

1 Introduction
The actinides (Z = 90 to 103) form the row of elements in the periodic chart across which the 5f atomic subshell is sequentially filled with electrons. The electronic structure provides the first hint of the apparently “anomalous” chemical behavior of these elements, particularly plutonium. As metals, there is a slight actinide “contraction” with the addition of each 5f electron because these electrons enter the conduction band, and the increased attractive nuclear force decreases the atomic size. However, at americium (95) each 5f subshell is occupied, and the atomic size increases dramatically as the 5f electrons become more localized and chemically less reactive. Subsequent additions of electrons lead to only a minor contraction of atomic radii. For the pure elements to the left of Pu, the 5f electrons are delocalized (bonding), and for those to the right, the electrons are localized (non-bonding). Pu is exactly in the middle of these two types of bonding behavior.
The electronic structure also leads to a rather complicated geochemistry (Runde, 2000). For the lighter actinides, the energy levels of the 6d and 5f orbitals are similar, thus leading to a tendency to provide more bonding electrons to chemical reactions. Plutonium can exist in five oxidation states between Pu(III) and Pu(VII). Coordination numbers vary between 3 to 12. Under typical oxidizing conditions encountered at the Earth's surface, Pu can exist as Pu(III), Pu(IV), Pu(V) and Pu(VI) as shown in the eH-pH diagram (Fig. 1). Typically, actinides in the (V) and (VI) oxidation states form actinyl ions: (UO2)2+, (PuO2)22+, (PuO2)+, (NpO2)+. These molecules readily form complexes in solution or polymerize to give many different solution species and crystal structures. As an example, U6+ forms hundreds of different mineral structures (Burns, 1999, 2005; Burns et al., 1996). The variety of oxidation states and strong tendency to form complexes, particularly with carbonates, provide a variety of mechanisms for the transport of actinides in the environment (Silva and Nitsche, 2002). Recent interest in actinide geochemistry has been motivated by the need to remediate contaminated sites, model the behavior of actinides in geologic repositories for high-level radioactive waste, and develop methods and materials that retard or prevent their release to the environment–a type of sequestration for actinides, particularly Pu.
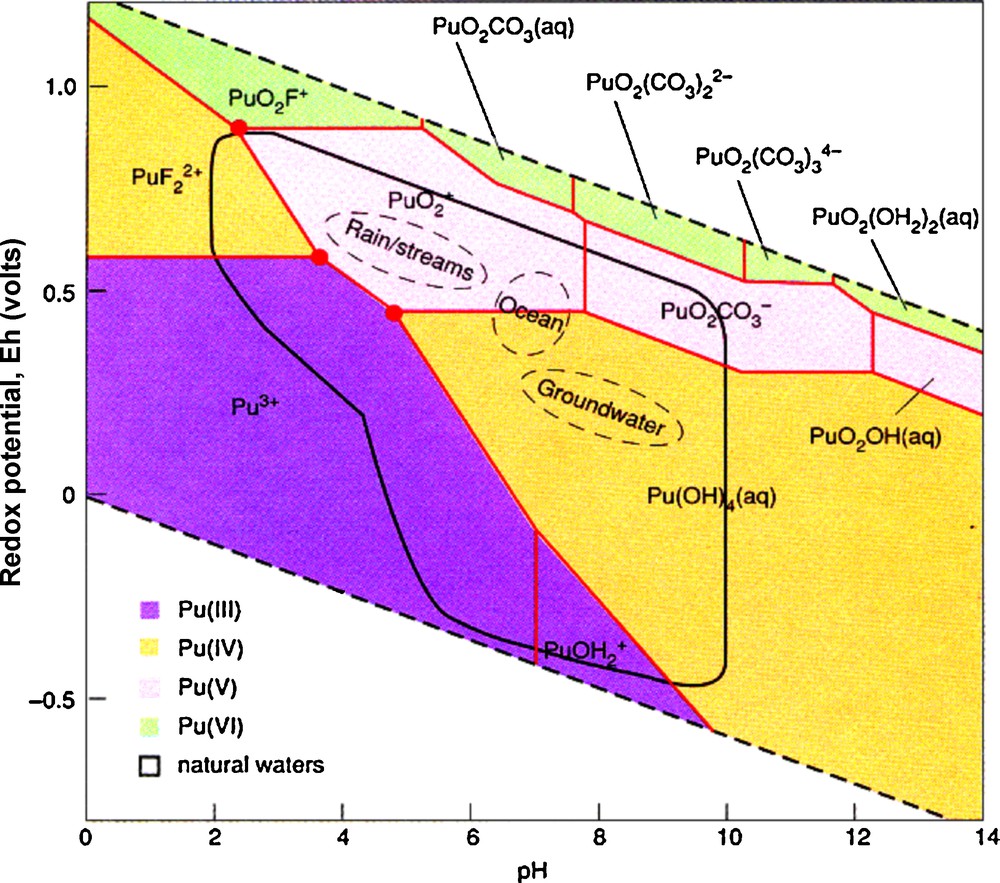
Eh-pH diagram for plutonium in water that contains hydroxide, carbonate and fluoride ions. The four colors indicate the different valence states of Pu. Note, the red dots or “triple points” where Pu may exist in three different oxidation states (from Bourdon et al. (2003) courtesy of Wolfgang Runde).
Diagramme Eh-pH pour le plutonium dans l’eau contenant des ions hydroxyde, carbonate et fluorure. Les quatre couleurs représentent les différents degrés d’oxydation de Pu. À noter les points rouges ou « points triples » où Pu peut exister sous trois degrés d’oxydation différents (d’après Bourdon et al. (2003), avec l’autorisation de Wolfgang Runde).
Importantly, all of the actinides are radioactive. The naturally occurring, long-lived actinides (232Th: half-life = 1.4 × 1010 yr; 235U: 7.04 × 108 yr; 238U: 4.47 × 109 yr) support radioactive decay chains that include other isotopes of actinium, thorium and protactinium. The man-made 237Np-decay series includes actinide isotopes of Ac, Th, Pa and U. Shorter-lived actinides (e.g., 239Pu: 2.4 × 105 yr), originally present at the time of the formation of the Earth, have decayed and are essentially non-existent in the Earth's crust. The now extinct 239Pu was the parent for the 235U series, and extinct 244Pu was the parent for the 232Th series. 241Pu is the parent of the man-made 237Np series. From the perspective of important nuclear properties, some of the actinide isotopes are fissile: 235U, 237Np, all of the Pu isotopes (there are 19), and 241Am + 243Am. 232Th and 238U are fertile and can be used to breed fissile 233U and 239Pu by neutron capture reactions and subsequent β-decay. In most nuclear reactors, 235U and 239Pu are the main sources of fission energy, and these same isotopes are the principal fissionable components of nuclear weapons. In geologic repositories for spent nuclear fuel, it is the long-lived actinides, principally the isotopes of U, 239Pu and 237Np, which account for most of the radiotoxicity after 1,000 years (Fig. 2). Additionally, after ∼700 years, most of the decay heat is generated by isotopes of Pu and 241Am (Wigeland et al., 2006). Thus, the chemical and nuclear properties of the actinides are key to the materials science of nuclear fuel and nuclear weapons design, as well as the design and evaluation of nuclear waste forms for the disposition or sequestration of actinides.
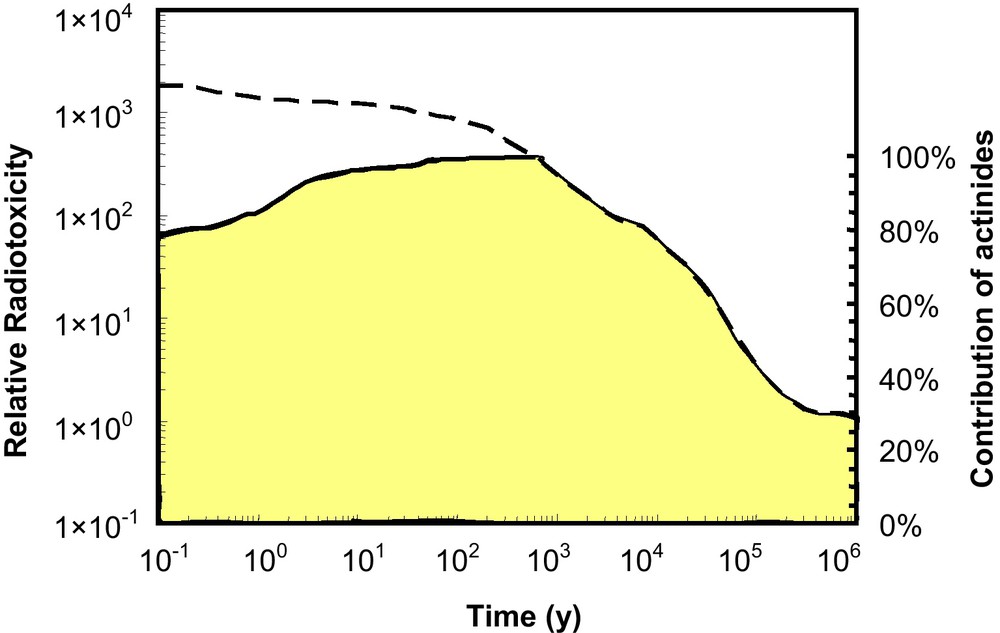
The upper curve is a calculated relative radiotoxicity (inhalation of spent nuclear fuel) with a burn-up of 38 MWd/kg U (Ewing, 2001). The relative toxicity is based on the comparison to the radiotoxicity of the amount of uranium ore that was originally mined to produce the fuel. The total toxicity includes fission and activation products plus the actinides and their decay products. The lower curve, shaded area, represents the contribution of the actinides and their daughter products to the total toxicity. Note, although there are long-lived fission products, such as 99Tc, 129I and 135Cs, their contribution to the total radiotoxicity is very low at times greater than 1000 years. This figure is only meant to illustrate the relative importance of different types of radionuclides over time, as it is unlikely that exposure to spent nuclear fuel would come via inhalation. Masquer
The upper curve is a calculated relative radiotoxicity (inhalation of spent nuclear fuel) with a burn-up of 38 MWd/kg U (Ewing, 2001). The relative toxicity is based on the comparison to the radiotoxicity of the amount of uranium ore ... Lire la suite
La courbe supérieure représente la radiotoxicité relative calculée (par inhalation de combustible nucléaire usé) avec un taux de combustion de 38 MWd/kg U (Ewing, 2001). La toxicité relative est calculée par comparaison avec la radiotoxicité de la quantité de minerai d’uranium extrait pour produire de combustible. La toxicité totale inclut les produits de fission et d’activation, ainsi que les actinides et leurs éléments fils. La courbe inférieure, délimitant la zone ombrée, représente la contribution à la toxicité totale des actinides et de leurs éléments fils. Bien qu’il existe certains produits de fission à longue durée de vie, comme 99Tc, 129I and 135Cs, leur contribution à la radiotoxicité totale est très faible pour des temps supérieurs à 1000 ans. Cette figure vise seulement à illustrer l’importance relative en fonction du temps des différents types de radionucléides, car il est peu vraisemblable que l’exposition au combustible nucléaire usé pourrait provenir d’une inhalation. Masquer
La courbe supérieure représente la radiotoxicité relative calculée (par inhalation de combustible nucléaire usé) avec un taux de combustion de 38 MWd/kg U (Ewing, 2001). La toxicité relative est calculée par comparaison avec la radiotoxicité de la quantité de minerai ... Lire la suite
Earth scientists are generally familiar with the properties and occurrence of the first four actinides: Ac(89), Th(90), Pa(91) and U(92). Uranium was discovered in 1789 by M. H. Klaproth and first isolated as an element by E. Péligot in 1841 and remains the subject of intense mineralogical and geochemical interest (Burns and Finch, 1999). Uranium and thorium are relatively abundant in the Earth's crust, from a few to tens of ppm, respectively, and form the backbone of most age-dating and uranium series techniques (Bourdon et al., 2003). Actinium and protactinium occur naturally, mainly in the decay chains of 232Th, 235U and 238U, although their crustal abundances are extremely low (10−10 to 10−6 ppm). The crustal abundances of Pu, Np, Cm and Am are essentially zero, although minute quantities of Pu and Np form in uranium deposits by neutron capture on 238U and subsequent β-decay:
The concentrations of Pu in uranium ores are between 10−13 to 10−12 g/g U ore. There is, however, the exceptional case of actinide creation in the natural fission reactors of Oklo, Gabon. More than 2 billion years ago, in uranium deposits of Oklo, the 235U concentration in the uranium ore was approximately 3.5 atomic percent, in the range of the enrichment factor for light water reactors. At this level of enrichment and with water as the moderator, sustained fission reactions occurred in more than a dozen natural reactors throughout the uranium deposit. Over 10 tons of 235U were fissioned and approximately 4 tons of Pu were created over several hundred thousand years (Janeczek, 1999). Although the Pu and Np that formed in these reactors has long since decayed away, the daughter products (e.g., 235U from 239Pu and 209Bi from 237Pu) are still present in the reactor zones (Jensen and Ewing, 2001). As will be discussed, the man-made production of Pu, Np, Am and Cm have far exceeded nature's modest efforts.
This review focuses on the man-made transuranium actinides that form at the highest concentrations in a nuclear fuel by (n,γ)-reactions and subsequent β-decay: Pu and the so-called “minor” actinides, Np, Cm and Am. The heavier actinides, Z = 97 to 103, generally have low yields and shorter half-lives and are not considered to pose a serious environmental hazard. Pu is certainly the most important of the transuranium elements, as it is the key element in a breeder fuel cycle, it is radiotoxic, and its diversion and proliferation due to its use in nuclear weapons are of great concern (Carter and Pigford, 1999).
2 Man-made transuranium elements
Since plutonium was isolated in microgram quantities in February of 1941 by Seaborg, Kennedy and Wahl, more than 1800 metric tonnes (mT) of plutonium have been created in nuclear reactors around the world. Approximately 300 mT are held in weapons programs, more than 200 mT have been separated from commercially generated spent nuclear fuel, mostly in the United Kingdom and France, and the balance, over 1200 MT, remains in spent nuclear fuel stored on-site at 236 nuclear power plants in 36 different countries (Albright et al., 1997; Carter and Pigford, 1999). Approximately 70 to 80 mT of new plutonium, generally left in the spent nuclear fuel, are added to the global inventory each year. Reactor-grade plutonium (> 60% 239Pu) with any degree of irradiation is a potential weapons material, and a nuclear device can be made with less than 10 kg of 239Pu (Mark, 1993). The good news is that as part of the first and second Strategic Arms Reduction Treaties, as well as unilateral pledges by both Russia and the United States, thousands of nuclear weapons have been dismantled since 1994. This will produce between 30 and 40 mT, pure and impure, of weapons-grade plutonium in each country, as well as hundreds of tons of highly enriched (in 235U) uranium. Depending on one's perspective, plutonium is either a valuable resource to be generated and used in a closed fuel cycle or a serious threat, contributing to the global proliferation of nuclear weapons. In a study by the National Research Council (Williams and Feiveson, 1990), the “excess” plutonium from dismantled nuclear weapons was described as “a clear and present danger to national and international security.”
In addition to the plutonium, “minor” actinides such as 237Np, 241Am + 243Am, and 244Cm are generated in reactors, and global production rates are 3.4, 2.7 and 0.35 mT per year, respectively. The long-lived actinides (e.g., 239Pu with a half-life of 24,100 years, 237Np with a half-life of 2.1 million years) are among the most important contributors to the calculated exposures to humans over the long periods envisioned for geological disposal, and after several hundred years, the radiotoxicity of disposed nuclear fuel is dominated by actinides, such as 239Pu and 237Np (see Fig. 2) (Hedin, 1997). The release of actinides from the spent nuclear fuel or other actinide-bearing solids is critical to the evaluation of the long-term performance of a geologic repository.
Thus, the renewed interest in expanding nuclear power production immediately raises the associated concerns of nuclear weapons proliferation and the geologic disposal of spent nuclear fuel or materials used to immobilize the actinides. As an example, a one GigaWatt (Gw) light water reactor generates approximately 200 kg of Pu per year, enough for more than 20 nuclear weapons (Williams and Feiveson, 1990). A ten-fold increase in global nuclear power production could place as much as five million kilograms of separated Pu into play in the global nuclear fuel cycle (Williams and Feiveson, 1990). Both issues, nuclear weapons proliferation and nuclear waste disposal, are inextricably tied to a consideration of the production and use of actinides in the nuclear fuel cycle and their fate after disposal in a geologic repository (Ewing, 2004).
3 Strategies for the disposition of plutonium
In the broadest sense, there are two strategies for the disposition of Pu (Ewing, 2004; Hedin, 1997; Williams and Feiveson, 1990): (1) the use of nuclear reactors or accelerators to “burn” or reduce the inventories of plutonium and the minor actinides. This involves reprocessing of nuclear fuels to reclaim fissile nuclides or the use of Pu from dismantled nuclear weapons for the fabrication of a mixed oxide (MOX) fuel, consisting of U and Pu, or the incorporation of fissile and non-fissile actinides into an inert matrix fuel (IMF). Inert matrix fuels do not contain fertile nuclides, such as 238U, that lead to the production of more Pu (238U becomes 239Pu by neutron capture and subsequent β-decay reactions) (Imaura et al., 2009). After a once-through burn-up, the MOX or IMF used fuels would be sent to a geologic repository; (2) direct disposal of spent nuclear fuel or actinide-bearing nuclear waste forms in a geologic repository. In the 1990s, the United States pursued a dual-track strategy in which the higher quality Pu from the pits of dismantled nuclear weapons would be used to fabricate a MOX fuel for once-through burn-up followed by direct disposal of the used MOX fuel. The “scrap” or less-pure Pu would be immobilized in a titanate ceramic, the dominant phase being a Hf-pyrochlore, (U,Pu,Hf,Gd)2Ti2O7. A considerable amount of research has been completed on phases suitable for Pu-immobilization, including pyrochlore and related structure types (Donald et al., 1997; Lumpkin et al., 2004; Lutze and Ewing, 1988). In April of 2002, the U.S. stopped almost all work on the Pu-immobilization strategy in favor of accelerated conversion of Pu into MOX fuel. This still leaves unresolved the fate of the “scrap” plutonium, several mT, that is not suitable for use in MOX fuel. Although research on the immobilization of Pu in crystalline ceramics has mostly ended in the United States, work continues in a number of other countries, mainly Russia, Great Britain and France.
Regardless of the strategy pursued, either for the use or disposal of actinides, particularly fissile Pu, the development of new materials either for storage (for tens to hundreds of years) or for disposal (for tens to hundreds of thousand years) is still required. The materials used for immobilization generally have relatively complex compositions, so that they can incorporate actinides (as well as neutron absorbers, such as Gd and Hf, and highly radioactive fission products, such as Cs and Sr), their synthesis must be accomplished remotely, the phases must be chemically durable, and their physical and chemical properties should not be degraded by α-decay event irradiation from the incorporated actinides.
Although the materials chemistry of Pu is complex, the disposal of Pu from dismantled nuclear weapons presents special opportunities: (a) as compared with other forms of high-level waste, the volumes are relatively small. For example, if Pu is immobilized in a typical waste-form with a waste loading of 10 wt.%, 100 mT of weapons Pu can be immobilized in a volume of several hundred cubic meters; (b) weapons plutonium is remarkably pure, consisting of a Pu-Ga alloy (0.5 to 2% Ga) coated with a corrosion-resistant layer, generally Ni. This high purity provides a materials engineer with a wide range of potential techniques for processing and the possibility of producing phase-pure waste forms at prescribed levels of waste loading. The absence of highly active fission products, such as 137Cs and 90Sr (the primary source of ionizing radiation), makes handling the material tractable using technologies comparable to those used to fabricate mixed-oxide reactor fuels; (c) although the half-life of 239Pu (24,100 y) is much longer than that of the much higher-activity fission products, a substantial amount of decay occurs over relatively short geological time-scales (e.g., containment of Pu for ten half-lives requires on the order of 240,000 y). Thus, immobilization over the time required for substantial radioactive decay is short relative to the durability of some geologic materials, measured in many millions of years.
In this review, we present some of the recent developments in the properties of materials considered for the immobilization of actinides, particularly plutonium (Table 1, Ewing and Weber, 2010). There have been a number of extensive reviews and comparisons of nuclear waste forms (Ewing, 1999, 2001; Lumpkin, 2006; Lumpkin et al., 2004). Some of the phases that have received the most attention for actinide immobilization include zircon (Ewing, 1999, 2001), titanates (Lumpkin, 2001; Lumpkin et al., 2004), and pyrochlore (Ewing et al., 2004b). In this article, we focus on the most recent results on the effects of radiation damage on pyrochlore as an actinide waste form.
Formes hôte pour déchets d’actinides.
| Simple oxides | |
| Zirconia | ZrO2 |
| Uraninite | UO2 |
| Thorianite | ThO2 |
| Complex oxides | |
| Pyrochlore | (Na,Ca,U)2(Nb,Ti,Ta)2O6 |
| Murataite | (Na,Y)4(Zn,Fe)3(Ti,Nb)6O18(F,OH)4 |
| Zirconolite | CaZrTi2O7 |
| Silicates | |
| Zircona | ZrSiO4 |
| Thoritea | ThSiO4 |
| Garneta | (Ca,Mg,Fe2+)3(Al,Fe3+,Cr3+)2(SiO4) |
| Britholite | (Ca,Ce)5(SiO4)3(OH,F) |
| Titanite | CaTiSiO5 |
| Phosphates | |
| Monazitea | LnPO4 |
| Apatitea | Ca4-xLn6+x(PO4)y(O,F)2 |
| Xenotimea | YPO4 |
a The long-term durability can be confirmed by studies of naturally occurring minerals (names given above).
4 α-decay event damage by actinides
A principal concern for actinide waste forms is the effect of the alpha-decay event on the crystalline structure of the waste form (Ewing et al., 1995, 2000, 2003, 2004b; Weber et al., 1997, 1998). In an α-decay event, the α-particle dissipates most of its energy (4.5 to 5.8 MeV for actinides) by ionization processes over a range of 15 to 22 μm, but it undergoes enough elastic collisions along its path to produce several hundred isolated atomic displacements. The largest number of displacements occurs near the end of the α-particle range. The more massive, but lower energy, α-recoil (86 keV 235U recoil from decay of 239Pu) dissipates nearly all of its energy in elastic collisions over a very short range, 30 to 40 nm, causing ∼1000 atomic displacements. The density of energy deposited into the cascade is high (up to 1 eV/atom) and occurs over an extremely short time (<10−12 s). Thus, a single α-decay event generates several thousand atomic displacements, significantly more than the 0.1 displacements generated per β-decay event. Because of the large number of atomic displacements during an α-decay event, there is a profound effect on the structure and properties of crystalline solids that incorporate actinides. The cumulative effect of dose will be time- and temperature-dependent because of relaxation and recrystallization of damaged areas (Chakoumakos and Ewing, 1985).
5 Pyrochlore
There are over 500 synthetic compositions (Chakoumakos, 1984), including actinides (Chakoumakos and Ewing, 1985), with the pyrochlore structure. A number of compositions with thorium and uranium (Laverov et al., 2001, 2002), as well as transuranium elements (e.g., Cm and Pu), have been synthesized (Kulkarni et al., 2000; Raison et al., 1999; Weber et al., 1985a, 1985b). Thus, it is not surprising that pyrochlore structure-types have received extensive attention as a potential host phase for actinides (Ewing et al., 2004b).
5.1 Structure
Pyrochlore is isometric (Fd3m, Z = 8, a = 0.9 to 1.2 nm), and the structural formula is ideally VIIIA2VIB2IVX6IVY (Roman numerals indicate the coordination number), where the A- and B-sites contain metal cations; X (= O2−) and Y (= O2−, OH−, F−) are anions (Chakoumakos, 1984; Subramanian et al., 1983). The structure can be described in a variety of ways, most commonly by describing the shapes and topology of the coordination polyhedra (Fig. 3a). Pyrochlore is closely related to the fluorite-structure (AX2), except that there are two cation sites and one-eighth of the anions are absent (Fig. 3c,d). The cations and oxygen vacancies are ordered. The loss of one-eighth of the anions reduces the coordination of the B-site cation from eight to six. The X-anion occupies the 48f position; and the Y-anion, the 8b (when the origin of the unit cell is placed at the B-site). All of the atoms in an ideal pyrochlore are on special crystallographic positions, except the 48f oxygen (O48f). The structure can also be visualized as a network of corner-linked BX6 octahedra (a B2X6-framework) with A-site cations filling the interstices (Fig. 3a). The A- and B-site coordination polyhedra are joined along edges, and the shapes of these polyhedra change as the positional parameter, x, of the O48f shifts to accommodate cations of different sizes (“x” is simply a positional coordinate within the unit cell for O48f). For x = 0.3750, the A-site coordination polyhedron is a regular cube, and the B-site polyhedron is distorted to a trigonally flattened octahedron (the topology of the fluorite structure). In this case, materials have a defect fluorite structure, and the occupancy of each anion site is 0.875. For x = 0.3125, the B-site is a regular octahedron and the A-site is a distorted trigonal scalenohedron and has the ideal pyrochlore structure (Chakoumakos, 1984; Subramanian et al., 1983). Thus, the 48f oxygen positional parameter, x, defines the polyhedral distortion and structural deviation from the ideal fluorite structure.
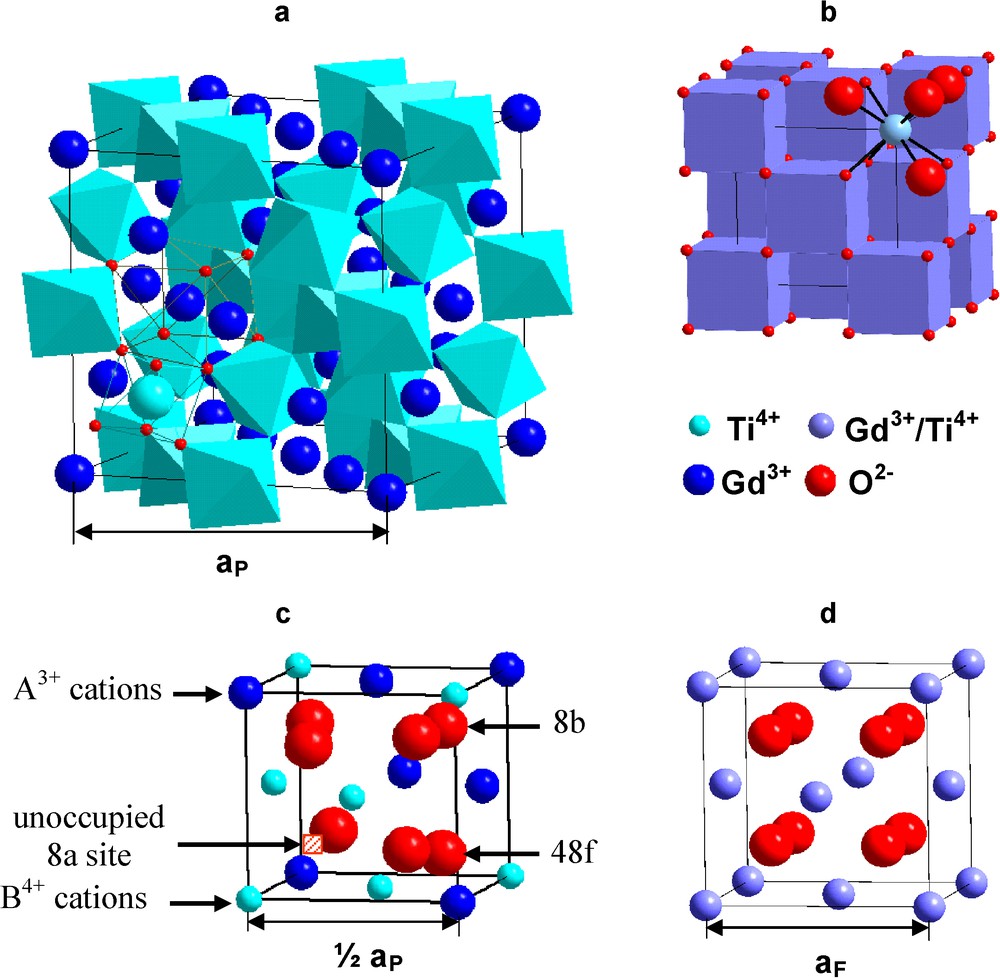
Pyrochlore structures described based on the polyhedral network (a) and the derivative of fluorite structure (c). Corresponding fluorite unit cell (b,d) are included for comparison. Note, than in this figure, the origin of the unit cell is set at the position of the B-site cation. There are two types of oxygens (8b = Y-anion; 48f = X-anion) and one-eighth of the oxygens are missing, at the 8a site. In the defect fluorite structure (d) all of the anion sites are partially-filled due to disordering of the anions over all of the anion sites and the occupancy at each site is 88%. Masquer
Pyrochlore structures described based on the polyhedral network (a) and the derivative of fluorite structure (c). Corresponding fluorite unit cell (b,d) are included for comparison. Note, than in this figure, the origin of the unit cell is set at the ... Lire la suite
Structure de prochlore décrite sur la base d’un réseau polyédral (a) ou à partir d’une structure fluorine (c). Les mailles correspondantes du réseau fluorine (b,d) sont également présentées pour comparaison. À noter que l’origine de la maille unitaire se situe à la position du site cationique B. Il existe deux types d’oxygène (8b = anion Y; 48f = anion X) et il manque 1/8 des oxygènes, au niveau du site 8a. Dans la structure fluorine défective (d), tous les sites anioniques sont partiellement occupés, en raison du désordre des anions sur tous les sites anioniques et le taux d’occupation sur chaque site est de 88 %. Masquer
Structure de prochlore décrite sur la base d’un réseau polyédral (a) ou à partir d’une structure fluorine (c). Les mailles correspondantes du réseau fluorine (b,d) sont également présentées pour comparaison. À noter que l’origine de la maille unitaire se situe ... Lire la suite
In ternary metal oxide systems, the pyrochlore structure-type, A2B2O7, is common because this isometric structure can accommodate a wide variety of combinations of A- and B-site cations (3+ and 4+ or 2+ and 5+ combinations of valence), as well as oxygen vacancies (Subramanian et al., 1983). The (3 + , 4 + ) pyrochlores are of greatest interest in nuclear waste management because of their ability to incorporate trivalent lanthanides and tri- and tetra-valent actinides (Subramanian et al., 1983). Of the most typical B-site compositions (e.g., Ti, V, Cr, Sn and Mo), the titanates have received the most attention because of their chemical durability. There is an extensive literature on the properties of lanthanide titanates (Ewing et al., 2004b). Data for actinide pyrochlores are limited; however, Chakoumakos and Ewing (1985) have used the pyrochlore cell geometry to analyze the potential of the pyrochlore structure to incorporate actinides. Actinides (3 + , 4 + , and 5 + ) are predicted to form the pyrochlore structure by substitutions on both the A- and B-sites. Higher valence states (e.g., Np6+ and Pu6+) can be incorporated into ideal or defect pyrochlores at the B-site.
5.2 Radiation effects
Radiation effects from α-decay events in many crystalline phases proposed for the immobilization of actinides result in amorphization, macroscopic swelling, and order-of-magnitude increases in dissolution rates (Ewing et al., 1995; Weber et al., 1998). A detailed study on the effects of α-decay on single-phase (Gd,Cm)2Ti2O7 pyrochlore determined that amorphization occurred at a dose of about 3.1 × 1018 α-decay events per gram and was accompanied by macroscopic swelling of about 5% and an increase, by a factor of 20 to 50, in the aqueous dissolution rate of the non-network forming Cm (Weber et al., 1986). The radiation-induced transformation to an amorphous state and the magnitude of the changes in swelling and dissolution rate are greatly dependent on the pyrochlore composition and irradiation conditions. Because self-radiation damage from alpha-decay can significantly affect the atomic-scale structure and the physical and chemical properties of actinide-bearing pyrochlore-based waste forms, long-term assessments of performance must take into account the effects of alpha-decay at relevant temperatures, dose rates, and times. In this regard, it is fortunate that systematic experimental studies using short-lived actinides and ion-beam irradiations, investigations of radiation effects in U- and Th-bearing minerals, and the development of new models of radiation damage processes over the past 20 years have led to significant improvements in understanding the processes of damage accumulation in pyrochlore and related defect-fluorite structures (Ewing et al., 2004b).
5.3 Ion beam simulation of alpha-decay damage
Ion beam irradiations can be used to simulate α-decay event damage under carefully controlled experimental conditions (e.g., ion mass and energy, temperature and fluence) (Chakoumakos and Ewing, 1985). Most experiments have been performed in-situ often using the IVEM-Tandem or HVEM-Tandem (now dismantled) facilities at Argonne National Laboratory. There, a 2 MeV tandem ion accelerator is interfaced to an intermediate-voltage electron microscope. During an ion beam irradiation, one can simultaneously observe microstructural changes in the pre-thinned sample using in situ transmission electron microscopy (TEM). The critical amorphization fluence (ions/cm2), Dc, is the fluence at which all of the diffraction maxima in the selected-area electron diffraction pattern disappear, and at this fluence, the sample is considered to be amorphous. Typically, the fluence (ions/cm2) is converted to units of displacements per atom (dpa) in order to facilitate the comparison of radiation effects on different materials subjected to different types of radiation (e.g., a high-energy ion vs. an α-decay recoil atom) (Chakoumakos and Ewing, 1985). The conversion requires knowledge of the minimum energy required to permanently displace an atom from its position in the structure (i.e., generally assumed to be ∼50 eV for all atoms in pyrochlore structure) and the use of a code, SRIM (The Stopping and Range of Ions in Matter). The radiation-induced transformation from the crystalline-to-amorphous state is a balance between the damage production and damage recovery processes; thus, the critical amorphization dose increases at elevated irradiation temperatures due to recovery processes. Complete amorphization will not occur if the amorphization rate is less than or equal to the damage recovery rate. The temperature at which the rate of damage recovery equals the damage rate is defined as the critical temperature, Tc, for amorphization of a given material under a specific set of irradiation conditions. Different mechanisms have been proposed for ion beam-irradiation induced amorphization processes (Weber, 2000), and the effects of ion species, energy, target mass and irradiation temperatures on Tc have been review by Wang et al. (2001). Depending on the purpose of the comparison, Tc and/or the amorphization dose at room temperature or 25 K may be used to characterize a material's stability under irradiation.
Systematic ion beam irradiations have been completed using a variety of ion sources: 600 keV Ar+, 600 keV Bi+, 1 MeV Kr+, 1.5 MeV Xe+ and 400 keV Au+ for pyrochlore-structured compounds, A2B2O7 (A3+ = La∼Lu and Y; B4+ = Ti, Sn, Hf, Zr, etc.). Surprisingly, the different pyrochlore compositions display a wide range of responses to ion beam-induced amorphization. All of the titanate pyrochlores were readily amorphized by ion beam irradiation at a relatively low damage level (see reference (Chakoumakos and Ewing, 1985) for a discussion of the conversion of ion fluence (ions/cm2) to displacements per atom (dpa)). Gd2Ti2O7 can be amorphized by 600 keV Ar+ at room temperature at ∼0.2 dpa (Wang et al., 1999b), a value that is consistent with the amorphization dose (∼0.16 dpa) for 244Cm-doped (3 wt.%) Gd2Ti2O7 (Weber et al., 1986). Also, an ion beam-induced pyrochlore-to-fluorite (a result of disordering of the cations on the A- and B-sites) structural transition was observed concurrently with the amorphization process for all of the titanate pyrochlores (Purton and Allan, 2002; Runde, 2000; Williford and Weber, 2001). The temperature dependences of critical amorphization dose for titanate pyrochlore single crystals under 1 MeV Kr+ ion irradiation are shown in Fig. 4. A significant difference in the radiation response of the titanate pyrochlores with different lanthanide elements occupying the A-site was observed. Generally, with the increasing ionic radius of the A-site cation, from Lu3+ (0.098 nm) to Gd3+ (0.106 nm), the critical amorphization temperature increases from 480 K (for Lu2Ti2O7) to 1120 K (for Gd2Ti2O7) (Fig. 4). However, with the increase in the ionic radius of the A-site cation from Gd3+(0.106 nm) to Sm3+(0.109 nm), the critical temperature decreased slightly from 1120 K to 1045 K. The fact that Gd2Ti2O7 has the highest Tc indicates that this composition is the most susceptible to ion irradiation-induced amorphization, as compared with the other rare-earth titanate pyrochlores. This is unfortunate, as the Gd2Ti2O7 composition was one of the main candidates for Pu-immobilization.
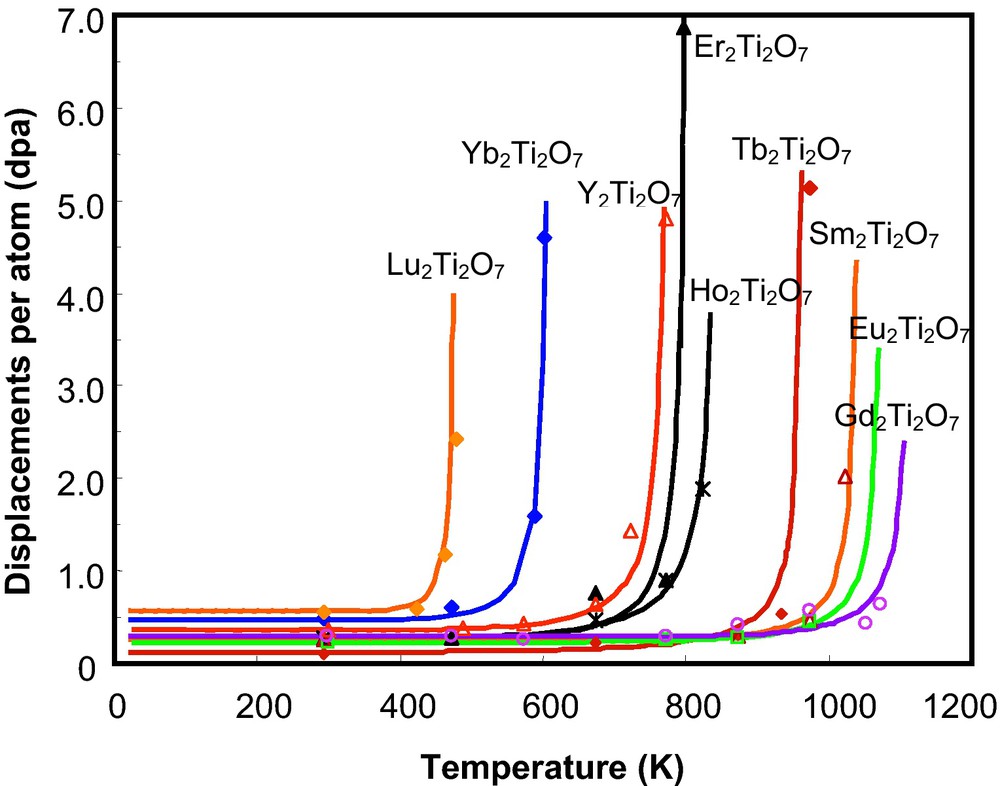
Temperature dependence of the critical amorphization dose of RE2Ti2O7 irradiated by 1 MeV Kr irradiation (Weber et al., 1985b). Note that each curve bends upward at elevated temperatures. For each material there is a unique temperature, Tc, above which the material cannot be amorphized and that temperature shifts dramatically for pyrochlore depending on the composition of the A-site cation.
Dépendance en température de la dose critique d’amorphisation de RE2Ti2O7 irradié par des rayonnements 1 MeV Kr+ (Weber et al., 1985b). À noter que chaque courbe s’incurve vers le haut à des températures élevées. Pour chaque matériau, il existe une température unique, Tc, au-dessus de laquelle le matériau ne peut plus être amorphisé et cette température se déplace de façon spectaculaire pour le pyrochlore, en fonction de la nature du cation en site A.
One of the recent and exciting outcomes from systematic studies of irradiation effects in different pyrochlore compositions was the discovery of the radiation “resistance” of Gd2Zr2O7 and Er2Zr2O7 (Ewing et al., 2004b; Sickafus et al., 2000; Wang et al., 1999a). These compositions can readily accommodate Pu on the Gd (or Er) and Zr-sites (Williforg and Weber, 2001). In the case of the Gd2(ZrxTi1-x)2O7 binary, there is a systematic increase in the radiation “resistance” (i.e., a decrease in Tc and increase in the amorphization dose at 0 K, Do, which is obtained by extrapolation) with increasing Zr-content under 1.0 MeV Kr+ irradiation (Wang et al., 1999a). Complete amorphization cannot be achieved for Zr-rich pyrochlore compositions with x ≥ 0.75 (Fig. 5). The end-member zirconate, Gd2Zr2O7, remained crystalline at a dose of ∼36 dpa under 1.5 MeV Xe+ irradiation at T = 25 K and at a dose of ∼100 dpa for 200 keV Ti+ implantation at room temperature (Lian et al., 2002). The high “resistance” of zirconate pyrochlore to ion beam-induced amorphization was also confirmed by Sickafus et al. (2000) for Er2Zr2O7. In contrast to the ordered pyrochlore Er2Ti2O7, Er2Zr2O7, which adopts a defect fluorite structure, readily accommodates radiation-induced structural disordering and remains crystalline at a dose of ∼140 dpa at room temperature (350 keV Xe+ irradiations). These results are consistent with recent molecular dynamics results that indicate amorphization occurs directly within displacement cascades in Gd2Ti2O7, while in Gd2Zr2O7, displacement cascades tend to produce only point defects (Purton and Allan, 2002). In addition to Gd2Zr2O7 and Er2Zr2O7, it has been demonstrated that Sm2Zr2O7 and Nd2Zr2O7 undergo an irradiation-induced pyrochlore to defect-fluorite structural transformation under irradiation with 1.5 MeV Xe+ ions (Lian et al., 2002), similar to the transition that occurs for the titanate pyrochlore compositions (Wang et al., 1999b); however, the resulting defect-fluorite structure remains resistant to amorphization at doses up to 7 dpa at 25 K (Lian et al., 2002). Of all of the rare earth Zr-pyrochlores, only La2Zr2O7 can be amorphized, but the critical temperature is low, ∼310 K (Lian et al., 2002, 2004b). Recent molecular dynamics simulations of 6 keV U displacement cascades in La2Zr2O7 at 350 K indicate the formation of a small number of point defects and a transition toward the defect-fluorite structure, consistent with the experimental observations (Charties et al., 2002).
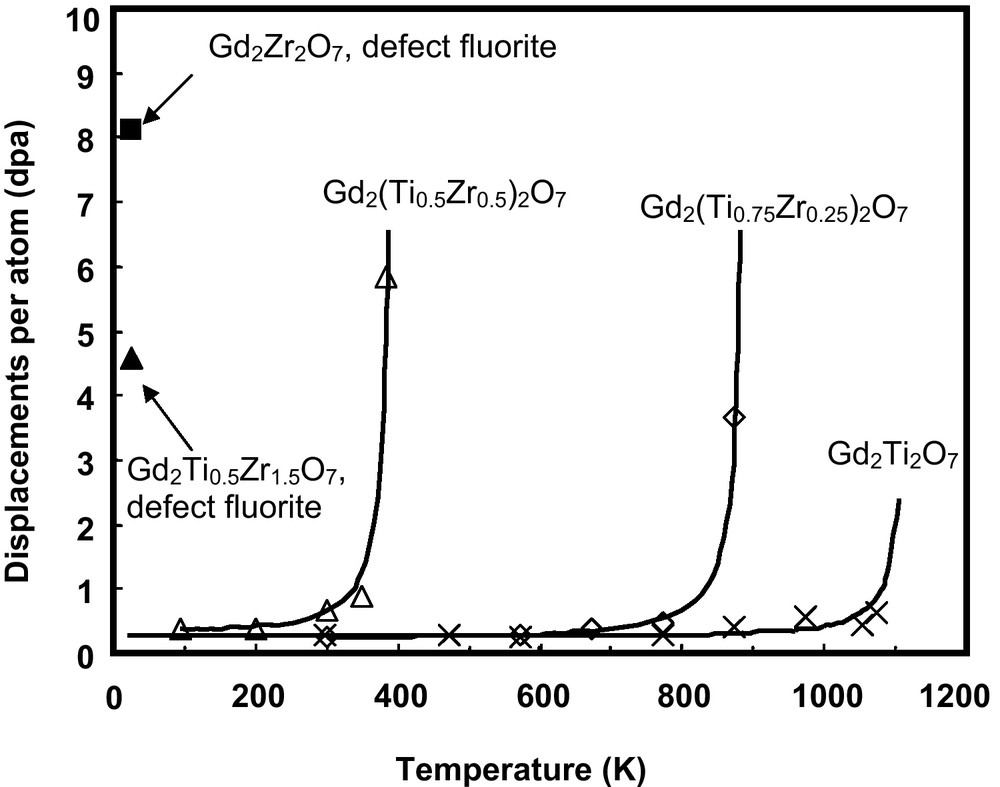
Temperature dependence of the critical amorphization dose of Gd2(Ti1-xZrx)2O7 irradiated by 1 MeV Kr+ (Purton and Allan, 2002). For Gd2Zr2O7, amorphization does not occur; instead, the structure disorders to a defect-fluorite structure type which is radiation “resistant”.
Dépendance thermique de la dose critique d’amorphisation de Gd2(Ti1-xZrx)2O7 irradié par des rayonnements 1 MeV Kr+ (Purton et Allan, 2002). Pour Gd2Zr2O7, on n’observe pas d’amorphisation; dans ce cas, la structure se désordonne vers une structure de type fluorine défective, qui est « résistante » aux rayonnements.
The radiation response of two Gd-pyrochlore compositions, Gd2Sn2O7 and Gd2Hf2O7, has been studied under a 1 MeV Kr+ irradiation (Fig. 6), and the radiation damage and microstructural evolution were examined by in situ TEM and ex situ high resolution TEM (Lian et al., 2004a). Gd2Sn2O7 is sensitive to ion beam-induced amorphization with a critical amorphization dose of ∼3.4 dpa at room temperature and has a Tc of ∼350 K (Lian et al., 2004a). In contrast, Gd2Hf2O7 does not become amorphous at a dose of ∼4.54 dpa at room temperature, but instead is transformed to a disordered fluorite structure, similar to that observed for zirconate pyrochlores (Lian et al., 2002, 2004a). Combined with the irradiation results of titanate and zirconate pyrochlores, these results highlight the significant effects of the type of cation, on both the A- and B-site, on the radiation response of the pyrochlore structure (Fig. 6).
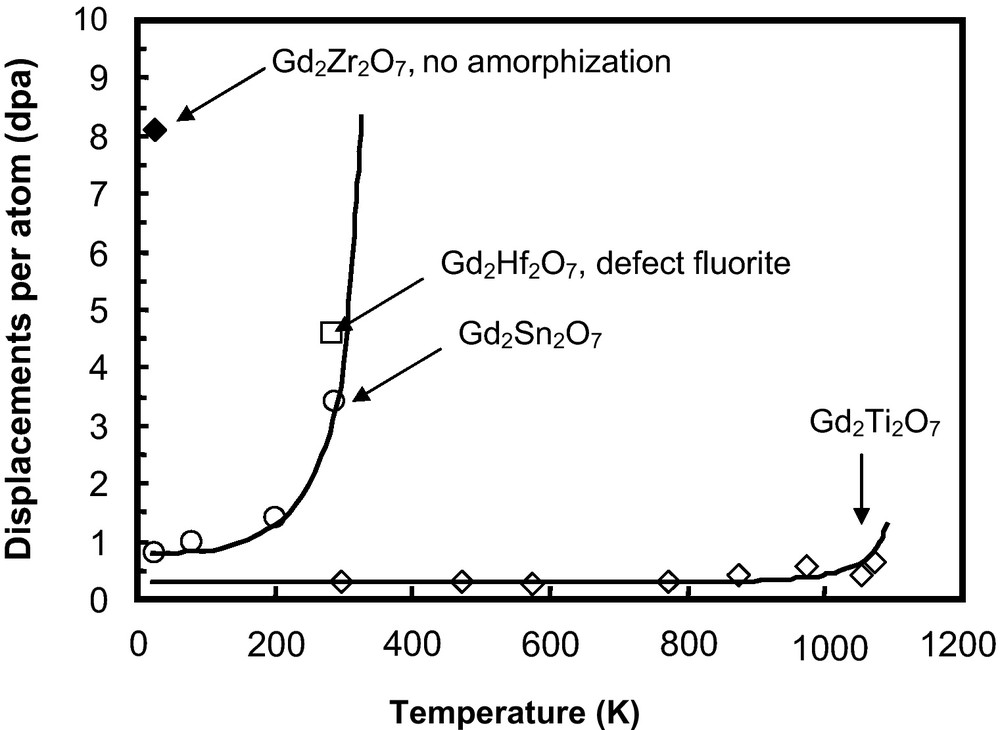
Effect of B-site cations on the radiation response of Gd-pyrochlore Gd2B2O7 (B4+ = Ti, Sn, Hf, and Zr) under 1 MeV Kr+ irradiation (Weber et al., 1985a).
Influence des cations des sites B sur la tenue sous irradiation du pyrochlore Gd2B2O7 (B4+ = Ti, Sn, Hf, and Zr) pour une irradiation de 1 MeV Kr+ (Weber et al., 1985a).
5.4 The effect of the cations
The radiation response of pyrochlore is highly dependent on composition, and this has been interpreted as being related to the ratio of the ionic radii of the A- and B-site cations, rA/rB, (Ewing et al., 2004a; Lian et al., 2002, 2003). Energy-minimization calculations (Sickafus et al., 2000) have suggested that the cation antisite defect is the most stable defect in the pyrochlore structure. As the A-site cation radius approaches that of the B-site cation radius, the material is more likely to adopt the fluorite structure-type. Thus, pyrochlore compositions with a lower cation radius ratio energetically favor the disordered, defect fluorite structure rather than the amorphous state (Helean et al., 2004). Generally, with the decreasing ionic radius ratio, the pyrochlore structure has a lower critical amorphization temperature, suggesting a higher radiation “resistance” to ion beam-induced amorphization. The change in radiation “resistance” to ion beam-induced amorphization is consistent in terms of the critical amorphization temperature, Tc, and the amorphization dose at ambient conditions. For example, the ionic size of Sn4+ (0.069 nm) is midway between the ionic radii of Ti4+ (0.0605 nm) and Zr4+(0.072 nm), and the ionic radius of Gd3+ is 0.1053 nm. Thus, it is expected that Gd2Sn2O7, with rA/rB = 1.526, would more likely disorder to the defect fluorite structure than Gd2Ti2O7 (rA/rB = 1.74). The critical amorphization dose at room temperature, Dc, and critical temperature, Tc, for Gd2Sn2O7 are ∼3.4 dpa and 350 K (Lian et al., 2004a), respectively, suggesting a much higher amorphization “resistance” as compared with that of Gd2Ti2O7 (∼0.2 dpa and 1120 K) (Wang et al., 1999b). In the binary system of Gd2(Ti1-xZrx)2O7, the critical amorphization dose increases dramatically with the decreasing cation radius ratio due to the substitution of Zr4+ for the smaller Ti4+, and complete amorphization cannot be induced by ion beam irradiation when x ≥ 0.75 (e.g., Gd2(Ti0.25Zr0.75)2O7) (Wang et al., 1999a). The radius ratio for Gd2(Ti0.25Zr0.75)2O7 is 1.523. Furthermore, because of the similarity of the ionic radii of Hf4+ (0.71 Å) and Zr4+ (0.72 Å), it is expected that Gd2Hf2O7 (rA/rB = 1.48) would exhibit an excellent “resistance” to amorphization, comparable to that of Gd2Zr2O7 (rA/rB = 1.46) (Lian et al., 2004a). Therefore, ion irradiation-induced defect fluorite in Gd2Hf2O7 is stable with respect to the amorphous state, similar to that observed for ion irradiated Gd2Zr2O7. Also, the thermally-driven, order-disorder structural transition occurs at about 1800 K for Gd2Hf2O7, consistent with that of Gd2Zr2O7 (1823 K) (Wuensch and Eberman, 2000). These results suggest a comparable tendency toward an order-disorder transition induced either by ion beam irradiation or high-temperature annealing for pyrochlore structure-types.
5.5 Bond-type effects
Although the ionic size of the cations plays an obvious and important role in determining the radiation response of different pyrochlore compositions, we have recently shown a significant influence of the electronic configurations of the A- and B-site cations (Lian et al., 2003). The effect of the cation electronic structure, i.e., the type of bonding, is closely related to the polyhedral distortion and structural deviation from the ideal fluorite structure, which may affect the dynamic defect recovery process. Gd2Ti2O7 has the highest critical amorphization temperature, Tc, among titanate pyrochlore compositions, suggesting that this composition is more sensitive to ion irradiation-induced amorphization as compared with other rare-earth titanate pyrochlores (Lian et al., 2003). This result is consistent with the fact that this composition has the greatest structural deviation from the ideal fluorite structure, as evidenced by its having the smallest 48f oxygen x parameter, as a result of the strong ionic character of Gd3+ due to the specific electronic configuration of the 4f sub-shell of Gd3+ (half-filled). The 48f oxygen positional parameter, x, defines the polyhedral distortions and the deviation from the ideal fluorite structure, and it is closely affected by the relative ionic size of cations on the A- and B-sites, their electronic configuration and structural disordering. With an increasing x value (B4+ as the origin of the unit cell, see Fig. 3c) and an increasing degree of structural disorder, the structure has greater distortion of the B-site coordination polyhedron and is closer to the ideal fluorite structure (Subramanian et al., 1983), and thus it is more sensitive to ion beam-induced amorphization. The effect of cation electronic configuration is further evidenced by the comparison between the radiation responses of Gd2Sn2O7 and Gd2(Zr0.75Ti0.25)2O7. Although the cation radius ratio of Gd2Sn2O7 (∼1.526) is similar to that of Gd2(Zr0.75Ti0.25)2O7 (∼1.523), there is a dramatic difference in the radiation “resistance”. No amorphization occurs in Gd2(Zr0.75Ti0.25)2O7 with an ion irradiation at 25 K; whereas, Gd2Sn2O7 can be amorphized at room temperature at a dose of ∼3.4 dpa. The covalent character of the <SnO> bond and the associated decrease in the <SnO> bond distance imply a lesser degree of distortion of the SnO6 coordination octahedron, resulting in a structure more compatible with the ordered pyrochlore superstructure (Kennedy et al., 1997). This leads to a greater susceptibility of Gd2Sn2O7 to ion beam irradiation-induced amorphization, as compared with Gd2(Zr0.75Ti0.25)2O7. First-principle calculations (Panero et al., 2004) using density functional theory have reported a significant covalency for the <SnO> bond and mainly ionic character for the <TiO> and <ZrO> bonds. The greater degree of covalent bonding between <Sn4+O> as compared with <Ti4+O> or <Zr4+O> results in defect formation energies otherwise unexpected solely due to the radius ratios of the cation species. For example, Y2Sn2O7 shows a 2–4 eV greater defect formation energy than otherwise predicted by the use of the average B-site cation size. This underscores the importance of the electronic configuration of cations on the crystal chemistry and the radiation “tolerance” of the pyrochlore structure.
6 Future research on nuclear waste forms
I have discussed the radiation response of different pyrochlore compositions in order to illustrate the present level of understanding of radiation effects in phases that may be used for the immobilization of Pu and the minor actinides. This fundamental understanding has emerged from systematic and complementary studies on alpha-decay damage (in actinide-doped and natural samples) and ion-beam irradiations of different pyrochlore compositions. It is now possible to predict the dose and, thus, the time dependence of amorphization in several actinide-host phases, such as pyrochlore, under repository conditions. As an example, while Gd2Ti2O7 (with a 10 wt.% loading of 239Pu) will become amorphous in less than 1000 years, Gd2Zr2O7 will not amorphize. A similar level of understanding must be obtained for the mechanisms of corrosion of the different actinide waste forms, but there is already substantial progress in this area (Lumpkin, 2001).
I believe that in the future, with a solid research base, we will be able to design nuclear waste forms for particular radionuclides and for the specific geochemical and hydrologic environments of a particular type of repository. With the present work as a starting point, we should use this understanding to improve the safety and efficiency of other materials used in the nuclear fuel cycle.
Acknowledgements
Much of the work summarized in this review is based on extended collaborations with LuMin Wang, ShiXin Wang and Jie Lian at the University of Michigan; Bill Weber at Pacific Northwest National Laboratory; Alex Navrotsky and Kate Helean at the University of California, Davis; Greg Lumpkin at ANSTO in Australia. Parts of this review are based on papers that have appeared in the Journal of Materials Research (Weber et al., 1997), Canadian Mineralogist (Burns et al., 1996), the Journal of Applied Physics (Lumpkin, 2001) and Earth and Planetary Science Letters (Ewing, 2005). This research and the collaborations have been sustained by support from the Office of Basic Energy Sciences of the U.S. Department of Energy, most recently from the Energy Frontier Research Center–Materials Science of Actinides (DE-SC0001089).