

Abridged English version
1 Introduction
The iron mines of Lorraine have an impact on water resources with associated major environmental consequences. In order to develop a new management scheme for this water resource, it is therefore necessary to predict any flow and water-quality modifications. The first step toward the modelling of the change of the water quality is to define both the pollution and the implicated chemical mechanisms.
2 Experimental study
The Lorraine iron-bearing deposit, dating from the Late Lias, is made of alternating marly levels (intercalations) and ferruginous limestone (mineralised layers) [4]. Moving upwards, transition from the marly facies to the lowest mineralised facies is progressive via a ‘basal’ layer. On top of the mineralised facies, the transition into the next overlying marl intercalation occurs via a level locally known as ‘crassin’. Our experimental studies were focused on ‘marls’, which, according to Hervé's results [10], release the largest quantity of sulphates. Thirteen samples were collected in the mine concessions, which are still being drained in the northern part of the Briey Basin. They were taken directly from the hanging walls of the layers or in the crosscuts.
A mineralogical study of these samples leads to distinguish two types of samples: ‘crassin’ and marl intercalations [8]. They are both rich in carbonate, quartz and goethite, but marl intercalations contain also pyrite, sulphates, phyllosilicates, plagioclases and microcline.
Closed reactor tests were carried out [7]. They consisted in bringing rock (5 mm<∅<10 mm) into contact with Dogger water (water supplying the mine) (Table 1) so as to characterise water–rock chemical equilibrium. The results of these experiments show that the two types of samples react differently. The marl intercalations release much more elements than the ‘crassin’. With the rock contact, the water is strongly enriched with SO4, Mg, Na, Mn, Ca, K and B, and, to a lesser degree, with Cl and Sr (Fig. 1).
Concentrations des éléments majeurs dans l'eau du Dogger
Concentrations observed in the Dogger water
| Élément | pH | Conductivité (mS cm−1) | B (μg l−1) | Ca (mg l−1) | HCO3 (mg l−1) | Fe (mg l−1) | K (mg l−1) | Mg (mg l−1) | Na (mg l−1) | SO4 (mg l−1) | Sr (mg l−1) |
| Concentration | 7,6 | 0,41 | <20 | 85,5 | 234 | <400 | 0,8 | 7,8 | 3,2 | 17,3 | 0,19 |
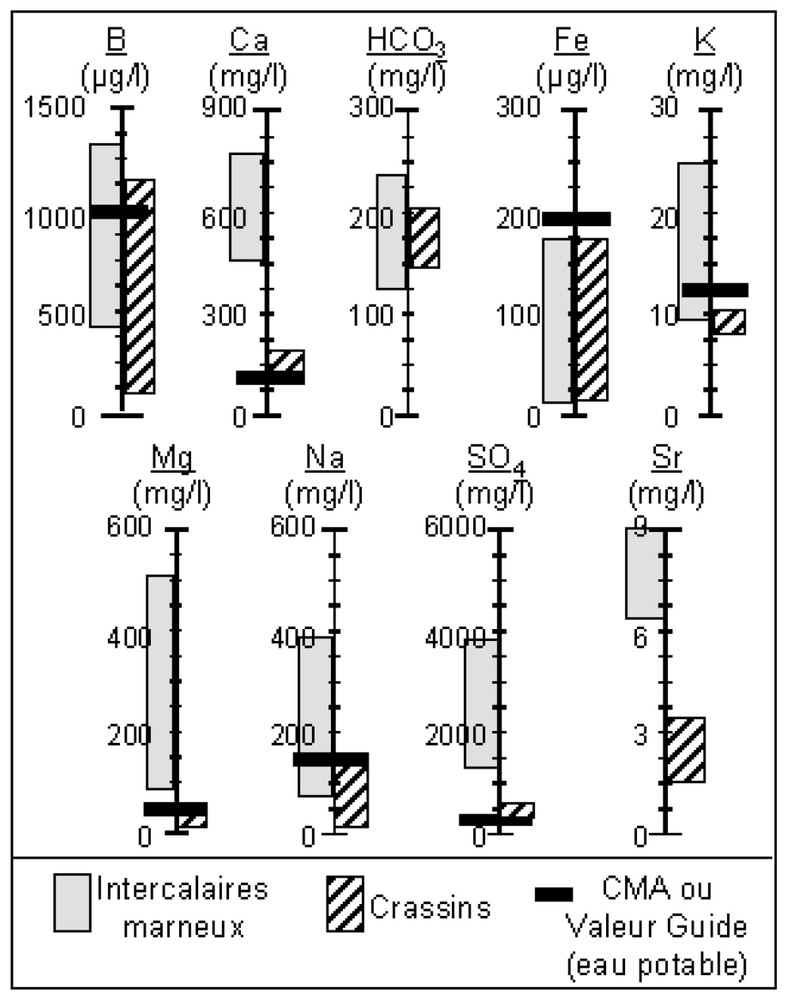
Gammes des concentrations observées dans l'eau à l'équilibre avec la roche et comparaison avec les concentrations maximales admissibles (CMA).
Concentrations observed in water in thermodynamic equilibrium with rocks and comparison with the Maximum Admissible Concentrations (CMA).
Two reactions can explain the observed SO4 concentrations: oxidation of pyrite and/or dissolution of gypsum. The first reaction needs oxygen as the oxidising agent (reaction (1)) and, combined with the dissolution of carbonates, releases carbon dioxide (reaction (2)):
| (1) |
| (2) |
A continuous measurement of gas composition was conducted during a closed reactor test. Neither O2 consumption nor CO2 production was detected (Fig. 2), but a concentration of 23.9 mmol l−1 of sulphate was measured. The quantity of consumed O2 can only explain 0.84% of the total produced sulphate. Moreover, another closed reactor test was made with desaerated water in a vacuum cell. High sulphate concentrations were observed without gaseous or dissolved oxygen. These experiments confirm that the high sulphate concentration in water during the flooding of abandoned workings is due to the dissolution of gypsum and not directly to pyrite oxidation.
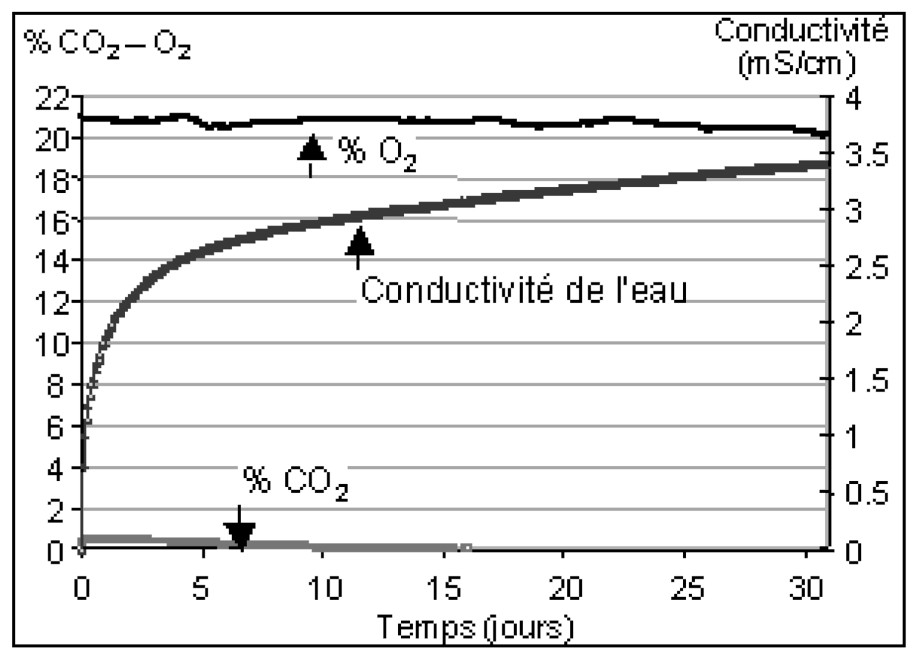
Évolution de la composition des gaz lors d'un essai en réacteur fermé. La conductivité traduit la quantité d'éléments en solution, donc l'avancement de la réaction.
Evolution of the gas composition during a closed reactor test. Conductivity expresses total dissolved elements, so the furtherance of the reaction.
3 Modelling
In order to understand the chemical mechanisms that could explain the composition of water, we built a kinetic chemical model. The formulation of the kinetics used is conventional, taking for example the following reaction:
Initially, we assumed standard conditions of temperature and pressure (25 °C, 1 bar), as used in our laboratory experiments. The values of K(T) for each one of the reactions were taken directly from the PHREEQC thermodynamic database [12]. The activity of the minerals and the water was considered as 1, whereas the activity of the gaseous species was assumed to be equal to their partial pressure. The activity coefficient of the ions is defined by the extended Debye–Hückel equation.
Different scenarios were tested by modifying the apparent kinetic constants (Table 2). The best agreement with experimental data was obtained with a model based on 12 chemical elements (Al, C, Ca, Fe, K, H, Mg, Na, O, S, Si, and Sr), 19 minerals dissolution and precipitation reactions, 25 aqueous speciation reactions, dissolution and degassing of O2 and CO2 and 4 cationic exchange reactions.
Réactions introduites dans le modèle géochimique et constantes associées
Reactions of the geochemical model and their constants
| LogK(T) T=25 °C | k pcp | b ∗ | |
| Réactions de dissolution/précipitation de minéraux | |||
|
|
−6,1 | 8,30×10−4 | 1 |
|
|
−18,002 | 1,76×10+3 | 1 |
|
|
−3,55 | 1,90×10−4 | 0,005<b<3,5 |
| CaCO3+H+↔Ca+++HCO3− | 1,849 | 1,05×10−4 | 0,08<b<4,2 |
| SrSO4↔Sr+++SO42− | −6,63 | 2,81 | 0,1<b<2,2 |
|
|
34,58 | 3,90×10+7 | 1 |
|
|
3,568 | 6,55×10−3 | 1 |
|
|
−7,669 | 8,67×10−6 | 0,7<b<3,4 |
|
|
−8,79 | 23,7 | 1 |
| FeOOH+H2O↔Fe(OH)3 | −13,56 | 3,02×10−10 | 1 |
|
|
−4,58 | 9,63×10−2 | 0,3<b<3,3 |
|
|
−29,128 | 5,13×10−4 | 1 |
|
|
−20,573 | 3,08×10−3 | 1 |
|
|
−37,997 | 7,39×10+6 | 1 |
|
|
−37,965 | 8,00×10+31 | 1 |
|
|
211,441 | 1,64×10−203 | 1 |
|
|
−3,98 | 1,89×10−6 | 1 |
|
|
−4,621 | 9,13×10−6 | 1 |
| SrCO3+H+↔Sr+++HCO3− | 1,058 | 9,25×10−8 | 1 |
| Réactions de complexation des ions | |||
| H2O↔H++OH− | 14 | 10+18 | 1 |
| Al(OH)3+H+↔Al(OH)2++H2O | 6,8 | 10+4 | 1 |
| Al(OH)3+H2O↔Al(OH)4−+H+ | −5,8 | 6,3×10+9 | 1 |
| HCO3−↔H++CO32− | −10,329 | 2,1×10+14 | 1 |
| Ca+++HCO3−↔CaCO3+H+ | −7,105 | 1,27×10+11 | 1 |
| Ca+++HCO3−↔CaHCO3+ | 1,106 | 10+4 | 1 |
| Ca+++H2O↔CaOH++H+ | −12,78 | 6×10+16 | 1 |
| Ca+++SO42−↔CaSO4 | 2,3 | 10+4 | 1 |
| Fe(OH)3+H+↔Fe(OH)2++H2O | 6,89 | 10+4 | 1 |
| Fe(OH)3+H2O↔Fe(OH)4−+H+ | −9,04 | 10+13 | 1 |
| H2O+O2↔H2 | −92,38 | 10+50 | 1 |
| CO2+H2O=H++HCO3− | −6,352 | 5×10+10 | 1 |
| H4SiO4↔H++H3SiO4− | −9,83 | 6,8×10+13 | 1 |
| H++SO42−↔HSO4− | 1,988 | 10+4 | 1 |
| K++SO42−↔KSO4− | 0,85 | 10+4 | 1 |
| Mg+++CO32−↔MgCO3 | 2,98 | 10+4 | 1 |
| Mg+++HCO3−↔MgHCO3+ | 1,07 | 10+4 | 1 |
| Mg+++H2O↔MgOH++H+ | −11,4 | 2,5×10+15 | 1 |
| Mg+++SO42−↔MgSO4 | 2,37 | 10+4 | 1 |
| Na++CO32−↔NaCO3− | 1,27 | 10+4 | 1 |
| Na++HCO3−↔NaHCO3 | −0,25 | 1,8×10+4 | 1 |
| Na++SO42−↔NaSO4− | 0,7 | 10+4 | 1 |
| Sr+++CO32−↔SrCO3 | 2,81 | 10+4 | 1 |
| Sr+++HCO3−↔SrHCO3+ | 1,18 | 10+4 | 1 |
| Sr+++SO42−↔SrSO4 | 2,29 | 10+4 | 1 |
| Réactions d'échange cationique | |||
|
|
1,439 | 8,55×10−3 | 1 |
|
|
0,548 | 0,44 | 1 |
|
|
1,349 | 3,93×10−2 | 1 |
|
|
−5,492 | 30,4 | 1 |
| Réactions de dissolution/dégazage | |||
| CO2(g)↔CO2 | −1,468 | 3×10−6 | 1 |
| O2(g)↔O2 | −2,96 | 1,97×10−7 | 1 |
∗ b varie d'un échantillon à l'autre pour les minéraux. Ces variations n'ont d'influence que si les réactions n'atteignent pas l'équilibre thermodynamique, mais en sont proches [8].
These simulations reproduce the concentrations observed in water at the end of the closed reactor tests (Fig. 3).
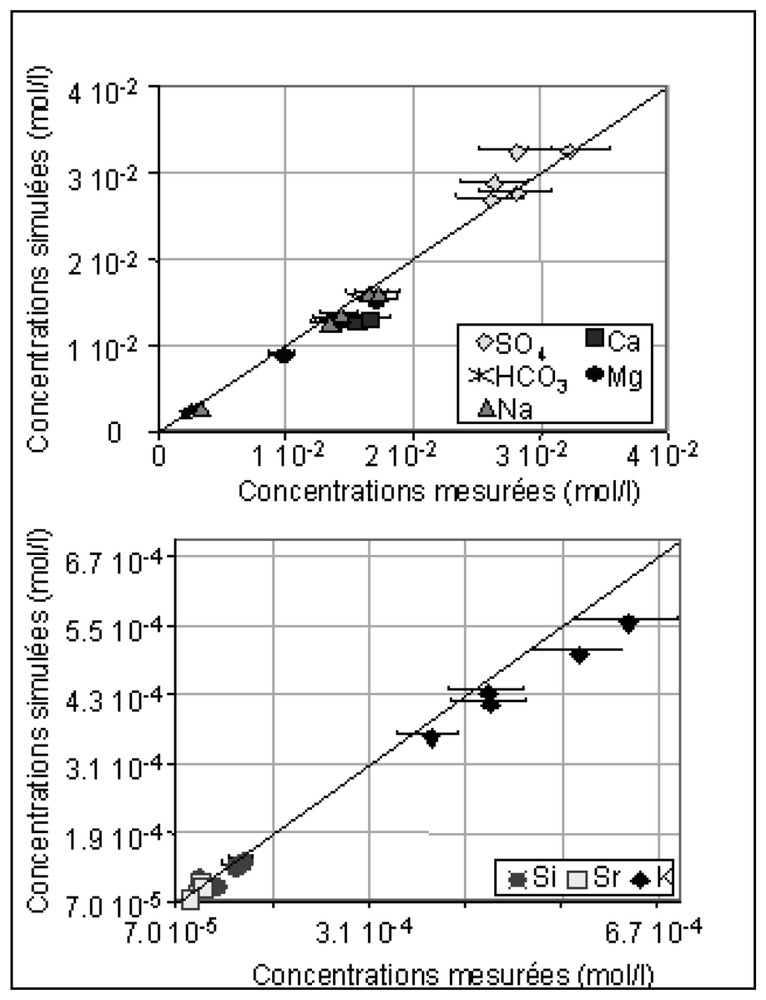
Corrélations entre les concentrations simulées et calculées.
Comparison between simulated and calculated concentrations.
4 Conclusions
The experimental study and the construction of a chemical model offer a better understanding of the mechanisms that control the water composition during flooding of the Lorraine iron mines.
The high sulphate concentrations result from the dissolution of gypsum. The gypsum is produced during the cutting of the mine galleries, through the oxidation of pyrite and neutralisation by carbonates. In the same way, strontium is released by the dissolution of celestite formed during the exploitation. During the workings, the local falling pH caused by pyrite oxidation favours local dissolutions of albite [9], illite and mica, and releases Mg2+, Na+ and K+ ions. These ions fix themselves onto cationic exchange sites. During the flooding of abandoned workings, exchange reactions within Ca2+, Mg2+, Na+, K+ and H+ occur at the surface of ferri-hydroxides and/or organic matter and control their concentrations in water. So, the production of Ca by dissolution of gypsum favours the release of Mg2+, Na+ and K+. The concentrations of Ca and Mg are also controlled by the precipitation of calcite and dolomite.
The concentration of Si is essentially controlled by the precipitation of kaolinite, the dissolution of chalcedony and the equilibrium with quartz.
1 Introduction
Le gisement de fer lorrain, un des plus vastes du monde, a été exploité pendant plusieurs siècles. Depuis la crise de la sidérurgie, dans les années 70, le manque de rentabilité de son extraction entraı̂ne la fermeture progressive des mines et un arrêt des pompages d'exhaure. Cet arrêt provoque un ennoyage des travaux miniers, dont l'une des conséquences est la détérioration de la qualité de l'eau souterraine. Ainsi, l'eau des mines ennoyées du bassin ferrifère lorrain (BFL) se caractérise par de fortes concentrations en sulfate, sodium, magnésium, manganèse et bore, qui la rendent impropre aussi bien pour une utilisation industrielle qu'à l'alimentation en eau potable (AEP). Cependant, les observations réalisées sur des mines ennoyées, depuis plus de 20 ans, montrent que cette contamination tend à diminuer progressivement. Cette décroissance s'effectue à un rythme propre à chaque bassin minier, que nous nous proposons de prévoir à l'aide d'un modèle hydrochimique.
L'objet de cet article est de présenter la première étape vers la construction d'un tel modèle, c'est-à-dire l'identification des réactions impliquées dans l'évolution chimique de l'eau pendant et au-delà de l'ennoyage. À cet effet, des expériences en laboratoire ont été réalisées et un modèle reproduisant les résultats expérimentaux a été construit.
2 Travaux expérimentaux
La formation ferrifère est constituée d'une alternance de calcaires ferrugineux, le minerai exploité, et de couches moins riches en fer appelées communément « intercalaires marneux ». Ces derniers sont formés d'une succession de siltites argileuses et carbonatées s'organisant en rythmes décimétriques à pluri-décimétriques et dont la composition est alternativement plus argilo-silteuse ou plus calcaire. Le passage d'un faciès marneux au faciès minéralisé sus-jacent se fait progressivement par un intermédiaire métrique ou « pied de couche ». Un niveau de calcaire coquillier grossier, ou « crassin », sépare parfois le minerai de l'intercalaire marneux [4].
Les expériences réalisées en 1980 par Hervé [10] sur les niveaux géologiques exploités dans la concession de Valleroy, minerais et toits compris, ont montré que les intercalaires marneux sont les principaux responsables de l'augmentation de la concentration en sulfate dans l'eau. Nous avons donc centré nos travaux sur ces niveaux géologiques.
Dans le secteur nord du bassin de Briey, qui n'est pas encore ennoyé, treize échantillons d'environ 30 à 40 kg ont été détachés directement au toit des couches minéralisées ou au niveau des travers bancs. Dans ce dernier cas, les échantillons proviennent donc du centre des intercalaires marneux. Plusieurs échantillons sont prélevés par couche, en des lieux distincts, dans le but d'évaluer la variabilité spatiale de la composition minéralogique au sein d'un même niveau stratigraphique. Les échantillons d'un même niveau sont distants de 0,2 à 4 km.
2.1 Étude minéralogique
Les échantillons ont fait l'objet d'une étude minéralogique complète comprenant l'observation de lames minces et sections polies au microscope à lumière transmise et au microscope à lumière réfléchie, une diffraction aux rayons X et une analyse chimique élémentaire. Une observation au microscope électronique à balayage (MEB) a également été effectuée sur quelques fragments rocheux [6].
Deux types d'échantillons ont été distingués : les « crassins » et les « intercalaires marneux » [8]. Ils contiennent tous deux des carbonates (calcite et, à moindre teneur, sidérite et dolomite), du quartz et de la gœthite. Les intercalaires marneux se caractérisent par la présence supplémentaire de pyrite, de sulfates, de phyllosilicates (chlorite ferrifère, illite, kaolinite et mica), de plagioclases et de microcline. La présence de la pyrite, détectée en traces par diffraction aux rayons X, a été confirmée par l'observation de sections polies dans tous les échantillons d'intercalaire marneux. Elle se présente en amas framboı̈des dispersés dans le ciment intergranulaire, sa forme la plus réactive. Des cristaux néoformés sont visibles à l'œil nu sur les plans de fractures des échantillons d'intercalaires marneux. Une analyse chimique élémentaire et une diffraction aux rayons X ont montré qu'il s'agit de gypse.
2.2 Essais en système fermé
Afin d'étudier les équilibres chimiques qui s'établissent entre l'eau et la roche lors de l'ennoyage des mines de fer lorraines, des essais en réacteur fermé ont été réalisés [7]. Environ 0,59 kg de roche broyée (diamètre des grains inférieur à 5 mm) et 0,28 kg d'eau du Dogger (Tableau 1), qui alimente le réservoir minier, ont été introduits dans des flacons d'un demi-litre en polypropylène. Chaque essai est doublé (flacons A et B). Les flacons sont régulièrement agités manuellement pour assurer l'homogénéité du mélange. Le système A reste fermé jusqu'à ce que la conductivité électrique de la solution B se stabilise. Cette dernière étant fonction de la quantité d'ions en solution, son augmentation traduit une solubilisation des éléments de la roche. On suppose que, lorsque la conductivité se stabilise, l'équilibre chimique eau-roche est atteint. L'eau est alors prélevée dans les flacons A pour analyse par ICP–MS.
Ces essais mettent en évidence deux degrés de réactivité chimique, qui correspondent aux deux types minéralogiques d'échantillons : les crassins et les intercalaires marneux. L'eau en contact avec les crassins ne se charge que faiblement : sa composition ne dépasse les concentrations maximales admissibles (CMA) pour l'alimentation en eau potable que dans le cas du sulfate (Fig. 1). Au contact des intercalaires marneux, l'eau s'enrichit en SO4, Mg, Na, Ca, K et B, et, dans une moindre mesure, en Sr, Mn et Cl. Quelles que soient les concentrations dans l'eau, le pH reste compris entre 7 et 8, confirmant le caractère neutre du drainage des mines de fer de Lorraine. Certains échantillons ont des teneurs en NO3 élevées correspondant vraisemblablement à des traces de l'explosif utilisé lors de l'exploitation, le nitrate-fuel composé de nitrate d'ammonium. Ces teneurs sont indépendantes des concentrations des autres éléments dans l'eau et peuvent être élevées ou faibles aussi bien pour des échantillons de crassin que de travers-banc.
2.3 Mise en évidence des réactions chimiques responsables de la mise en solution du SO4
D'après l'étude minéralogique des échantillons, deux minéraux peuvent être à l'origine de la sulfatation de l'eau : la pyrite et le gypse.
Lorsque le pH de l'eau est supérieur à 7, l'oxydation de la pyrite nécessite l'oxygène comme oxydant (réaction (1)). En présence de carbonates, comme c'est le cas dans nos expériences, cette réaction s'accompagne d'une réaction de dissolution des carbonates (réaction (2)) qui maintient le pH autour de 7 et déplace l'équilibre de la réaction (3) vers la production de CO2 gazeux. C'est le drainage minier neutre (par exemple, [2,3,11, etc.]). Lors de l'exploitation minière, le milieu n'est pas saturé en eau de par les pompages d'exhaure. De ce fait, le sulfate et le calcium produits par les réactions (1) et (2) précipitent sous forme de gypse (réaction (4)) (par exemple, [2,3,5,13, etc.]) et explique l'observation, dans nos échantillons, de cristaux de gypse néoformés sur les surfaces de fracture [6]. Lors de l'ennoyage, ce gypse peut constituer une source de sulfate par dissolution dans l'eau.
| (1) |
| (2) |
| (3) |
| (4) |
La présence de nitrate dans la roche, liée à l'explosif utilisé, pourrait également conduire à une réaction d'oxydation de la pyrite (réaction (5)). Dans ce cas, un dégagement d'azote serait observé et la consommation d'ion H+ déplacerait les équilibres des réactions (2) et (3) vers la consommation de CO2 gazeux :
| (5) |
Pour déterminer, parmi ces réactions, lesquelles interviennent dans la sulfatation de l'eau, lors de l'ennoyage, les pressions partielles d'O2 et de CO2 ont été mesurées en continu dans la fraction gazeuse au cours d'un essai en réacteur fermé.
Au cours de cette expérience, aucune modification significative de la composition des gaz n'a été observée (Fig. 2). La pression partielle d'O2 ne montre aucune tendance générale et sa variation a une amplitude inférieure à 1 %. La pression partielle de CO2 décroı̂t lentement jusqu'à être inférieure au seuil de détection de l'appareil de mesure. Les évolutions de ces pressions partielles peuvent aussi bien être liées à la dérive de l'appareil qu'à de vraies tendances.
Au terme de l'essai, la concentration de l'eau en SO4 est de 23,9 mmol l−1, ce qui correspond à une production totale dans la colonne de 160 mmol de SO4. La réaction (1) indique que 1,75 mol d'O2 sont consommées lorsqu'une mole de SO4 est produite. Si tout le SO4 provient de l'oxydation de la pyrite par l'oxygène, alors 280 mmol d'O2 ont été consommées. Or, si l'on suppose qu'au début de l'essai l'eau est en équilibre avec l'atmosphère, alors le dispositif expérimental contient seulement 19 mmol d'O2 sous forme gazeuse ou dissoute. Cette quantité d'O2 est donc insuffisante pour expliquer la concentration en SO4 observée selon l'hypothèse d'une oxydation de la pyrite. La variation de pression partielle d'O2 de 1 %, variation maximale observée, ne pourrait expliquer que la production de 1,35 mmol de SO4 par oxydation de la pyrite, soit moins de 1 % de la teneur en SO4 observée.
Dans une seconde expérience, réalisée dans une cellule sous vide, la mise en contact d'échantillons d'intercalaire marneux avec de l'eau désaérée, dépourvue d'oxygène, a entraı̂né la dissolution de 21,9 mmol· l−1 de SO4. Ceci confirme les résultats précédents et montre que l'oxydation de la pyrite n'est pas la réaction dominante productrice de SO4 au moment de l'ennoyage. L'eau joue le rôle d'une barrière à la diffusion de l'oxygène vers les sites réactifs et l'ennoyage limite donc les réactions d'oxydation par l'oxygène.
En ce qui concerne l'oxydation de la pyrite par le nitrate, la réaction (5) implique une production d'azote. Expérimentalement, dans un système fermé, la mise en solution de 160 mmol l−1 de SO4 par oxydation de la pyrite par le nitrate aurait dû s'accompagner d'une production de 112 mmol de N2. Cependant, la pression partielle de N2 n'a pas été mesurée. En revanche, la consommation d'ions H+ de la réaction (5) aurait dû se traduire par une augmentation de pH et/ou une consommation de CO2. Le pH étant resté compris entre 7,6 et 7,8 durant toute l'expérience, seule une dissolution de 64 mmol de CO2 et une précipitation de calcite aurait pu fournir les ions H+ nécessaires. Or, initialement, le dispositif ne contenait que 0,27 mmol de CO2. Par ailleurs, la présence de nitrate est occasionnelle dans les échantillons étudiés, et nous pouvons penser que la quantité d'explosif résiduel est faible en comparaison de la quantité de roche mise en jeu dans les zones foudroyées. Par conséquent, nous avons considéré cette réaction comme non dominante pour expliquer l'évolution chimique de l'eau.
À l'inverse de l'oxydation de la pyrite par l'oxygène, la dissolution de sulfates néoformés, comme le gypse, ne fait intervenir aucun gaz dissous. Dans les échantillons d'intercalaire marneux, la quantité totale de soufre sous forme sulfate a été déterminée par une méthode gravimétrique selon la norme NF ISO 11048. Elle est toujours égale ou inférieure à la limite de détection (=21 mmol de SO4 par kg de roche). Or, dans chaque essai, les quantités de gypse nécessaires pour expliquer les concentrations en SO4 observées sont inférieures à cette limite de détection. La dissolution du gypse peut donc être considérée comme la principale réaction responsable de l'augmentation des concentrations en SO4 lors de l'ennoyage.
2.4 Bilans de matière
Les échantillons prélevés dans la mine ont également fait l'objet d'expériences de lessivage en colonne. Chacune des six colonnes du dispositif de lessivage a été remplie avec 16,3 kg de roche broyée (20<∅<50 mm) et 8 kg d'eau du Dogger, soit le même rapport en masse eau–roche que dans les essais en réacteur fermé. Dans une première étape, le système reste fermé afin d'approcher l'équilibre chimique eau–roche. Dans une seconde étape, le système est ouvert, afin d'observer l'évolution chimique en conditions de lessivage. Chaque colonne est alors alimentée à sa base par un débit de 200 ml j−1 d'eau du Dogger. L'eau déborde par une ouverture située au sommet de la colonne où des échantillons, régulièrement prélevés, sont analysés. La conductivité et la température de l'eau dans la colonne sont mesurées en continu.
Dans les colonnes, le pH oscille entre 7,2 et 8,2. Les concentrations de HCO3 et Si restent constantes. En revanche, la conductivité diminue progressivement jusqu'à redevenir à peu près égale, après 200 jours de lessivage, à celle de l'eau du Dogger. De la même manière, les concentrations en SO4, Ca, Mg, Na, K, Mn, B et Sr diminuent au fur et à mesure du lessivage, jusqu'à ce que l'eau, dans la colonne, ait une composition semblable à celle de l'eau d'alimentation du système. Par conséquent, la quantité d'éléments extractibles dans la colonne est limitée et définie par la quantité de roche totale. Dans la mine, une fois le remplissage achevé, les infiltrations d'eau atmosphérique entraı̂neront un lessivage similaire à celui des expériences en colonne. Si la quantité d'éléments extractibles dans ces roches est limitée, alors la pollution de l'eau provoquée par l'ennoyage doit s'atténuer naturellement au fur et à mesure du lessivage du réservoir minier.
L'extrapolation, puis l'intégration en fonction du temps des concentrations mesurées ponctuellement en sortie de colonne permettent d'estimer la quantité extractible de chaque élément dans les divers échantillons. Les masses extractibles de chaque élément, exception faite du Na, représentent moins de 10 % de la quantité totale de ces mêmes éléments mesurée dans la roche. La précision des analyses étant de 5 à 10 %, la variation de la composition de la roche se situe à la limite de détection des techniques mises en œuvre. Ces expériences ne permettent donc pas de déterminer directement les minéraux responsables de l'évolution de la qualité de l'eau.
3 Modélisation géochimique
Afin de représenter les réactions chimiques entre l'eau et la roche, nous avons construit un simulateur numérique qui s'appuie sur un modèle cinétique basé sur la thermodynamique chimique. Considérons une réaction de dissolution–précipitation d'un minéral AnB, représentée par l'équation suivante :
La vitesse totale de réaction (en mol s−1) est définie par la différence entre les vitesses de dissolution (vdiss) et de précipitation (vpcp) :
L'activité des minéraux, (AnB), comme celle de l'eau, est considérée égale à 1 (pôle pur). Celle des composés gazeux est supposée égale à leur pression partielle (gaz parfait). L'activité d'un ion dans l'eau, (A+), est définie par : (A+)=γA+[A+], où [A+] est la concentration de l'ion A+ en solution et γA+ est son coefficient d'activité. Pour calculer le coefficient d'activité des ions, nous avons utilisé la formulation de Debye–Hückel. Les valeurs des constantes d'équilibre K(T) dans les conditions de température et de pression des expériences (25 °C, 1 bar) sont directement issues de la base de données PHREEQC [12] (Tableau 2). Dans notre modèle, les réactions de spéciation des ions dans l'eau ont une écriture cinétique. Leurs constantes kpcp ont été fixées de manière à rendre compte de la grande rapidité de ces réactions par rapport à celles de dissolution–précipitation des minéraux.
Différents scénarios réactionnels ont été testés. Pour chacun, une composition minéralogique probable a été calculée en cohérence avec les données de l'étude minéralogique. Tous les minéraux identifiés en diffraction X ont été systématiquement introduits dans la composition de la roche. Les constantes de cinétiques apparentes ont été ensuite ajustées par essais/erreurs afin de reproduire les résultats expérimentaux (Tableau 2).
Le modèle chimique retenu comprend 12 éléments chimiques (Al, C, Ca, Fe, K, H, Mg, Na, O, S, Si et Sr), 19 réactions de dissolution/précipitation de minéraux, 25 réactions de complexation des ions dans l'eau, les réactions de dissolution/dégazage de l'O2 et du CO2 et 4 réactions d'échange cationiques. Appelo et al. [1] ont montré que des réactions d'échanges impliquant les ions Ca++, Mg++, Na+, K+ et H+ pouvaient s'opérer à la surface des oxyhydroxydes de fer et/ou de la matière organique.
Ce modèle permet de reproduire le pH avec une précision de ±0,1 unité pH et les concentrations en SO4, Fe, HCO3, Mg, Si, Na, K et Sr observées dans les expériences en réacteur fermé avec une précision de ±10 %. Les concentrations en Al simulées sont de l'ordre de 10−8 mol l−1, soit, tout comme les concentrations mesurées, inférieures à la limite de détection de la méthode d'analyse (=2×10−6 mol l−1). La Fig. 3 illustre les corrélations entre les résultats analytiques et ceux obtenus par la simulation. En tenant compte des incertitudes des analyses de laboratoire, les points correspondant au SO4, Fe, HCO3, Mg, Si, Na, K et Sr se situent aux environs immédiats de la première bissectrice, ce qui traduit la bonne adéquation entre les concentrations mesurées et simulées.
4 Conclusion
En exploitant nos résultats expérimentaux et les données bibliographiques, un modèle chimique cinétique a été construit. Il permet de reproduire les concentrations en SO4, Ca, Mg, Na, K, Sr, B et Si observées dans l'eau à l'issue des essais en réacteur fermé. Ces résultats permettent de proposer des schémas réactionnels simples et validés par l'expérimentation, replacés dans un scénario en deux phases pour expliquer l'évolution de la chimie de l'eau lors de sa mise en contact avec les intercalaires marneux du BFL.
La première phase correspond à l'ouverture de la mine et à son exploitation, qui place le milieu en conditions oxydantes. Il y a oxydation de la pyrite. Cette réaction entraı̂ne une dissolution des carbonates, qui conduit à la précipitation de néo-cristaux de gypse, et en moindre quantité de célestite. Les baisses locales de pH provoquées par l'oxydation de la pyrite favoriseraient les dissolutions lentes de l'albite [9], du clinochlore, du microcline et de l'illite libérant des ions Na+, Mg2+ et K+. Ceux-ci seraient fixés sur des sites d'échanges cationiques.
La seconde phase correspond à l'ennoyage des travaux miniers. La dissolution du gypse néoformé explique les fortes concentrations en SO4 et Ca dans l'eau. Comme l'eau limite fortement la diffusion de l'oxygène jusqu'aux sites réactifs, l'ennoyage entraı̂ne un arrêt des réactions d'oxydation dans le réservoir. La concentration en oxygène dissous est donc un facteur limitant pour les dissolutions de la sidérite et de la pyrite. La précipitation de calcite et de dolomite tend à abaisser les teneurs en Ca et Mg. Des échanges cationiques à la surface des oxyhydroxydes de fer et/ou de la matière organique [1] contrôleraient les concentrations en Na, Mg, K et Ca, et constitueraient également un tampon pH. Ainsi, l'augmentation de la concentration en ions Ca++, liée à la dissolution du gypse, entraı̂nerait indirectement une mise en solution des ions Na+, Mg++ et K+. La baisse de pH liée à l'équilibre calco-carbonate serait tamponnée par la fixation de H+ sur ces mêmes sites d'échange. La concentration en Si est essentiellement contrôlée par les réactions de dissolution de la calcédoine, de précipitation de kaolinite et de dissolution/précipitation du quartz. La dissolution de célestite permet de reproduire les concentrations en Sr observées.
Ces résultats constituent une base de travail pour la construction d'un modèle hydrochimique des réservoirs miniers [6]. Un tel modèle permet d'estimer le temps nécessaire pour que les eaux du BFL reviennent à une concentration proche de la potabilité. Il nécessite un couplage aux phénomènes de transport et sera développé dans une publication ultérieure.
Vous devez vous connecter pour continuer.
S'authentifier