

1 L'allogreffe de cellules souches allogéniques comme traitement des déficits primitifs du système immunitaire
Les déficits primitifs du système immunitaire peuvent être schématiquement sous-divisés en deux grands groupes : les défauts de développement lymphocytaire et les défauts fonctionnels lymphocytaires ou de l'immunité innée.
Parmi les défauts du développement lymphocytaire, les déficits immunitaires combinés sévères (SCID) représentent les formes les plus sévères. En effet, les enfants atteints ont une espérance de vie limitée à quelques mois [1,2]. L'injection de cellules souches hématopoïétiques (CSH) capables de se différencier en lymphocytes représente une stratégie à la fois évidente et simple. Une particularité des greffes allogéniques effectuées chez ces patients dépourvus d'immunité adaptative est liée au fait qu'il n'est pas nécessaire de recourir à une myéloablation ; cet avantage n'est pas négligeable, compte tenu du jeune âge de ces patients, souvent atteints d'infections graves au moment du diagnostic. Les données du registre européen [3] (Fig. 1) montrent que la transplantation des CSH à partir d'un donneur génoidentique confère plus de 80% de chance de survie, avec correction du déficit immunitaire combiné sévère, avec un recul médian de 15 ans ; ce résultat est aussi à mettre en relation avec une incidence de la réaction aiguë du greffon contre l'hôte (GVH), particulièrement faible dans ce contexte, sans que l'on puisse l'expliquer aisément. En l'absence d'un donneur HLA génoidentique (fratrie), l'urgence thérapeutique impose le recours à des donneurs familiaux HLA partiellement compatibles, tels qu'un parent. Dans ce cas, la probabilité de survie à long terme des patients greffés est significativement inférieure et, selon les maladies, comprise entre 30% et 75% [3,4]. Malgré une amélioration significative de la survie de ces patients liée à une meilleure prévention de la GVH et des infections, elle reste insuffisante. Parmi les trois paramètres qui influencent la survie – GVH, rejet de greffe et cinétique du développement lymphocytaire –, ce dernier constitue le paramètre clé du devenir à long terme des patients greffés. En effet, une période de 3–4 mois, au minimum, est nécessaire pour détecter une nouvelle lymphopoïèse T. On ne sait pas à l'heure actuelle ce qui explique la longueur de cette période, pendant laquelle les patients, faute de système immunitaire fonctionnel, sont à risque de décès par infection. Sur le long terme, on observe parfois une diminution du nombre de lymphocytes T, liée à une absence d'implantation des CSH du donneur dans la moelle osseuse, responsable d'un arrêt de la fabrication de lymphocytes T après quelques années. De ce fait, l'avantage initial (transplanter sans chimiothérapie) peut se révéler comme un désavantage à long terme, indépendamment du fait que la greffe ait été réalisée en condition de compatibilité HLA ou non. De plus, en l'absence de chimiothérapie pré-greffe, on observe fréquemment la persistance d'un déficit lymphocytaire B nécessitant un traitement substitutif par immunoglobulines [5].
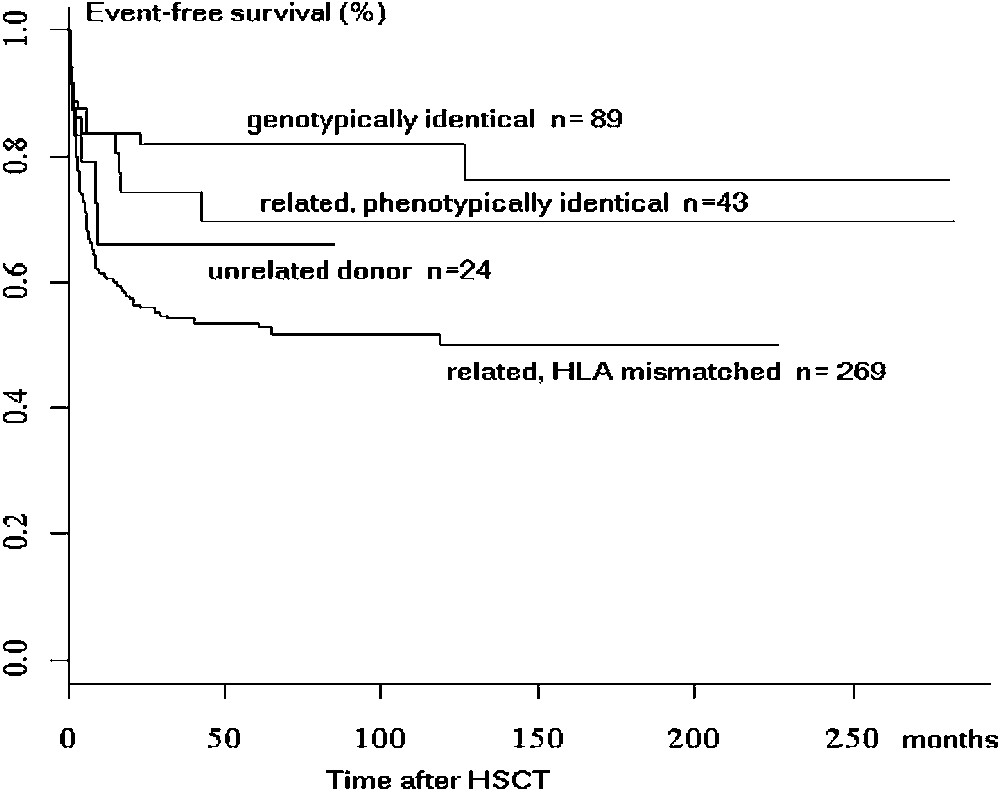
Probabilité de survie des patients SCID après transplantation des CSH selon la compatibilité du donneur–receveur. Figure extraite de la publication de Corinne Antoine, Lancet 2003 (361) 553–560.
2 Les pistes de recherche
Étant donné l'impact clinique du déficit immunitaire persistant dans les mois qui suivent la greffe et du déclin possible de l'immunité à long terme, il est d'une importance cruciale de mettre au point des stratégies thérapeutiques capables, d'une part, d'accélérer la reconstitution immunitaire et, d'autre part, de permettre la persistance de CSH du donneur sans recourir à une chimiothérapie conventionnelle, très toxique. La découverte de l'interleukine 7, cytokine clé de la différentiation lymphocytaire, a fait espérer que son utilisation au moment de la greffe puisse induire une accélération notable de la différentiation T. En fait, nous n'avons pas pu mettre en évidence ce rôle d'accélérateur de cette cytokine dans toutes les conditions expérimentales testées [6]. Une alternative est représentée par l'injection des lymphocytes T du donneur dépourvus d'alloréactivité [7–9] à l'égard du receveur ou de lymphocytes T spécifiquement dirigés contre les agents infectieux les plus communément détectés chez ces patients immunodéprimés [10,11]. Le contrôle in vitro et/ou in vivo de l'alloréactivité des lymphocytes T du donneur reste un objectif difficile à atteindre, en dépit des nombreuses technologies testées [12–14], car il persiste un risque résiduel de GVH qui empêche l'injection du nombre important de lymphocytes T.
Une seconde stratégie, probablement plus spécifique, consiste à isoler, à l'aide des tétramères HLA peptides, du pool des lymphocytes T du donneur ceux qui sont spécifiquement dirigées contre des virus tels que cytomégalovirus, adénovirus ou le virus d'Epstein Barr. L'injection in vivo de ces lymphocytes T spécifiques obtenus uniquement par sélection immunologique, sans besoin de cultures longues et complexes, paraît envisageable, compte tenu de l'efficacité biologique de cette technique.
Une approche alternative consiste en l'autogreffe de CSH génétiquement corrigées.
3 Résultats cliniques de la thérapie génique appliquée à deux formes différentes de SCID
Depuis 1999, quatre essais cliniques de thérapie génique ont été effectué en Europe, avec, comme objectif, la correction de deux formes de SCID : la forme récessive liée à l'X par défaut de la chaîne γ commune (γc) des récepteurs de cytokines hématopoïétiques (SCID-X1) et le défaut de l'enzyme adénosine deaminase [15–17]. Ces essais utilisent un vecteur rétroviral défectif codant pour le gène d'intérêt capable de corriger ex vivo les cellules souches et les précurseurs hématopoïétiques. Une trentaine de patients, dont l'âge est compris entre un mois et un an, ont été traités à ce jour, avec pour 28 d'entre eux une correction du phénotype pathologique compatible avec une vie normale et sans traitement anti-infectieux associé. L'analyse des deux patients chez lesquels la thérapie génique n'a pas permis la restauration de la différenciation T a montré (1) qu'en présence d'hypersplénisme, les précurseurs corrigés ne peuvent pas migrer correctement dans le thymus [18] et (2) qu'il existe une valeur « seuil » du nombre de cellules précurseurs corrigées nécessaire pour obtenir une normalisation de la différentiation T. Cette valeur est de l'ordre de cellules hématopoïétiques par kilogramme de poids corporel. Parmi les 28 patients pour lesquels la thérapie génique a corrigé le déficit immunitaire au long cours (jusqu'à 7,5 ans actuellement), on a observé l'apparition d'un répertoire T polyclonal et fonctionnel, comme le démontre la capacité de ces lymphocytes T à être activés in vitro et in vivo contre des agents infectieux. Cette reconstitution T est complète pour la grande majorité de ces 28 patients. Elle s'explique par la présence d'un avantage sélectif conféré aux cellules corrigées par l'expression γc en comparaison des cellules précurseurs non transduites. Cependant, il faut souligner une différence entre les deux protocoles de thérapie génique réalisés : la nécessité d'une chimiothérapie dans le cas du défaut en adénosine deaminase (ADA), rendue nécessaire par le moindre avantage sélectif apporté par la présence de l'enzyme ADA comparé au déficit en γc. L'absence de chimiothérapie dans le traitement du SCID-X1 explique l'observation d'un faible pourcentage de lymphocytes B transduits (). Toutefois, on observe la présence d'immunoglobulines sériques, qui permettent à la majorité des patients traités (5/8 dans notre protocole) de ne pas recevoir de traitement par immunoglobulines.
Ces résultats sont très encourageants, mais le recul, encore insuffisant (1–7,5 ans), et le nombre encore limité de patients traités empêche de tirer des conclusions définitives quant à l'efficacité à long terme de la thérapie génique. La survenue dans notre essai clinique de trois cas de lymphoprolifération monoclonale incite à modifier la technique utilisée afin d'essayer d'augmenter la sûreté de cette approche [19].
4 Effets secondaires liés à l'utilisation de vecteurs rétroviraux de type C
Trois événements graves et inattendus sont survenus chez trois patients inclus dans notre essai clinique 30 à 34 mois après l'injection des cellules hématopoïétiques autologues (CD34+) génétiquement corrigées. Chez ces trois patients, une lymphoprolifération T monoclonale très similaire à une leucémie aiguë lymphoblastique (LAL) s'est développée. La monoclonalité du processus, le nombre très élevé des lymphocytes circulants au moment de sa découverte clinique, la présence de blastes et, pour deux des trois malades, la détection de translocations chromosomiques constituent des éléments de similitude avec un processus leucémique. Deux des trois patients sont en vie, en rémission complète après chimiothérapie. Du fait de ces effets secondaires graves, l'essai clinique a été arrêté. De nombreuses études ont été entreprises pour en comprendre la physiopathologie. L'élément clé de ces lymphoproliférations est représenté par des événements de mutagenèse insertionnelle, provoqués par l'insertion du provirus à proximité d'oncogènes [20]. L'effet enhancer du LTR du provirus peut provoquer l'expression aberrante d'un oncogène localisé à proximité de l'insertion du provirus dans le génome cellulaire. D'autres facteurs de risque ont été évoqués : jeune âge des patients, nature du déficit immunitaire, l'expression non contrôlée d'une sous-unité d'un récepteur hématopoïétique (bien que non mis en évidence, du fait du blocage de différenciation propre aux SCID) et, enfin, l'accumulation chez ces patients de précurseurs lymphoïdes en cycle, ce qui augmente la probabilité de transduction, et l'éventuelle activation d'oncogènes physiologiquement exprimés à ce stade de différenciation.
L'ensemble de ces facteurs de risque constitue autant de pistes de recherche investiguées avant de pouvoir envisager la reprise d'une thérapie génique fondée sur une approche modifiée.
5 Les modifications envisagées pour diminuer ou prévenir les effets toxiques de la thérapie génique
La thérapie génique des SCID a démontré être à la fois un puissant moyen thérapeutique et un traitement à risque. Des efforts importants ont été entrepris par de nombreuses équipes pour modifier les vecteurs utilisés de façon à préserver leur efficacité tout en réduisant leur toxicité. De plus, des efforts ont été entrepris pour mettre au point des modèles animaux prédictifs d'événements oncogéniques [21].
Une modification incluse dans deux protocoles cliniques consiste à inactiver l'effet enhancer du rétrovirus et à introduire un promoteur interne « faible » capable d'induire, en principe, la transcription exclusive de l'ADN thérapeutique. D'autres modifications sont envisagées comme : l'introduction d'un gène « suicide », comme la thymidine kinase, et l'introduction de séquences d'ADN « insulateurs » capables d'isoler la cassette transcriptionnelle introduite avec le vecteur du gène cellulaire [22]. À plus long terme, des stratégies de recombinaison homologues pourraient permettre une approche par correction des mutations. Des progrès ont été accomplis, même si beaucoup reste à faire avant une éventuelle application thérapeutique [23].
6 Conclusions
La thérapie cellulaire des maladies héréditaires monogéniques du système hématopoïétique, notamment des déficits immunitaires héréditaires, a fait d'énormes progrès, qui ont permis une amélioration significative de la survie des maladies traitées. Cependant, la toxicité de cette approche reste non négligeable, en particulier lorsque les allogreffes sont accomplies en situation d'incompatibilité partielle pour le système HLA. C'est pourquoi une approche fondée sur l'utilisation de cellules souches autologues génétiquement modifiées est développée. La thérapie génique des maladies héréditaires pourrait représenter une alternative aux greffes allogéniques HLA, partiellement compatibles pour certaines pathologies, à condition de maîtriser les risques inhérents aux conséquences possibles de l'insertion de rétrovirus dans le génome cellulaire.
Remerciements
L'ensemble de ce travail a pu être réalisé grâce à l'engagement de tout le personnel du service d'immunologie–hématologie pédiatrique et du département de biothérapie de l'hôpital Necker-Enfants-Malades. Cette étude a été subventionnée par l'Institut national de la santé et de la recherche médicale (Inserm), l'Assistance publique–Hôpitaux de Paris (AP–HP), l'Union européenne – contrat Concert no. LSHB-CT-2004–005242 – et l'Agence nationale de la recherche (ANR).