

1 Introduction
Genomes vary in sequence as a result of evolution and hence polymorphism data allows us to elucidate the evolutionary history of species. Moreover, genome-wide investigation of the patterns of polymorphism in a large sample of individuals is the first step to assess the relationship between genotype and phenotype within a population. Large-scale polymorphism surveys and analyses were first reported for a small number of species including Drosophila melanogaster, Arabidopsis thaliana and Homo sapiens.
For A. thaliana, the exploration of its genetic diversity began with a study in which 876 fragments (approximately 1% of the genome) across the genomes of 96 isolates, sampled worldwide, were compared by Sanger sequencing [1]. Then, to capture the genome-wide common sequence variation, a high-density array resequencing strategy was used on a subset of 20 accessions [2,3]. Major conclusions from these first studies were that most sequence variants are found worldwide although population structure and isolation by distance are evident. Linkage disequilibrium (LD) decays over about 10 kb. Hence, the average linkage disequilibrium in A. thaliana is not very different from that in humans, perhaps surprising, given the selfing nature of this organism. This result might reflect that outcrossing events are common and the effective population size is large. Today, a project with the goal of describing the whole-genome sequence variation in 1001 accessions of A. thaliana is underway (http://1001genomes.org/) [4,5]. The main motivation for the 1001 genomes project is to have a deeper insight into the genotype-phenotype relationship of this species by using a genome-wide association studies (GWAS) strategy.
In parallel, the release of the reference human genome sequence in 2001 provided the foundation for cataloguing human genetic variation [6]. A few years later, the International HapMap project released a catalogue of polymorphic sites containing approximately 3.1 million single nucleotide polymorphisms (SNPs) [7]. This resource allowed the mapping of thousands of genomic regions linked to disease susceptibility by genome-wide association studies. Today, an international collaboration aims to sequence 2500 human genomes from the five major population groups: Europe, East Asia, South Asia, West Africa and the Americas (http://www.1000genomes.org/). The pilot phase of this project was recently completed [8]. As for A. thaliana, the objective of this project is to identify large sets of functional polymorphisms that underlie phenotypic variation in multiple human populations.
The complete genome sequence of S. cerevisiae was a milestone in the field of genomics in the 1990s [9,10]. The S. cerevisiae laboratory strain S288c became the pioneer eukaryotic genome. Ever since, the hemiascomycetous yeasts (the subphylum of fungi that includes S. cerevisiae) have been used as model organisms for evolutionary and comparative genomics. Because of the structure of their genome (small and compact), these organisms represent a powerful model for comparative genomics and studies of genome evolution [11]. The availability of genome sequence data represents an unprecedented opportunity to evaluate DNA sequence variation and genome evolution in a phylum spanning a broad evolutionary range. The wealth of these data on interspecific sequence differences stands in contrast to our limited knowledge of variation within yeast species. Nevertheless, significant progress has been made over the last few years to characterize the polymorphic variation within yeast species.
Here, we review recent studies that explored the genetic diversity within large collections of yeast isolates of the same species. Yeast population genomics to date have mainly been focused on two species: S. cerevisiae and S. paradoxus. These comprehensive genome analyses have increased our understanding of the population structures as well as the evolutionary history of the species. We first begin with a discussion of the two complementary approaches that focused on either individual or species genomic variation. We then discuss how these polymorphism resources lay the foundation for dissecting the relationship between genotype and phenotype.
2 From comparative genomics to population genomics
By comparing genome sequences at different phylogenetic distances, diverse questions can be addressed. At very large evolutionary distances, broad insights about the gene content as well as evolution of the genome structures can be gleaned. At moderate phylogenetic distances, sequence comparisons allow a deeper insight into the functional parts of the genome and help our understanding of the relationship between genotype and phenotype. Together, comparative and population genomics provide a comprehensive and complementary view of genome sequence variation. Because of their wide ecological, geographical, clinical and industrial distribution and their genome size, yeasts are ideal model organisms to understand genome evolution in different species as well as in natural populations.
Since 2000, yeasts (or more precisely hemiascomycetes) have been at the forefront of comparative genomics [12]. Today, more than 40 hemiascomycetes species are either completely or partially sequenced [11]. Among these sequenced organisms, there are species evolutionary distant from S. cerevisiae but also closely related species such as those belonging to the Saccharomyces sensu stricto complex. Because of the variable phylogenetic distance between these organisms, yeast genome comparisons were very fruitful. As an example, the comparison of the Saccharomyces sensu stricto species led to the mapping of conserved regulatory motifs along the Saccharomyces genome [13–15]. Sequencing of species such as Kluyveromyces waltii (recently renamed Lachancea waltii) and Ashbya gossypii provided clear proof of a major event in the history of the hemiascomycetous subphylum: the whole genome duplication event in the lineage leading to S. cerevisiae [16,17]. Finally, comparisons of more distant species represented a powerful model for studies of genome evolution [18,19].
Nevertheless, a single genome sequence is not representative of a species. It is now known that S. cerevisiae strains can vary in Ty element [20,21], gene content [22] and similar findings have been found for other species, including humans. Of particular importance is the observation that some genes with fundamental effects on life-history traits are not even present or functional in the S288c reference strain [23]. In most of the comparative genomic studies, it is, therefore, sometimes difficult to know if the observed genomic variation is either strain or species specific.
Now, new molecular methods allow the characterization of genetic variation at the whole-genome level between and within a species. The generation of high-density arrays suitable for whole-genome variation detection was the first major technological breakthrough [2,24]. Today, next-generation sequencing technology provides a novel opportunity for collecting genome-scale sequence data [8]. In the near future, this current technological revolution in sequencing will allow the exploration of large numbers of new species as well as to have a better insight into natural populations. All these strategies lay the foundation for the evolving field of population genomics.
3 Saccharomyces cerevisiae population genomics
Previous studies have aimed for a global view of variation across the S. cerevisiae genome through a variety of different means. Nucleic acid polymorphism among S. cerevisiae isolates has been documented using different molecular markers [25,26], identifying single features polymorphisms (SFPs) [27], by multilocus sequence analysis [28–31] or using selected segregating sites [32]. While these strategies offer some insight into allelic variation between S. cerevisiae strains, they provide polymorphism information for only a very small fraction of the genome, limiting our ability to make inference about the general structure of a population. Hence, the next step was to obtain a precise measure of the diversity within S. cerevisiae species and to compare whole genomes of a large population at the nucleotide level.
A first exploration of the yeast whole-genome variation at the nucleotide level focused on seven commonly used laboratory S. cerevisiae strains: A364A, W303, FL100, CEN.PK, ∑1278b, SK1 and BY4716 [33]. Genomic polymorphism maps were generated using high-density arrays. A major conclusion of this study was that all the studied laboratory strains except SK1 are derivatives of the S288c reference strain. In fact, these genomes are mosaics of large regions identical to S288c interspersed with small regions of high sequence divergence.
Nevertheless to have a better insight into natural population variation, two major projects generated genome-wide maps at the nucleotide level of large S. cerevisiae collections, sampled from a diverse array of sources (beer, bread, vineyards, immunocompromised individuals, various fermentations and soil) and from different continents (Fig. 1). Two different strategies were used: high-density arrays [24] and low coverage whole genome sequencing [22]. In the first study, a total of 1.89 million single nucleotide polymorphisms (SNPs) grouped into 101,343 distinct segregating sites were identified in a sample of 63 S. cerevisiae strains. In the second study, the Saccharomyces Genome Resequencing Project (SGRP) described 235,127 SNPs and 14,051 indels from 1-4 fold, or more, coverage whole genome sequences of 36 S. cerevisiae and 35 S. paradoxus strains. The SGRP created a genome assembly for each strain by developing an iterative process called Parallel ALignment and ASsembly (PALAS). This process implemented an imputation method to computationally infer missing (or poor quality) information (both nucleotide and INDELs) from related strains by means of a method based on ancestral recombination graphs. This sequence survey also discovered 38 new hypothetical open reading frame (mostly subtelomeric) that are absent in the S288c reference genome.
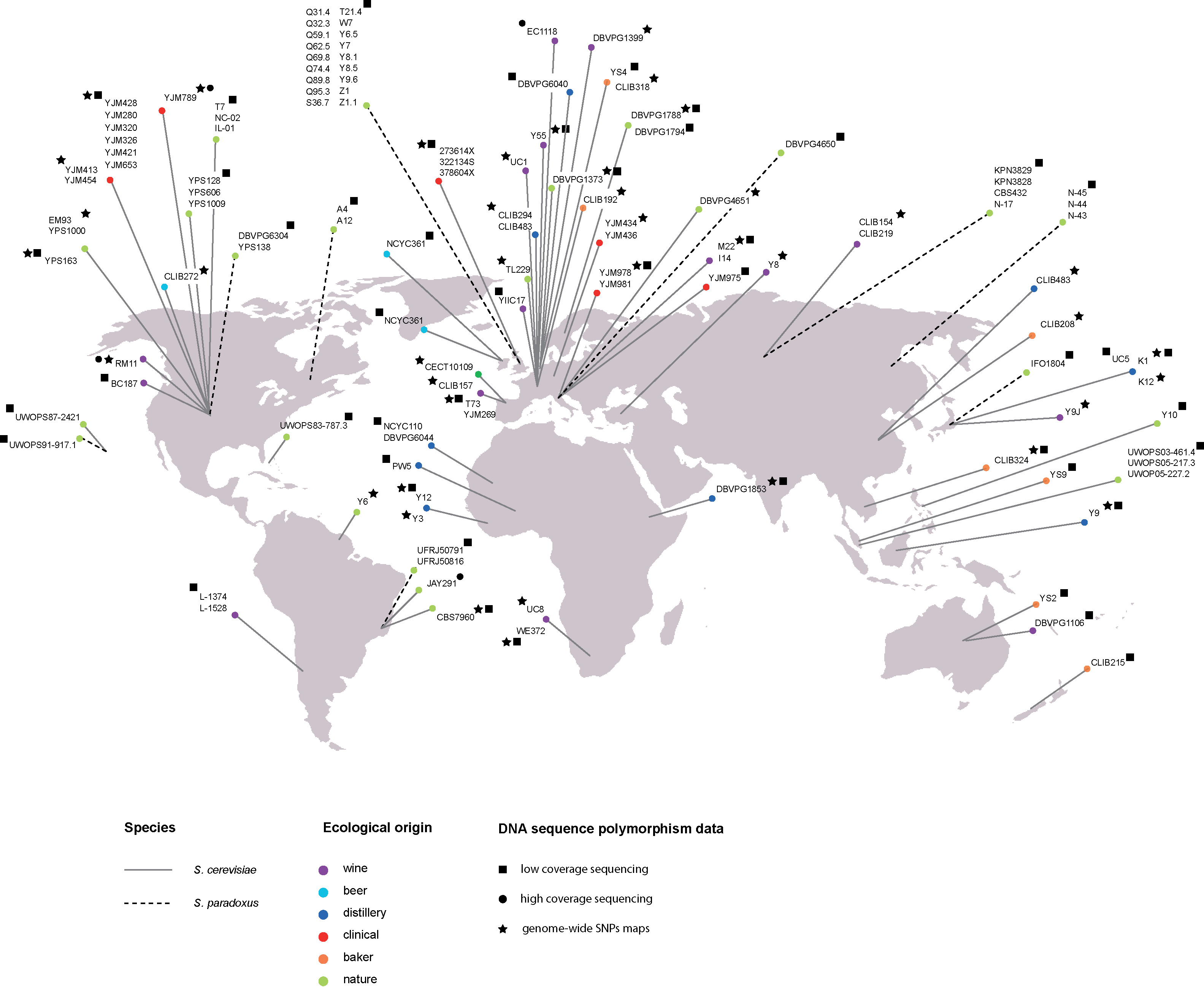
Exploration of the intraspecific diversity of yeast isolates. Geographical and ecological origins of yeast strains that were used in large-scale polymorphism surveys. The line denotes the species and circle color denotes ecological niche as specified in the key.
Both analyses provided a deep insight into the population structure as well as the evolutionary history of this yeast species. From the sequence polymorphisms survey [24], the authors observed clear population structure at the level of major ecological subgroups. These data strongly supported the presence of different clusters such as strains from vineyards. These clusters represent separate domestication events, but S. cerevisiae as a whole is not domesticated. The SGRP analysis proposed the presence of five “clean” lineages (specific to geographic location or ecological niches), with the majority of segregating sites private to one population and uniformly distributed along the genome, as well as many recombinant (mosaics) strains originated from the various clean lineages. An alternative scenario to the domestication process was also proposed, with humans having utilised natural existing variants for different fermentation processes and offered opportunity to interbreeding (generating the mosaics). Surveying additional strains is needed to fully resolve the roles of ecology versus geography in the genetic differentiation of this species.
The determination of linkage disequilibrium (LD) – non random association of alleles at two or more loci – also provided information about recombination and evolutionary history of the species. Linkage disequilibrium falls to half of its maximum value at about 11 kb [24]. Nevertheless, the architecture of linkage disequilibrium is variable within the different subpopulations: laboratory, wine, clinical and distillery strains. Most of the laboratory strains are derivatives of the sequenced reference strain S288c. Their genomes are mosaics of large regions identical to S288c interspersed with small regions of high sequence divergence. As a result, high linkage disequilibrium can be observed. Linkage disequilibrium falls to half of its maximum value at about 23.8 kb. By contrast, the low level of linkage disequilibrium (about 2.5 kb) in the wine strains probably reflects the extended length of time since the most recent common ancestor of these strains, and perhaps a higher frequency of outcrossing events. Finally, the linkage disequilibrium decay and the fact that segregating sites are located every ∼100–200 bp suggest that S. cerevisiae could be a good model organism for genome-wide association studies.
In parallel to these large-scale polymorphism surveys, genomes of several S. cerevisiae strains were sequenced at high coverage (Fig. 1). These studies focused on strains from different genetic backgrounds: a clinical strain (YJM789), wine strains (RM11-1a and EC1118) a strain involved in biofuel production (JAY291) and laboratory strains (SK1, Y55, W303) [22,23,34,35]. Exploration and extensive analysis of these complete genomes was very fruitful. The genome of the wine yeast EC1118 differs from other S. cerevisiae strains by three large regions originated from either a closely related or a non-Saccharomyces species [23]. These introgressions encompass 34 genes and potentially play a key function in fermentation, such as metabolism of sugars and nitrogen. Similarly, the genome of the JAY270 strain pointed out specific gene polymorphisms and introgressions important for bioethanol production. These could explain desirable phenotypes such as ethanol and cell mass production as well as high temperature and oxidative stress tolerance [34]. In addition to this genotype-phenotype relationship exploration, whole genome sequences allowed a better insight into the yeast life cycle. Based on the distribution of segments of shared genealogy among three strains: YJM789, RM11 and S288 C, it was estimated that only 314 outcrossing events have occurred during approximately 16 million cell divisions [36]. This results show that outcrossing is relatively infrequent in S. cerevisiae: roughly once every 50,000 divisions.
4 Saccharomyces paradoxus: a rising evolutionary model
In the field of yeast population genomics, S. paradoxus is becoming an attractive model organism. S. paradoxus is the closest known species to S. cerevisiae and is remarkably similar in terms of genome organization and physiology. Most of the S. paradoxus strains were isolated from the bark and surrounding soil of oak trees, where it can co-exist with its sibling species S. cerevisiae [37,38]. S. paradoxus has been isolated from many locations worldwide and it is considered as a non domesticated wild species.
Initially, multilocus sequence analysis was used to infer phylogenetic relationships between S. paradoxus geographic subpopulations [28,39,40]. These studies indicated that strains within this species are both genetically divergent and partially reproductively isolated. In addition, three highly diverged geographic subpopulations were identified: Europe, Far East Asia and America lineages.
Subsequently, a sequence variation study scaled up on the entire third chromosome (approximately 2.3% of the genome) [41]. The authors analysed the sequence of 20 isolates from two subpopulations: 12 from Europe and eight from Far East Asia. This allowed a precise quantification of the life cycle. Mutational and recombinational diversity along this chromosome clearly shows that S. paradoxus is primarily asexual (like S. cerevisiae) [41,42]. In both subpopulations, the sexual cycle occurs approximately every 1000 asexual generations. Moreover, these population genomic data have allowed the identification of recombination hotspots. Interestingly, multiple recombination hotspots are conserved between S. paradoxus and S. cerevisiae probably as a result of the low frequency of sex in these two yeasts [42].
The SGRP initiative sequenced 35 S. paradoxus strains, representative of the major geographical subpopulations, with the aim of drawing comparisons with S. cerevisiae (Fig. 1). Half of the strains were isolated from the same geographical location (England) as representatives of a single recombining population. Genome sequences revealed a previously unknown fourth diverged lineage from Hawaii. The sequence divergence between lineages is variable: 1.2% (Far Eastern/European), 2.3% (American/Hawaii) and 3.7% (European-Far Eastern/North America-Hawaii). Genetic diversity in S. paradoxus is considerable higher than in S. cerevisiae. The large majority of S. paradoxus SNPs are private to each subpopulation resulting in a marked population structure that follows geographic boundaries. The South American isolates (previously regarded as S. cariocanus) show the highest Ty counts [22], consistent with the rapid accumulation of chromosomal translocation in this lineage [43]. Genome-wide analysis also confirmed the presence of a large region (23 kb) introgressed from S. cerevisiae into the European subpopulation [28], with identical break-points in all strains, perhaps originated from a single event. Finally, this study also showed that S. paradoxus strains have limited phenotypic variation compared to S. cerevisiae [22]. This observation stands in contrast with the genetic diversity. A possible explanation could be that S. cerevisiae strains exhibit a higher phenotypic variation because they occupy a larger ecological niche.
5 Dissecting the functional variants
The genetic differences among individuals lead to broad quantitative phenotypic variation [44]. Dissecting the genetic mechanisms underlying this natural phenotypic variation is a major challenge in modern biology. There are two major approaches to mapping the quantitative differences in phenotypic traits: linkage analysis and genome-wide association studies. In the first case, the causative loci are mapped using the segregating progeny of crosses between genetically divergent strains. The aim is to identify segregating genetic variants that contribute to phenotypic variation in progenies. In contrast, genome-wide association studies use a large sample of unrelated individuals from the same population.
In the past decade, S. cerevisiae has become a primary model for dissecting the complex architecture of quantitative traits using linkage mapping. The genetic basis of a number of interesting phenotypes have been studied in yeast, including growth at high temperature [45], gene expression [46], and response to drugs [47]. Nevertheless, these studies focused on pairs of specific strains and systematically used the S288c laboratory strain (or its derivative BY). They were instrumental and laid the foundation of yeast forward genomics but the whole genetic diversity of a large population has not yet been systematically explored. The new Saccharomyces population genomic datasets help to characterize the genetic diversity of the species. In fact, these data can facilitate the mapping of functional variants in multiple ways. The characterised genomic relationships among strains can guide an accurate experimental design for both linkage and association mapping.
Recent studies have used strains obtained from multiple lineages and sampled a larger fraction of the variation. Two strains, representative of diverged wine and North America oak populations, were recently used to accurately dissect the variation in the efficiency of sporulation [48]. The authors found that the observed difference is mostly explained by allelic variation of three transcription factors: IME1, RME1, and RSF1. Interestingly, this study nicely illustrates how genetic interactions between transcription factors might have an impact on phenotypic diversity.
A new collection of genetically tractable parentals [49] and F1 segregants from four highly diverged S. cerevisiae are also available [50]. Using this set of F1 segregants, linkage analysis clearly showed that subtelomeric regions play a major role in quantitative natural variation, supporting their importance in adaptive variation.
Furthermore, 16 strains were selected and crossed in all pairwise combination to create a large library of F1 hybrids for genetic analysis such as heterosis [51]. Finally, SNPs dataset aid the prediction of functional and deleterious variants using a conservation based approach like the one implemented by SIFT [52].
6 Conclusions and perspectives
The advent of next generation sequencing technologies has changed the face of genomics and given access to population genomic data. Although yeast has proven important in developing essential tools for analysing multiple individuals, this field is only just beginning. A new set of genome sequenced 27 strains is now available (http://www.genetics.wustl.edu/jflab/data4.html) and deep coverage (40–50X) of 42 SGRP S. cerevisiae and S. paradoxus strains will soon be released (Fig. 1). Expanding the genome-wide sequence datasets will increase the number of variants and add power for genome-wide association approach. In addition, the availability of multiple S. paradoxus wild isolates characterised at the genome level offer an attractive model to study ecology and evolution. Furthermore, most of the genetic and molecular techniques as well as the QTL mapping are readily available in S. paradoxus [53] and a deletion collection is underway.
The characterization of SNPs is only the first step toward characterising the genetic make-up of a natural population. The high coverage sequencing data will allow characterization of heterozygous diploid genomes without the need of creating homozygous derivatives. This is a major step forward given the majority of Saccharomyces isolates are diploids or have a more complex ploidy configuration (e.g. aneuploidy, polyploids). However, the ability to sporulate and generate viable gametes remains an essential feature for subsequent forward and reverse genetics analysis and should be considered during selection of strains. The high genome coverage will enable detection of copy number differences, perhaps relevant for genome-wide association studies.
A major challenge that also remains is to produce a complete de novo end-to-end assembly. The subtelomeric regions are particularly difficult due to their repetitive nature [54], intrinsic high genomic instability [55] and functional divergence [56]. Resolving the structure of chromosome ends is crucial as they contain many genes involved in secondary metabolisms and, therefore, play key roles in individual variation. The generation of full genome assemblies will also yield a complete picture for other polymorphisms such as structural variants and their impact in evolution and fitness can be determined.
Finally, parallel explorations of new species as well as individuals from the same species will offer a precise view of the evolution of the genotype-phenotype relationship in yeast. Today, yeast population genomic studies are underway in many other species including Schizosaccharomyces pombe, Lachancea kluyveri and Candida albicans. These new datasets will allow specific analysis within species as well as drawing of important parallels between species. Yeasts with their compact and well-characterised genome are likely to play a key role in the rising population genomic field.
Disclosure of interest
The author declare that he has no conflicts of interest concerning this article.
Acknowledgements
The authors are grateful to Dr C. Nieduszynski and M. Hawkins for critical reading of the manuscript. GL is funded by The Biotechnology and Biological Sciences Research Council (grant number BB/F015216/1). JS was supported by a CNRS PEPS grant.