology of extraction and to contribute to the description of this polymer. Methanolysis as well as hydrolysis under various experimental conditions were performed and molecules from these different extracts have been identified using Gas-Chromatography-Mass-Spectrometry (GC-MS).
2 Material and methods
Plant material. Cork samples (reproduction cork collected at the second crop) were collected from Quercus suber L., provided by ‘Melior Liège’ (Fréjus, Var, France). They were grounded and sieved (0.5 mm diameter) before extraction.
Extraction of the wax fraction. 5 g of cork powder were mixed with 750 ml of the solvent tested (hexane, dichloromethane, chloroform, ethyl acetate, acetone and ethanol). The wax fraction was then saponified in 15 volumes of KOH 2 M dissolved in water, and refluxed for 4 h. Both saponified and unsaponified wax fractions were analysed by GC-MS as described in paragraph GC-MS analysis.
Cork depolymerisation. Pre-extraction of cork: cork was pre-extracted sequentially in chloroform, ethanol and water, air-dried and used as such for the following reactions. All the experiments of pre-extraction and extraction were performed twice.
Alcalin methanolysis: 5 g of pre-extracted cork were mixed with ten volumes of sodium methanoate, 3% in methanol, were added. The mixture was refluxed for 4 h then acidified to pH 5 with sulphuric acid 47.5% [10] and extracted with the same volume of chloroform for 1 h. The organic fraction was washed twice with 100 ml of distilled water, the solvent was evaporated and the extract was finally dried over anhydrous sodium sulphate.
Acid methanolysis: 5 g of pre-extracted cork were added to 750 ml of chloridric acid, 0.4 M in methanol and stirred for 4 h at 80 °C. The mixture was then extracted as described above.
Basic hydrolysis: 5 g of pre-extracted cork were added to 750 ml of KOH dissolved in water or ethanol and refluxed for 4 h. The following KOH concentrations were tested: 1, 2 or 5 M. The mixture was then extracted as described above.
Acidic hydrolysis: 5 g of pre-extracted cork were added to 750 ml of chlorydric acid in methanol or water and refluxed for 4 h. Different chlorydric acid concentrations were tested: 1, 2 or 5 M. The mixture was then extracted as described above. The same experiment was performed at room temperature or refluxed.
After each extraction, the volatile fraction was obtained from the polymeric fraction by distillation with temperature ranging from 100 to 150 °C.
GC-MS analysis. Ethylic esters were obtained by mixing 60 mg of the extract with 2 ml of BF3, 14% in methanol and 2 ml of anhydrous methanol. The mixture was incubated at 100 °C for 2 h and then extracted with 8 ml of chloroform/distilled water (50/50). The solvent is evaporated and the sample was sylilated at 50 °C for 2 h using N,O-bis-trimethylsilyltrifluoroacetamide (BSTFA) and dimethyl formamide (Shanchun et al., [26]). 1 μl was directly injected into the gas chromatography. GC-MS was performed on an Agilent (Agilent, DE, USA) Gas chromatograph 5973N interfaced to an Agilent MSD II detector. The chromatograph was equipped with a HP5 capillarity column (30 m × 0.25 I.D., 0.23 μm film thickness, J&W Scientific, CA, USA). The injection was performed at 300 °C in the split less mode and helium was used as carrier gas with a constant flow rate of 1 ml/min. A temperature gradient ranging from 150 °C to 325 °C was realized (4 °C/min). Electron impact mass spectra were measured at acceleration energy of 70 eV.
3 Results and discussion
3.1 Extraction and partial characterisation of waxes from cork
Most of studies on cork have used pre-extractions with different solvents in order to eliminate molecules which are not covalently bound to the polymer. This fraction is mainly composed of waxes. Different solvent were used: hexane, dichloromethane, chloroform, ethyl acetate, acetone and ethanol. Extraction yields were calculated as g of extract (dry weight)/g of cork (dry weight). The best extraction yield was obtained with chloroform (6.5 ± 0.5%) while the weakest were obtained when polar solvents (acetone and ethanol) were used (respectively 2.5 ± 0.2% and 3.2 ± 0.3%). Furthermore, extraction with these solvents may lead to polar molecule extraction such as phenols. These results are similar to those obtained previously [5]. GC-MS analysis has allowed us to identify several molecules before and after saponification of the wax extract. We identified three different triterpens from the samples extracted with chloroform: friedelin, friedelanol and betulin, with friedelin as the major compound (Table 1). These molecules were identified by their base peak m/z as well as by retention time and fragment m/z [11–16]: for friedelin, base peak m/z 426, fragment m/z (%) 341 (10), 302 (36), 205 (57), 123 (84), 109 (77), 95 (91), 81 (74), 69 (100), 55 (77) for friedelanol (sylilated) base peak m/z 500, fragment m/z (%) 485 (23), 410 (7), 395 (8), 347 (26), 321 (14), 305 (17), 257 (16), 237 (71), 231 (36), 197 (31), 147 (38), 121 (56), 95 (100), 62 (66), 50 (68) and for betulin, base peak 442 m/z and fragment m/z (%) 363 (10), 234 (22), 207 (48), 189 (100), 175 (45), 161 (32), 135 (74), 107 (58), 95 (79), 81 (68), 69 (50), 55 (61), 41(60). In this study, cerin has not been identified in the wax fraction. Previous works [17] have shown that cerin was the less abundant terpen in waxes of cork from various sites in Spain and that the proportions of terpens (friedelin, betulin and cerin) varied depending on crop localisation. The results found here may be explained by the geographic origin of our samples: Conde et al. [17] have worked on reproduction cork which has been collected from various sites in Spain while we studied cork collected in South Eastern France. Friedelin and betulin have been extracted from cork and from various other tissues of higher plants as reported by previous studies [12,15,18,19]. However, friedelanol has never been isolated from cork before. This molecule has been found in waxes from bell pepper [13] or in leaves and root bark of Celastraceae and of Hippocrataceae respectively [12] as well as in the rhizomes of Polygonaceae [14]. This molecule, which biosynthesis requires oxidosqualene, is known to be a precursor of friedeline [12].
Relative composition of waxes extracted with chloroform and saponified under neutral or acid conditions (neutral and acid fractions, respectively)
| Molecules extracted | Molecule/chloroformic extract (%, w/w) |
| Neutral fraction | |
| Triterpenes | 52 |
| Friedeline | 30 |
| Friedelanol | 12 |
| Betuline | 10 |
| Fatty alcohols | 10 |
| Sterols | 8 |
| Non-identified | 18 |
| Acid fraction | |
| Fatty acids | 4 |
| Diacids | 8 |
| 1,22-docosanedioic acid | 2 |
Saponification of the wax extract led to the hydrolysis of esters and the acid or neutral fractions obtained were analysed using GC-MS. The acid and neutral fractions represent 12 and 88% of the wax extract respectively (Table 2). Furthermore, we showed that molecules extracted under acid conditions were monoacids (palmitic, thapsic, stearic, eicosanoic, docosanoic, tetracosanoic acids) and diacids (eicosanedioic, docosanedioic, tetracosanedioic acids) while extraction under neutral conditions led to the identification of long chain alcohols (eicosanol, docosanol, tetracosanol) as well as sterols and triterpens (β-sitosterol, stigmasterol, friedelanol, friedelin and betulin). These results seem to show that sterols such as β-sitosterol and stigmasterol may be esterified with fatty acids since these molecules have not been directly identified after extraction with chloroform but are observable after saponification.
Relative composition of suberin monomers (%, w/w) using different conditions of extraction
| Suberin monomers | Basic methano lysis | Acidic methano lysis | Acidic hydrolysis [HCl] | Basic hydrolysis in ethanol [KOH] | Basic hydrolysis in water [KOH] | ||||||
| 1 M | 2 M | 5 M | 1 M | 2 M | 5 M | 1 M | 2 M | 5 M | |||
| Fatty alcohols | 2.4 | – | 1 | 0.7 | 3.9 | 2.6 | 2.4 | 4.8 | 1.9 | 2.5 | 2.8 |
| Fatty acids | 0.4 | – | 0.5 | 0.5 | 1.8 | 1.2 | 0.3 | 0.2 | 0.9 | 1.5 | 0.5 |
| Fatty diacids | 15.8 | 10.5 | 10 | 14.6 | 9 | 16.2 | 9.2 | 9.3 | 17.9 | 20 | 21.1 |
| 1,18-octadecen-9-dioic acid | 5.3 | 4.5 | 5 | 4.5 | 3.1 | 5.5 | 2.5 | 2.3 | 6.7 | 11 | 9.2 |
| 1,22-docosanedioic acid | 3 | 3.4 | 3.5 | 3.5 | 3.4 | 6.5 | 3 | 2.6 | 4.8 | 5.1 | 5 |
| Monohydroxylated fatty acids | 41.9 | 44.6 | 48 | 47.5 | 37.6 | 73.8 | 80.2 | 80.3 | 74.1 | 76 | 71.8 |
| 18-hydroxyoctadecen-9-oic acid | 19.9 | 14.1 | 15 | 13.5 | 8.3 | 29.9 | 26.9 | 30.8 | 24.3 | 26 | 28.5 |
| 22-hydroxydocosanoic acid | 11.9 | 18.9 | 23.2 | 16.9 | 20.2 | 43.9 | 41.4 | 45.9 | 34.6 | 42 | 32.1 |
| 9,10,18-trihydroxyoctadecanoic acid | 14.3 | 6.8 | 1.4 | 1 | 6.5 | 3.7 | 5.9 | 3 | 3.1 | 3.5 | 2.3 |
| 9,10-dihydroxy-1,18-octadecanedioic acid | 7.6 | 0.8 | 1.4 | 1 | 7.5 | 2.5 | 2 | 2.3 | 2.1 | 3.5 | 1.5 |
| 9,10-epoxy-18-hydroxyoctadecanoic acid | 11.9 | 18.8 | 18.6 | 17.3 | 14.8 | – | – | – | – | – | – |
| 9,10-epoxy-1,18-octadecanedioic acid | 6.8 | 18.3 | 19.5 | 19.5 | 18.9 | – | – | – | – | – | – |
The identification of certain diacids in the chloroform extract which has been saponified is in agreement with Lopes et al. [20]. The authors have shown that some of them (C16:0, C18:0, C20:1) were easily extracted from cork using alcalin methanolysis with a sodium methanoate concentration of 0.01 M. Thus the localisation of certain diacids in suberin structure remains unclear: they may be either covalently linked to suberin but easily extractable or free in the cork structure and extractable without hydrolysis.
3.2 Partial depolymerisation of cork suberin
In most of previous studies, alcalin methanolysis has been extensively used in order to depolymerise suberin. Here, cork suberin was partially depolymerised using various methodologies (methanolysis, alcalin or acidic hydrolysis under different experimental conditions) to test whether these techniques lead to different yields and/or different molecules extracted. Whatever the technique used, three fractions were obtained in various proportions (Fig. 1): a polymeric fraction, a depolymerised fraction which contains some suberin monomers and a volatile fraction. Both depolymerised and volatile fractions were analysed by GC-MS.
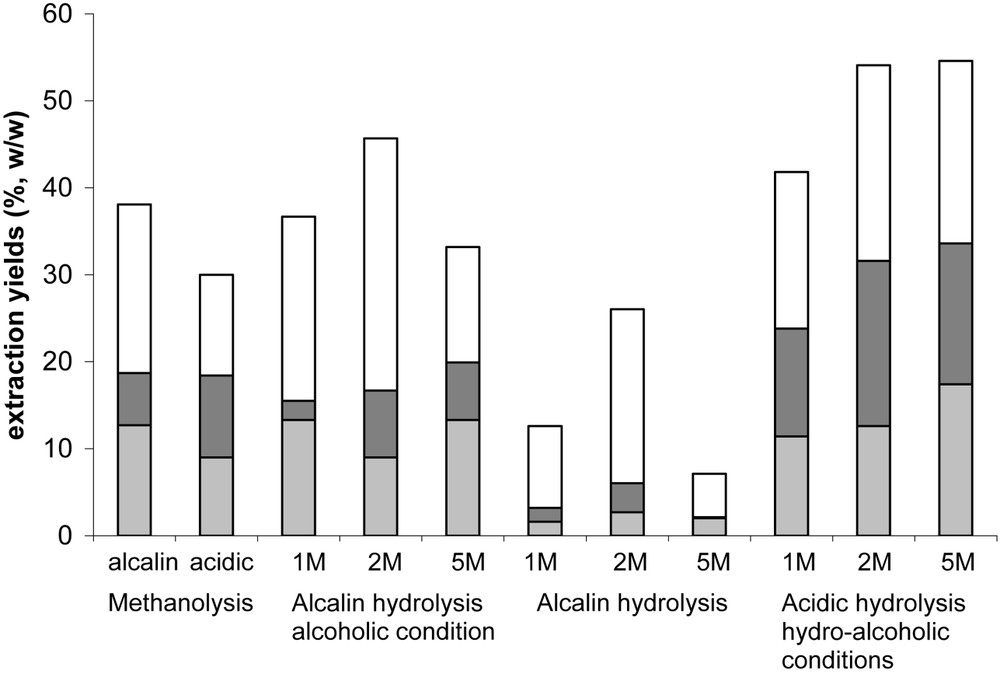
Percentages of the different fractions obtained using different methodologies of extraction: monomeric (), polymeric (□) and volatile (■). Concentrations in KOH (alcalin hydrolysis) or HCl (acidic hydrolysis) are specified.
The quantification of suberin monomers was performed after calibration of GC-MS system as described by Lopes et al. [20]: only one monomer (docosanoic acid, docosanol, 16-hydroxyhexadecanoic acid and hexacosanedioic acid) of each main chemical group was used for calibration since all the monomers from suberin are not commercialised. The identification of the molecules was performed using both software NIST 05, Software Version 2.0 (Agilent Technologies, CA, USA) and literature [7,20–22].
Depolymerisation yields were higher using basic or acidic hydrolysis in alcoholic medium. The optimum concentrations of KOH and HCl for depolymerisation were 2 M. 5 M for both KOH and HCl may be considered as drastic conditions which may provoke a major degradation of monomers. Hydrolysis and methanolysis led to the same monomers observed in previous studies [4,7,23]: ω-hydroxylated acids, epoxy acids and diacids are the main components respectively about 40–80, 20–30 and 10–15% depending on the methodology used. Alcalin hydrolysis in alcoholic medium allowed the best extraction yields for monohydroxyacids (about 80%) with 22-hydroxydocosanoic acid as the main compound (Table 2).
Since monomer proportion was the highest using alcalin hydrolysis in aqueous medium (low extraction yields for polymerised and volatile fraction), we tested room temperature as another experimental condition in order to obtain non-degraded monomers. We found that alcalin hydrolysis using room temperature gave very interesting results since similar yields of depolymerisation were obtained compared to reflux conditions: 25 and 22%, respectively. Furthermore the extract composition varies depending on the methodology used: when extraction was performed at room temperature, higher yields of extraction were observed for dihydroxyacids or trihydroxyacids while ω-hydroxy fatty acids were found in a less extent (Table 3). These experimental conditions seem to be less drastic and favour the extraction of certain molecules such as polyhydroxylated acids. Furthermore, non-oxygenated diacid extraction was more favoured at room temperature than at reflux (32 and 22%, respectively), which can be correlated with our previous results where certain diacids were found to be directly extractible with chloroform.
Relative composition of suberin monomers using acidic hydrolysis at room temperature (RT) or at reflux
| Experimental conditions | ||
| Suberin monomers | RT | Reflux |
| Fatty acids | 0 | 0 |
| Fatty alcohols | 1.5 ± 0.1 | 2.5 ± 0.15 |
| dicarboxylic acids | 32 ± 2 | 20 ± 2.5 |
| 1,18-octadecen-9-dioic acid | 9.5 ± 0.3 | 11 ± 1 |
| 1,22-docosanedioic acid | 3.9 ± 0.15 | 5.9 ± 0.2 |
| ω -hydroxylated fatty acids | 53.5 ± 2.4 | 76 ± 3.8 |
| 18-hydroxyoctadecen-9-oic acid | 22 ± 1.4 | 26 ± 2.1 |
| 22-hydroxydocosanoic acid | 16.5 ± 1.3 | 42 ± 2.5 |
| 9,10-dihydroxyoctadecane-1,18-dioic acid | 6 ± 0.8 | 3.5 ± 0.3 |
| 9,10,18-trihydroxyoctadecanoic acid | 7 ± 1.2 | 3.5 ± 0.7 |
The polymeric fraction found here has been described before [20,24]. This fraction is probably due to partial depolymerisation of suberin.
However the volatile fraction pointed out in this study has never been shown. The proportion of this fraction is greater when acidic hydrolysis in alcoholic medium was used. Its composition has been analysed by GC-MS after both acidic and basic hydrolysis: low molecular-weight molecules which are probably resulting from the transformation of the aliphatic or aromatic compounds of suberin, were identified (Table 4). For instance, the three hydroxyacids identified (glycolic, 4-hydroxybutanoic and 6-hydroxyhexanoic acid) are probably formed by β-oxidation of long chain hydroxyacids while benzyl alcohol, benzoic acid and anisole would come from the transformation of aromatic compounds from suberin or lignin. Interestingly, 2,6-heptanediol has been identified in this fraction as the major molecule (20–40%) and for each extraction tested. Because of its structure, this molecule can hardly be a result of chemical degradation (no carbonyl and/or carboxylic functions). 2,6-heptandiol may be a natural compound which may come from the biological transformation of various oxygenated acids from suberin. For instance, nonadioic acid has already been identified in suberin by Lopes et al. [20] and may be an oxidative product of this transformation. Beta-oxidation and decarboxylation of nonadioic acid may lead to heptanedione which may be reduced in 2,6-heptanediol. Thus, this molecule may be either a product of degradation from plant metabolism or a structural compound of suberin since high amount can be found and because of its diol structure.
Relative composition of the volatile fraction extracted under different experimental conditions
| Molecules identified | Molecules/extract (% w/w) | ||
| Alcalin hydrolysis | Acidic hydrolysis (hydro-alcoholic) | ||
| Alcoholic conditions | – | ||
| Glycolic acid | / | / | 6 |
| Butanoïc acid | / | 10 | 10 |
| 3-ethoxy-1-propanol | / | / | 9 |
| Butanol | / | / | 10 |
| 4-hydroxybutanoic acid | 6 | / | / |
| 6-hydroxyhexanoic acid | 11 | / | / |
| 2,6-heptanediol | 37 | 40 | 20 |
| 2-heptanone | / | 10 | 15 |
| Cyclohexanone | 7 | / | 4 |
| Benzylic alcohol | 6 | / | 2 |
| Benzoïc acid | / | / | 5 |
| Anisole | / | 5 | / |
| Non identified | 33 | 35 | 19 |
4 Conclusions
The findings reported here have underlined the variability of chemical composition of cork which can depend, for instance, on the geographical localisation of the sample. Further investigations should compare the molecular composition of cork using chosen environmental parameters (climate, edaphic conditions ...). Among the triterpens already identified in suberin (friedelin and betulin), we have found out friedelanol which has never been described before in this polymer.
Furthermore, 2,6-heptanediol has been depicted in the volatile fraction obtained after depolymerisation. Extraction conditions which lead to incomplete depolymerisation and using enzymes such as cellulases, pectinases [25] or lipases and phenoloxidases, should be considered. This technique may indeed provide information about the sequence of depolymerisation of these molecules and thus their potential localisation in the net structure of this polymer.