

Abridged English version
Clay minerals identification is systematically performed using X-ray diffraction (XRD) and oriented clay deposits onto glass slide. Infrared (IR) spectroscopy is a quick, simple, relatively inexpensive method giving useful complementary data on the crystal chemistry of clay minerals. Clay minerals show various chemical compositions according to numerous cationic substitutions. Chemical analyses are often performed at macroscopic scale and the deduced structural formulae do not necessarily reflect the actual chemistry of clays identified by XRD. Even though the relationship between clay structure and IR spectrum is complex and has only been partially rationalized up to now, the spectrum reflects the crystal chemistry of clay minerals. Indeed, compared with XRD, IR allows distinction between minerals that exhibit a range of chemical composition within a group (see, for example, [1] for micaceous minerals, [5] for smectite, and [6] for kaolinite). In the case of kaolin minerals, IR is also useful to distinguish between different polymorphs [8].
The aim of this paper is to show how to take benefit easily to the complementarity of XRD and IR techniques, by using the same sample preparation: the clay oriented deposit onto glass slide.
Clay samples were chosen to give a short overview of the capability of the technique. Two smectitic samples already described in literature were used: SAz-1, which is a montmorillonite from the Source Clay Repository of the Clay Minerals Society, and Ölberg, which is a Fe-rich montmorillonite [7]. For kaolin minerals, kaolinite KGa-2 from the Source Clay Repository of the Clay Minerals Society, and dickite from Ste Claire [4] were used.
The Fourier transform infrared (FTIR) spectra were obtained with a resolution of 4 cm−1 in the 2500–5000 cm−1 range with a Nicolet Magna 760 FTIR spectrometer equipped with a KBr beam splitter and a DTGS KBr detector. The spectra of samples were collected in transmission through the oriented deposits onto glass slide previously used for XRD studies. For each sample, 64 scans were collected.
Oriented clay deposits were obtained using the standard procedure [2]. Deposits were 30 to 50 μm thick. The orientation distribution of particles was not measured.
Fig. 1 shows the FTIR spectra, in the νOH region, of the SAz-1 and Ölberg smectites obtained from KBr pellet and from oriented deposit onto glass slide. For each sample, the spectra obtained by the two techniques are similar. Both bands between 3500 and 3700 cm−1 and near 3400 cm−1 are indicative of smectite. The broad band centered near 3400 cm−1 is due to interlayer water. The band between 3500 and 3700 cm−1 is due to structural OH vibrations in smectite, with frequencies depending on the OH-surrounding octahedral cations, and to a lesser extent, to the chemistry of the tetrahedral sheet. The shoulders and the broadness of the structural OH band are mainly due to contributions of several structural OH groups occurring in these smectites, and peak-fitting analysis would be necessary to determine the main contributions of them. However, the position of the maximum of the band is clearly indicative of the chemical composition of smectites. In case of SAz-1, the absorption band near 3620 cm−1 indicates an Al-rich smectite, while for the Ölberg clay, the absorption band near 3560 cm−1 indicates an iron-rich smectite [8].
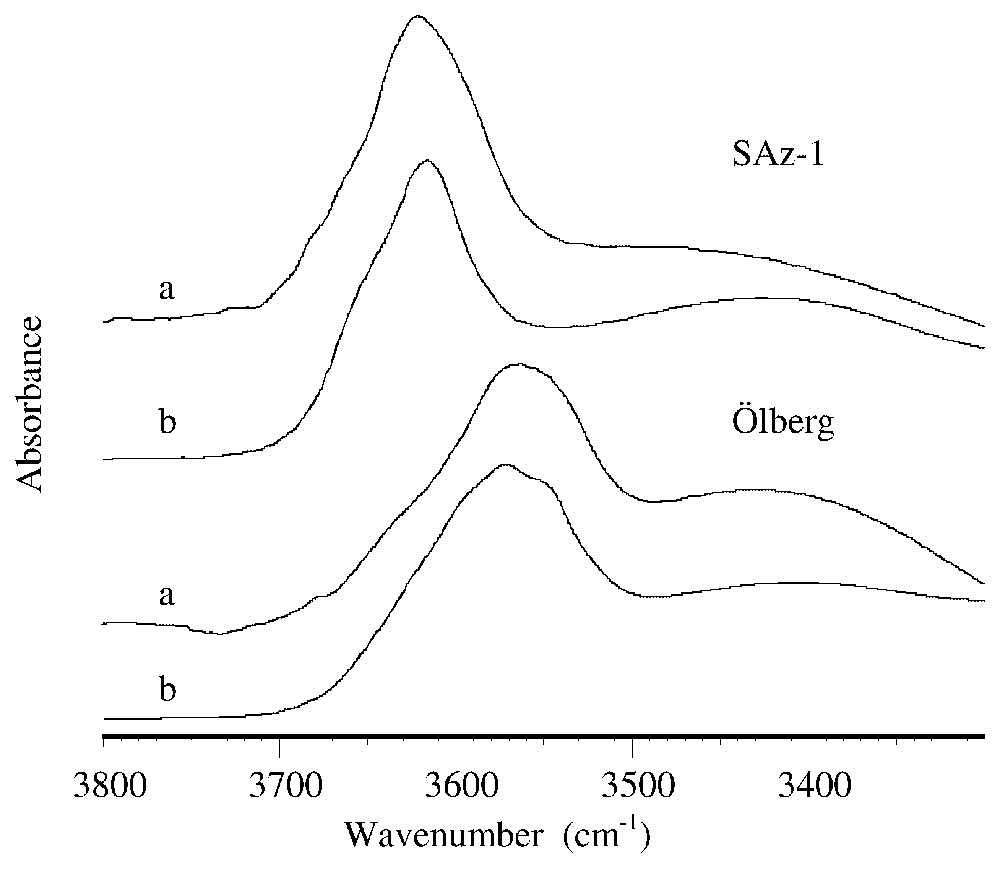
Spectres FTIR des smectites SAz-1 et Ölberg, obtenus (a) à partir de pastilles de KBr; (b) à partir de dépôts orientés sur lame de verre.
FTIR spectra of the smectitic samples SAz-1 and Ölberg, obtained (a) from KBr pellets and (b) from oriented deposits onto glass slide.
Moreover, the (νOH+δOH) combination bands, occurring in the >4000 cm−1 region, can also be studied by this technique. In Fig. 2, the Ölberg IR spectrum (obtained using clay deposits onto glass slide) zoomed in the >4000 cm−1 region appears very similar to that obtained from powder by DRIFTS.

Spectre FTIR de la smectite Ölberg, à partir de dépôts orientés sur lame de verre, dans le domaine 4500–3000 cm−1. En encadré, la zone agrandie des bandes OH de combinaison: (a) dépôt orienté sur lame de verre; (b) spectre de poudres obtenu par réflexion diffuse, pour comparaison.
FTIR spectra of the smectitic sample from Ölberg from oriented deposit onto glass slide in the 4500–3000 cm−1 region. The zoomed part represents the region of the OH combination bands : (a) oriented deposit onto glass slide and (b) spectrum obtained from powder by DRIFTS given for comparison.
In Fig. 2, the FTIR spectra of kaolinite KGa-2 and of dickite Ste Claire obtained from KBr pellet are very similar to those obtained from clay deposits onto glass slide. The two kaolin polymorphs, kaolinite and dickite, can then be readily distinguished using IR spectra of clay deposits, by differences in position and relative intensity of the bands [8].
The results presented above show that it is possible to use clay deposits onto glass slide to obtain FTIR spectra. The use of XRD sample preparations for IR studies implies that: (i) the same clay particles can be studied by both FTIR and XRD techniques, which is not so trivial especially for heterogeneous samples; (ii) the FTIR spectrum is obtained in a very short time (only the acquisition time, 1–2 min), because no specific preparation of sample is necessary.
Of course, this technique does not substitute itself for the pressed pellet (most often KBr) technique. Indeed, the region <2000 cm−1, and notably the δOH and Si–O vibrations zone, cannot be studied due to the glass absorption. However, the νOH region study can sometimes be enough to answer the objectives (ex: distinction between kaolin polymorphs, occurrence of kaolinite in mixture,...). Spectra in the NIR region can also be obtained by this technique. NIR region is less known. However, this region can be self-sufficient to determine the crystal chemistry of clays [6].
A limitation of the use of clay deposits onto glass slide, compared to the pressed pellet technique, is due to the particle orientation relative to the intercepting beam of IR radiation. Absorption of radiation by a vibrating interatomic bond will be at maximum when the polarizing vector associated to the bond is parallel to the electric vector of the beam (i.e. perpendicular to the beam direction), and will be at a minimum when the two vectors are mutually perpendicular. This pleochroism is in fact especially exhibited by trioctahedral clay minerals, such as talc, which have their O–H dipole perpendicular to their layer plane and then to the electric vector of the IR beam in oriented samples. Dioctahedral clay minerals show less change between random orientation and normal incidence, because their O–H dipole lies at 75–80° to the crystallographic axis. Texture effects can then partly explain that some differences in relative intensities are observed between spectra obtained from KBr pellets and from clay deposits onto glass slides (Figs. 1 and 3).
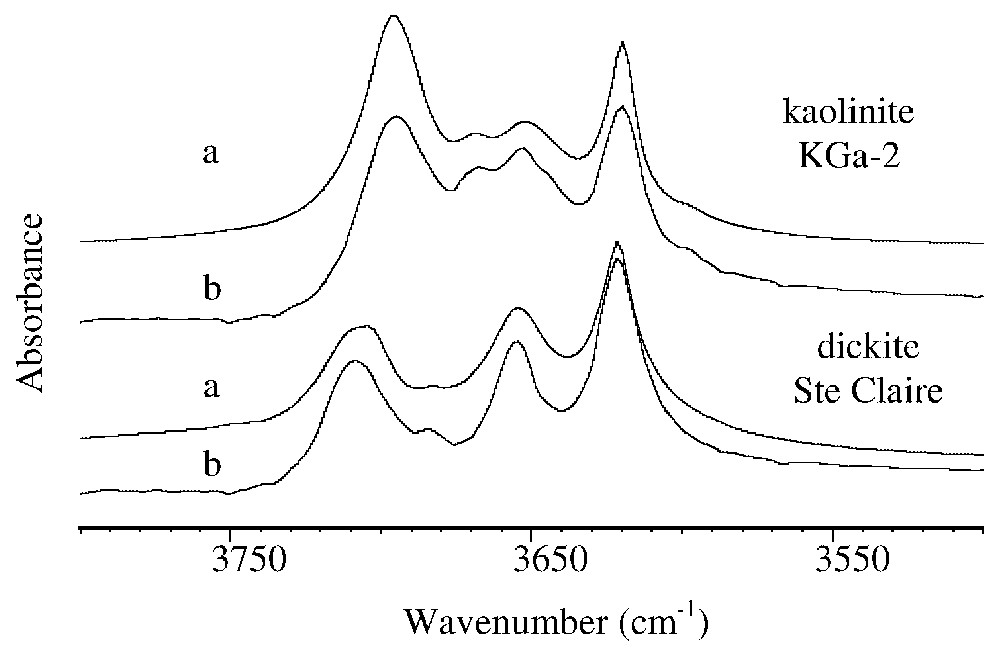
Spectres FTIR de la kaolinite KGa-2 et de la dickite de Sainte-Claire obtenus (a) à partir de pastilles de KBr, (b) à partir de dépôts orientés sur lame de verre.
FTIR spectra of kaolinite KGa-2 and of Sainte-Claire dickite obtained (a) from KBr pellets and (b) from oriented deposits onto glass slide.
By an other way, this approach will be efficiently used for IR studies of petrologic thin sections, absorption bands of resin occurring at wavenumbers <3200 cm−1.
1 Introduction
L'identification des minéraux argileux est classiquement faite par diffraction des rayons X (DRX), et le plus souvent à partir de dépôts de suspensions sur lames de verre. Très performante, cette technique ne permet que difficilement d'obtenir des informations sur la chimie des minéraux argileux. Or, les minéraux argileux ont des chimies très variables, dues aux nombreuses substitutions cationiques dans les différents sites structuraux. Les analyses chimiques d'argile sont le plus souvent réalisées à des échelles macroscopiques ou mésoscopiques (microsonde, MEB) et les formules structurales déduites de ces données ne reflètent pas toujours la réalité cristallochimique de ces minéraux. La spectroscopie infrarouge (IR) est une méthode simple, rapide et peu coûteuse, donnant des informations cristallochimiques complémentaires nombreuses. Bien que l'attribution des signaux IR soit complexe, délicate et seulement partiellement résolue jusqu'à aujourd'hui, la spectroscopie IR permet, très généralement, de distinguer entre eux des minéraux montrant une grande variabilité chimique au sein d'un groupe (voir [1] pour les micas, [5] pour les smectites et [6] pour les kaolinites). Pour les kaolins, la spectroscopie IR est efficace pour identifier les polymorphes [8].
L'objectif de cet article est de montrer qu'il est possible de réaliser une étude conjointe des minéraux argileux par DRX et spectroscopie IR en utilisant la même préparation : un dépôt de suspension argileuse sur lame de verre.
2 Matériels et méthodes
Les échantillons ont été choisis pour donner une brève vue d'ensemble des possibilités de cette technique. Deux smectites bien décrites dans la littérature ont été utilisées (SAz-1, qui est une montmorillonite de la Source Clay Repository of the Clay Minerals Society, et Ölberg, qui est une montmorillonite riche en fer [7]) et deux kaolins (la kaolinite KGa-2 (Source Clay Repository of the Clay Minerals Society) et la dickite de Sainte-Claire [4]).
Les dépôts d'argile ont été réalisés en utilisant la procédure standard pratiquée pour la DRX [2]. Environ 4 ml d'une suspension argileuse (60 g l−1) sont déposés sur une lame de verre et séchés à l'étuve (90 °C) pendant une nuit. Le dépôt obtenu correspond à 10 à 15 mg d'argile par cm2, et à une épaisseur de 30 à 50 μm. Le degré d'orientation des particules d'argile n'est pas mesuré, mais il est considéré comme équivalent à celui obtenu par une compaction à 550×105 Pa [3].
Les spectres infrarouge sont obtenus à l'aide d'un spectromètre à transformée de Fourier Nicolet Magna 760, équipé d'une séparatrice en KBr et d'un détecteur KBr DTGS. Les spectres sont enregistrés entre 2500 et 5000 cm−1, avec une résolution de 4 cm−1. Le bruit de fond est enregistré en transmission à travers une lame de verre propre.
Des spectres IR de poudres ont été réalisés pour comparaison en mode transmission, à l'aide d'un spectromètre à transformée de Fourier Nicolet 510, la résolution étant de 4 cm−1. Le spectromètre est continuellement sous balayage d'air sec pendant l'acquisition (64 scans). Les échantillons sont préparés sous forme de pastilles de KBr pressées (1 mg d'argile et 150 mg de KBr). Pour obtenir des spectres dans le domaine proche IR (au-dessus de 4000 cm−1), le spectromètre Nicolet Magna 760 a été équipé d'une séparatrice en CaF2, d'un détecteur PbSe, et d'un accessoire de réflexion diffuse (DRIFT) Spectra-Tech.
3 Résultats
Les spectres FTIR, dans le domaine des vibrations νOH, des échantillons de smectites SAz-1 et Ölberg, obtenus à partir de pastilles de KBr et dépôts orientés sur lames de verre, sont présentés sur la Fig. 1. Pour chaque échantillon, les spectres obtenus à l'aide des deux techniques sont très semblables. Les bandes observées entre 3500 et 3700 cm−1, d'une part, et vers 3400 cm−1, d'autre part, sont caractéristiques des smectites. La bande majeure est due aux vibrations des groupements (OH) structuraux des smectites ; sa position varie en fonction de la chimie de la couche octaédrique et, dans une moindre mesure, de la chimie de la couche tétraédrique. La largeur et les épaulements de cette bande sont ainsi dus aux différents environnements structuraux des groupements (OH) au sein des structures cristallines des smectites. Seule la décomposition des spectres expérimentaux en composantes élémentaires permet d'identifier la contribution de chaque environnement [5]. Cependant, la position des maxima de ces bandes donne des indications claires sur la chimie des smectites. Ainsi, pour la smectite SAz-1, la bande située à 3620 cm−1 caractérise une smectite alumineuse, tandis que, pour l'échantillon Ölberg, la bande à 3560 cm−1 indique qu'il s'agit d'une smectite ferrique [8]. La bande large culminant à 3400 cm−1 est due à l'eau interfoliaire.
Par ailleurs, le spectre (IR) obtenu à partir des agrégats orientés permet également d'étudier les bandes de combinaison νOH+δOH, au-delà de 4000 cm−1. Sur la Fig. 2, le spectre IR de l'agrégat orienté, dilaté dans ce domaine de nombre d'ondes, apparaı̂t très similaire au spectre de poudre obtenu par réflexion diffuse (DRIFTS).
Les spectres IR de la kaolinite KGa-2 et de la dickite de Sainte-Claire, obtenus à partir de pastilles de KBr et d'agrégats orientés déposés sur lame de verre, sont présentés sur la Fig. 3. Les spectres obtenus par les deux méthodes sont très semblables et permettent aisément de distinguer les deux polymorphes par la position et l'intensité relative des bandes [8].
4 Discussion et conclusions
Les résultats présentés montrent qu'il est possible d'obtenir un spectre IR à partir d'un dépôt d'argile sur une lame de verre, utilisé préalablement pour la DRX. Grâce à cette technique, (i) un spectre IR est obtenu en un temps très court (1–2 min) correspondant à sa seule acquisition (pas de préparation spécifique) ; (ii) le spectre IR est obtenu à partir du même matériel que celui ayant servi à la DRX, ce qui est parfois difficile à réaliser, notamment pour les échantillons hétérogènes.
Bien entendu, cette technique ne remplace pas totalement celle des échantillons pressés dans des pastilles de KBr. En effet, les bandes des groupements Si–O et δOH, situées au-dessous de 2000 cm−1, ne peuvent pas être étudiées à cause de l'absorption par la lame de verre. L'étude de la région des bandes νOH est cependant souvent très suffisante pour répondre aux questions minéralogiques et/ou cristallochimiques posées (par exemple présence ou absence de kaolinite, distinction entre les polymorphes des kaolins, etc.). Les spectres des minéraux argileux dans le proche infrarouge peuvent aussi être obtenus par cette technique. Bien que moins couramment utilisée, cette région spectrale permet aussi de déterminer la cristallochimie des argiles [6].
Une limitation à l'utilisation des dépôts d'argile sur lame de verre tient à l'orientation des particules par rapport au faisceau IR. L'adsorption du rayonnement IR par une liaison interatomique est en effet maximum, quand le vecteur polarisation associé au mode de vibration est parallèle au vecteur champ électrique du faisceau (c'est-à-dire perpendiculaire à la direction de propagation de ce dernier), et sera minimum quand le vecteur sera perpendiculaire au vecteur champ électrique. Ainsi, pour un dépôt orienté d'argile trioctaédrique, comme le talc, cet effet pléochroı̈que sera maximum pour les modes de vibration νOH, puisque leurs vecteurs polarisation, étant parallèles à l'axe cristallographique , seront perpendiculaires au vecteur champ électrique du faisceau IR. Les bandes νOH seront donc d'intensité faible à nulle. Pour les minéraux dioctaédriques, l'angle entre les dipôles O–H et l'axe variant entre 75 et 80°, les effets dus à l'orientation des particules seront moins forts. Ils sont, cependant, probablement en partie à l'origine des différences d'intensité observées entre les spectres IR obtenus à partir de pastilles de KBr et d'agrégats orientés (Figs. 1 et 3).
Les dépôts d'argile sur lame de verre, facilement obtenus, pourront par ailleurs se substituer, pour des études en spectroscopie IR spécifiques, à des préparations plus difficiles à réaliser, comme les dépôts sur supports transparents aux IR (AgCl, CaF2), films autosupportés, ... .
L'approche présentée ici pourra, d'autre part, être efficacement utilisée pour l'étude par spectroscopie IR de lames minces de roche (épaisseur 20 μm), préparées pour des études pétrographiques et collées sur lames de verre, les bandes d'absorption des résines se situant au-dessous de 3200 cm−1.