

1 Introduction
In recent years, there has been a growing awareness of the importance of interactions between the biosphere and the outermost regions of the geosphere; those regions within a few metres of the Earth's surface essential for the support of life and, hence, now termed the ‘critical zone’ (Richter and Mobley, 2009). Undoubtedly, the organism having the most dramatic impact on this critical zone is Homo sapiens, through the exploitation and utilization of Earth resources (see Craig et al., 2001 for an overview). Indeed, as long ago as the mid-1990s, Hooke (2000) estimated that as much material was being moved around at the Earth's surface by human activities as by all natural processes combined.
In terms of processes in the critical zone, interactions involving microbes are particularly important. Such interactions may involve other living matter and organic compounds, but it is the interactions with minerals which are of greatest interest to geoscientists and which are volumetrically the most significant. For example, in Fig. 1 processes specifically associated with metal-microbe interactions are illustrated, the metals being largely derived from mineral sources. Mineral-microbe interactions necessarily occur at the mineral surface, so that understanding mineral surfaces is crucial, and so is understanding the interaction of organic molecules with such surfaces as a step towards understanding more complex bio-mineral systems. In all of these areas, it is a molecular-scale understanding that is required in order to predict and model behavior at all scales.
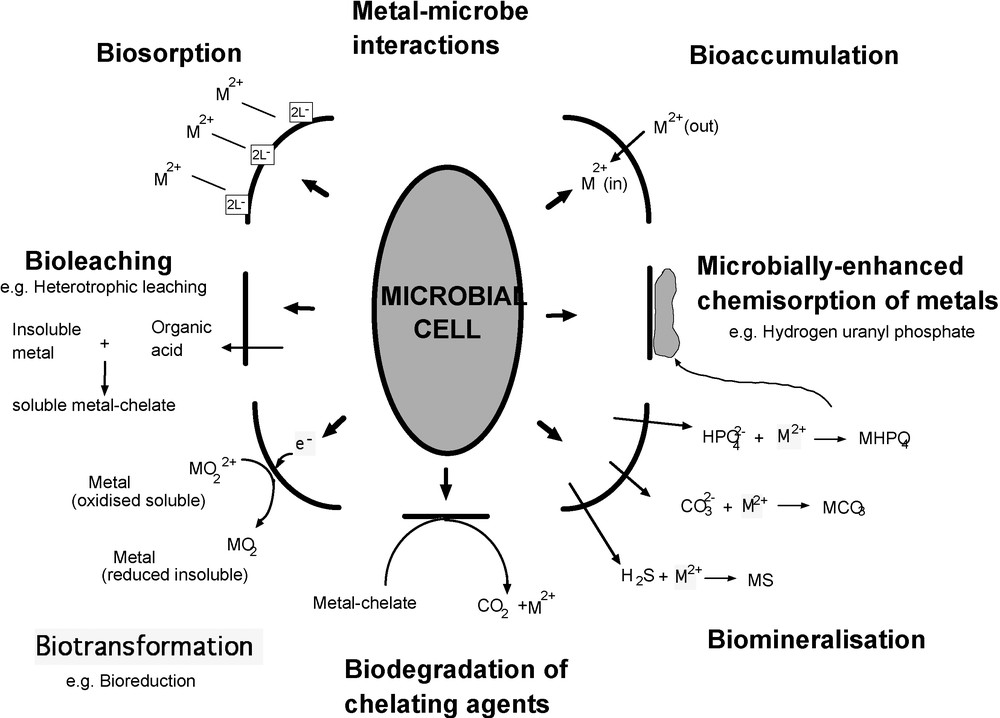
Diagram illustrating the diverse types of interaction that can take place between a microbe and a metal in solution or in a solid mineral phase.
Diagramme illustrant les différents types d’interaction qui peuvent prendre place entre un microbe et un métal en solution ou dans une phase minérale solide.
In this article, some of the advances in understanding mineral-(organic)-microbe interactions are reviewed by centering on the example of iron. Microbial redox reactions involving iron minerals are the focus, along with biofilms, their role in precipitation of iron minerals, and their influence on transport of fluids carrying metals in solution. Processes involving iron in turn impact on the behaviour of other elements, including highly toxic contaminants such as Cr, As, U. These are important areas of study in ‘environmental mineralogy’, being central to understanding the release, transport and bioavailability of these and many other contaminants. A complementary aspect of such studies concerns the potential for technological applications of mineral-microbe interactions and of synthetic biominerals in environmental ‘clean-up’, or in industrial processes such as catalysis. In the following discussions, the emphasis is on describing work in our own laboratories, but it is important to emphasise that many other important contributions are now being made in this field.
2 Mineral-microbe interactions and redox processes
Redox transformations of many mineral phases can be linked directly to microbial respiration. These direct processes commonly involve highly specialised microorganisms that use the mineral surface as a source of electrons for the respiration (reduction) of oxygen or nitrate, or couple the oxidation of organic matter (or some other suitable electron donor) to the reduction of the mineral phase itself, normally in the absence of oxygen as an electron acceptor. The range of elements that can be harnessed for respiratory electron flow during these transformations is large, with redox changes that may be complex and difficult to dissect in experimental systems. For this reason, the microbial redox cycle for iron is a useful model system to examine. Iron is particularly important, being the fourth most abundant element in the Earth's crust, and one that has a key role in many biological systems. It is dominated by only two oxidation states: ferrous iron (Fe(II)) and ferric iron (Fe(III)), which are involved in many environmentally important oxidation and reduction processes.
2.1 Microbial oxidation of Fe(II)
Geomicrobiological studies have shown that there are at least three distinct mechanisms for Fe(II) oxidation. Under acidic conditions, Fe(II) can be oxidised aerobically (Johnson, 1998), whereas under anaerobic conditions at neutral pH, Fe(II) oxidation by phototrophs or denitrifying microorganisms has been recognised (Kappler and Newman, 2004; Weber et al., 2006). The latter two processes are environmentally important in areas ranging from the controls of metal/radionuclide solubility in geochemical systems to the processes of precipitation of banded iron formations (Kappler et al., 2005; Weber et al., 2006). However, they have only recently been characterized and have not been studied in the same detail as the oxidation of Fe(II) at low pH. Hence, we will focus here on the latter process.
Aqueous Fe(II) is very unstable in the presence of oxygen at neutral pH; however, at low pH (< 4.0; Stumm and Morgan, 1981) ferrous iron is oxidised very slowly by oxygen and is an excellent source of electrons for aerobic “acidophilic” prokaryotes. Although the free energy associated with the oxidation of Fe(II) is quite small, the relatively large amounts of ferrous iron in many acidic environments means that this process can play a defining role in controlling the chemistry at such sites. This is especially true where pyrite (FeS2) is present, for example at former sites of coal or metal mining, because when this mineral is exposed to moisture and air it oxidizes spontaneously with either molecular oxygen or ferric iron acting as the oxidant. A typical overall reaction can be written:
The mineral arsenopyrite (FeAsS), closely related to pyrite, is particularly environmentally hazardous, being a source of arsenic pollution as well as acid mine waters. Arsenopyrite oxidation has been shown to be 3 to 5 times faster than pyrite oxidation (McGuire et al., 2001; McKibben et al., 2008). Recent work involving both abiotic and microbial (Leptospirillum ferrooxidans) oxidative dissolution experiments, with mineral surface characterisation using Environmental Scanning Electron Microscopy (ESEM), X-ray Photoelectron Spectroscopy (XPS) and Auger Electron Spectroscopy (AES), has demonstrated the effectiveness of microbial involvement in accelerating the arsenopyrite breakdown process (Corkhill et al., 2008; see Fig. 2). As proposed for pyrite oxidation (Rimstidt and Vaughan, 2003), it is suggested that arsenopyrite oxidation is an electrochemical process with three steps: cathodic reaction, electron transport, and anodic reaction (Corkhill and Vaughan, 2009). The cathodic reaction involves an aqueous species that accepts electrons from an Fe(II) site on the mineral surface. The most important aqueous species in nature (and acid mine drainage environments) are O2 and Fe3+. These react with arsenopyrite according to the following generalised equations (after Walker et al., 2006 and Yunmei et al., 2007):
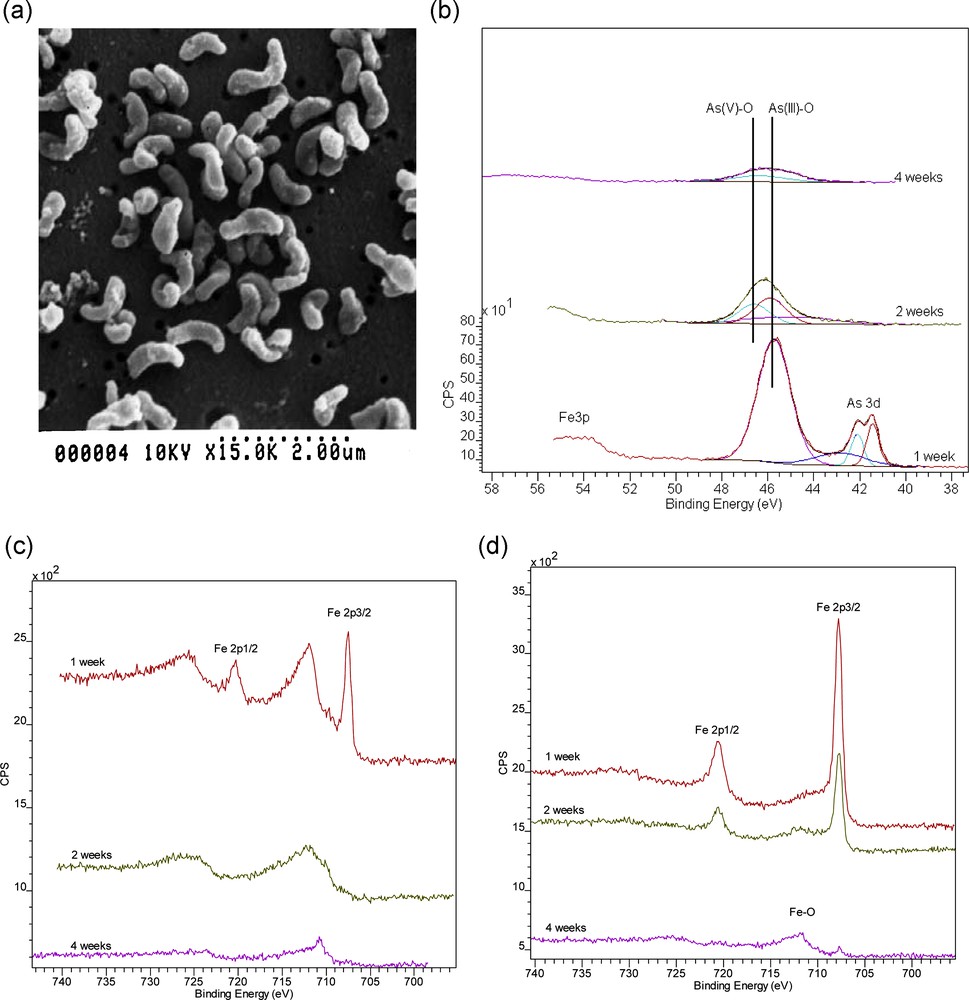
Oxidative breakdown of arsenopyrite (FeAsS) involving the organism Leptospirillum ferrooxidans: a: ESEM image of Leptospirillum; b: As 3d XPS data for the arsenopyrite surface oxidized in the presence of Leptospirillum for 1, 2 and 4 weeks; c: Fe2p XPS data for the arsenopyrite surface oxidized for periods of 1, 2 and 4 weeks in the presence of Leptospirillum; d: the same experiment in the absence of Leptospirillum. The Fe2p spectra show development of Fe3+ and Fe oxide peaks over time with much more rapid oxidation in the biotic system. The As3d spectra show evidence for the later onset of As3+ to As5+ oxidation (after Corkhill et al., 2008).
Dégradation de l’arsénopyrite (FeAsS) par oxydation sous l’action de l’organisme Leptospirillum ferrooxidans ; a : image de Leptospirrillum au microscope à balayage environnemental ; b : données de spectroscopie des photoélectrons RX pour As3d, dans le cas où la surface de l’arsénopyrite est oxydée en présence de Leptospirillum pendant 1, 2 et 4 semaines ; c : données de spectroscopie des photoélectrons RX pour Fe2p, dans le cas où la surface de l’arsénopyrite est oxydée en présence de Leptospirillum pendant 1, 2 et 4 semaines ; d : même expérience en l’absence de Leptospirillum. Les spectres Fe2p montrent le développement des pics de Fe3+ et d’oxydes de fer au long du temps, avec oxydation beaucoup plus rapide dans le système biotique. Les spectres As3d indiquent un démarrage plus tardif de l’oxydation d’As3+ en As5+ (selon Corkhill et al., 2008).
These reactions indicate that iron is not oxidised, while arsenic and sulphur are oxidised. The studies of Yunmei et al. (2004, 2007) and McKibben et al. (2008) show that the oxidation rate of arsenopyrite depends on dissolved oxygen and Fe(III); hence, the rate determining step of arsenopyrite oxidation is the transfer of electrons from the mineral to the oxidant. This important step transfers electrons from the anodic site to the cathodic site. It is here that acidophilic bacteria play an important role, transferring an electron from Fe(II) in the mineral surface to oxygen, creating energy and Fe(III).
In the anodic reaction, sulphide mineral oxidation involves the removal of seven electrons from mono-sulphide to form sulphate (Corkhill et al., 2008), and the removal of seven electrons from arsenide (As-1) to form arsenate (As5+) (Corkhill et al., 2008; Schaufuss et al., 2000).
This means that the sulphur and arsenic atoms must pass through a series of oxidation states during the oxidation process, leading to the many varied reaction products which have been identified in the literature. The most important reaction at the anodic site is likely to be the attachment of oxygen to arsenic, which has been shown in many cases to oxidise the most rapidly (e.g., Corkhill et al., 2008; Costa et al., 2002; Nesbitt et al., 1995; Schaufuss et al., 2000). When arsenic atoms are removed from the mineral surface, sulphur atoms from the (As-S)2 dianion are left with dangling bonds (Schaufuss et al., 2000). Based on an ab initio study of pyrite oxidation, Rosso et al. (1999) also suggest the presence of sulphur dangling bonds and propose that Fe-O bonds form in pyrite at the expense of negative charge of sulphur atoms, which might increase the susceptibility of surface sulphur atoms to hydrophilic attack. This would allow for the subsequent formation of S-O bonds. It is possible that these anodic reactions also occur for arsenopyrite, but further research, including ab initio computational studies of the arsenopyrite surface, is required to resolve the detailed mechanism of arsenopyrite oxidation.
2.2 Microbial reduction of Fe(III)
In the absence of oxygen in the subsurface, Fe(III) minerals can also make excellent electron acceptors for the anaerobic growth of specialist dissimilatory Fe(III)-reducing microbes, including a very wide range of Archaea and Bacteria (Lovley et al., 2004). The environmental relevance of microbial Fe(III) reduction has been well documented (Lovley, 1991; Thamdrup, 2000), with Fe(III) being the dominant electron acceptor in many subsurface environments (Lovley and Chapelle, 1995). As such, Fe(III)-reducing communities can be responsible for the majority of the organic matter oxidized in such environments (Lovley, 1993), while dramatically influencing the mineralogy of sediments. For example, the reductive dissolution of insoluble Fe(III) oxides can result in the release of potentially toxic levels of Fe(II), and also of trace metals that were bound by the Fe(III) minerals. Depending on the chemistry of the water, a range of reduced minerals can also be formed including magnetite (Fe3O4), siderite (FeCO3) and vivianite (Fe3(PO4)2.8H2O), resulting in a change in structure of the sediments. Fe(III)-reducing microorganisms can also impact on the fate of other high valence contaminant metals through direct enzymatic reduction, and also via indirect reduction catalysed by biogenic Fe(II). Metals reduced by these mechanisms include U(VI), Cr(VI) and Tc(VII) and, in such cases, reduction results in immobilization of these potentially toxic and mobile metals in sediments (Lloyd, 2003).
2.2.1 Overall mechanisms of Fe(III)reduction
The mechanisms of Fe(III)-reduction have been studied in most detail in two particular Fe(III)-reducing bacteria; Shewanella oneidensis and Geobacter sulfurreducens. Research on these organisms has been given added impetus through the availability of the genome sequences, and suitable genetic systems for the generation of deletion mutants for both of these organisms (Coppi et al., 2001; Myers et al., 2000). Although the terminal reductase has yet to be identified unequivocally in either organism, the involvement of c-type cytochromes is implicated in electron transport to Fe(III) in several studies (Beliaev et al., 2001; Gaspard et al., 1998; Lloyd et al., 2003; Magnuson et al., 2000; Myers and Myers, 1993, 1997). In some examples, activities have been localized to the outer membrane or surface of the cell (see Figs. 3 and 4), consistent with a role in direct transfer of electrons to Fe(III) oxides that are highly insoluble at circumneutral pH (DiChristina et al., 2002; Gaspard et al., 1998; Lloyd et al., 2002; Myers and Myers, 1992, 2001). A simplified schematic of the electron transfer chain that traverses both the periplasm and outer membrane of the Gram-negative Geobacter sulfurreducens cell is shown in Fig. 4. Geobacter outer membrane (OmcB, OmcD and OmcE) and periplasmic (PpcA and PpcB) c-type cytochromes are shown, and analogous proteins have been identified in Shewanella species. In addition to the proposed direct transfer of electrons to Fe(III), soluble “electron shuttles” are also able to transfer electrons between metal-reducing prokaryotes and the mineral surface (Fig. 3). This mechanism alleviates the requirement for direct contact between the microorganism and mineral. For example, humics and other extracellular quinones are utilized as electron acceptors by Fe(III)-reducing bacteria (Lovley et al., 1996), and the reduced hydroquinone moieties are able to abiotically transfer electrons to Fe(III) minerals. The oxidized humic is then available for reduction by the microorganism, leading to further rounds of electron shuttling to the insoluble mineral (Nevin and Lovley, 2002). Very low concentrations of an electron shuttle, e.g., 100 nM of the humic compound analogue anthraquinone-2,6-disulfonate (AQDS), can rapidly accelerate the reduction of Fe(III) oxides (Lloyd et al., 1999). Recent studies with Shewanella species have also shown that flavin molecules (FMN and riboflavin) are secreted to act as endogenous electron shuttles in this organism (Marsili et al., 2008; von Canstein et al., 2008). The environmental significance of such processes, however, remains to be confirmed. The addition of small amounts of electron shuttle to cultures of Fe(III)-reducing bacteria is, however, a very useful strategy to accelerate dramatically the rate of Fe(III) reduction in laboratory studies. A final mechanism of electron transfer to Fe(III) minerals that has also received much attention is the formation of conductive nanowires projecting from the surface of Geobacter and Shewanella species (Gorby et al., 2006; Reguera et al., 2005). The environmental relevance of these potentially important extracellular structures remains to be confirmed and is being addressed in ongoing research in many laboratories worldwide (see Gralnick and Newman, 2007, Shi et al., 2009 and Weber et al., 2006 for additional general reviews in this area).
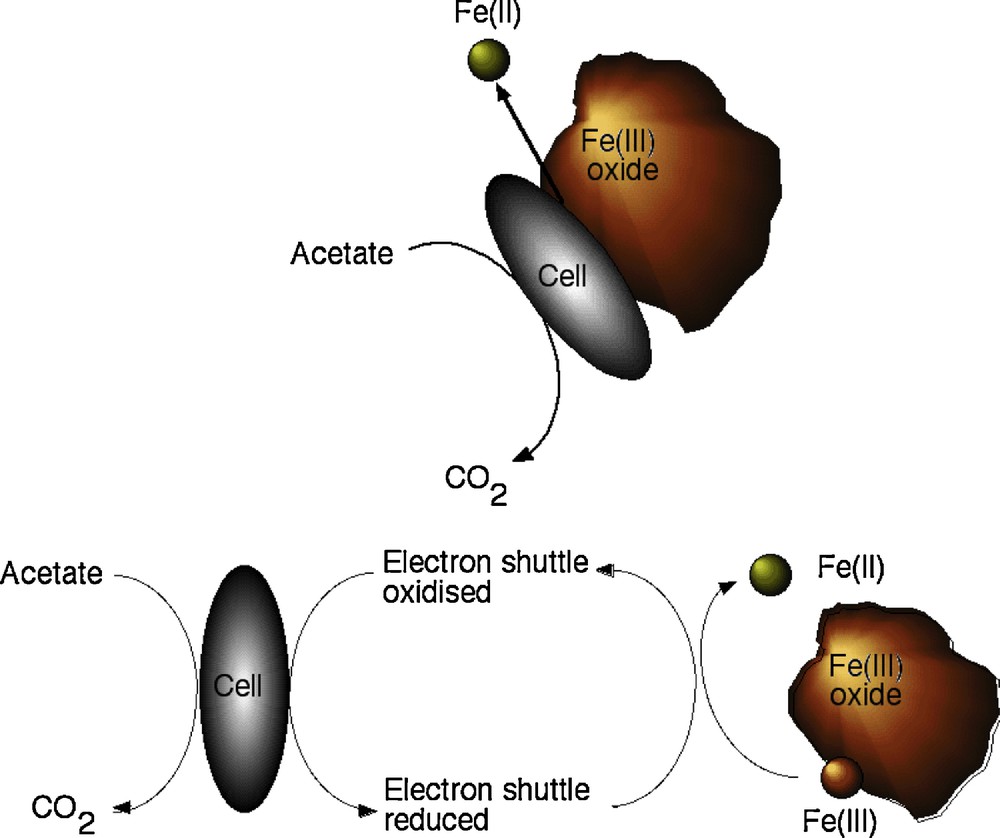
Schematic ‘cartoon’ diagram illustrating (top) direct reduction of an Fe(III) oxide particle through contact with the cell of a bacterium such as Geobacter or Shewanella with concomitant oxidation of organic matter (acetate) and (bottom) the same process involving an electron shuttle (such as a humic compound or AQDS) (after Lloyd, 2008).
Diagramme schématique en « bande dessinée » montrant (dans la partie supérieure), la réduction directe d’une particule d’oxyde de Fe(III) au contact d’une cellule de bactérie telle Geobacter ou Shewanella, avec oxydation concomitante de matière organique (acétate) et (dans la partie inférieure) le même processus comportant un électron « navette » (tel un composé humique ou AQDS) (selon Lloyd, 2008).
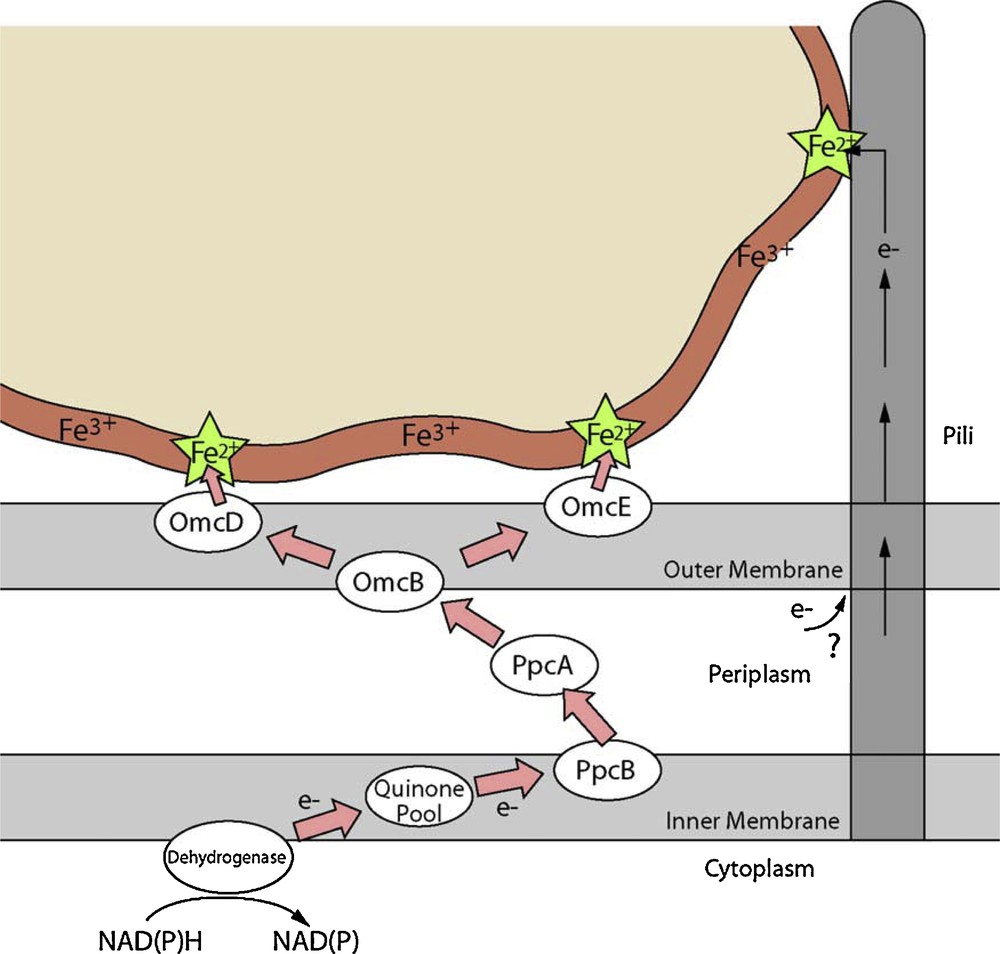
Schematic ‘cartoon’ diagram showing the proposed biochemical transformations and electron transfer paths at the interface between a Fe(III) mineral and Geobacter sulfurreducens. Similar mechanisms are present in other Fe(III)-reducing bacteria including Shewanella species.
Diagramme schématique en « bande dessinée » montrant les transformations biochimiques et les itinéraires de transfert d’électrons proposés, à l’interface entre un minéral ferrifère Fe(III) et Geobacter sulfurreducens. Des mécanismes similaires existent pour d’autres bactéries réductrices de Fe(III) incluant l’espèce Shewanella.
2.2.2 Fe(III) reduction: the mineral substrate
The process of Fe(III) reduction is also dependent upon the nature of the mineral substrate, and a range of techniques have proved useful to investigate the interactions of Fe(III)-reducing bacteria with the mineral surfaces that they respire. For example, hydrated films of poorly crystalline Fe(III) oxides offer good models for the highly bioavailable mineral phases that are the ideal substrates for these organisms in natural environments. Here, environmental scanning electron microscopes are particularly useful in visualising the interactions of the microbial cells with the hydrated samples (Wilkins et al., 2007a, 2007b), showing clearly the impact of electron transfer at the cell surface on the Fe(III) mineral phase (see Fig. 5). Interestingly, probing the topmost few nanometres of the surface with X-ray Photoelectron Spectroscopy (XPS) in this study (see Fig. 5) failed to identify Fe(II), and this was confirmed by challenging the samples with redox active chemical probes, highly sensitive to exposed Fe(II) (Wilkins et al., 2007a, 2007b). These observations suggest electron transfer is possible deeper into the mineral substrate, effectively increasing the amount of electron acceptor available to the organism, which in turn has important implications for the growth yield of these organisms in the subsurface.
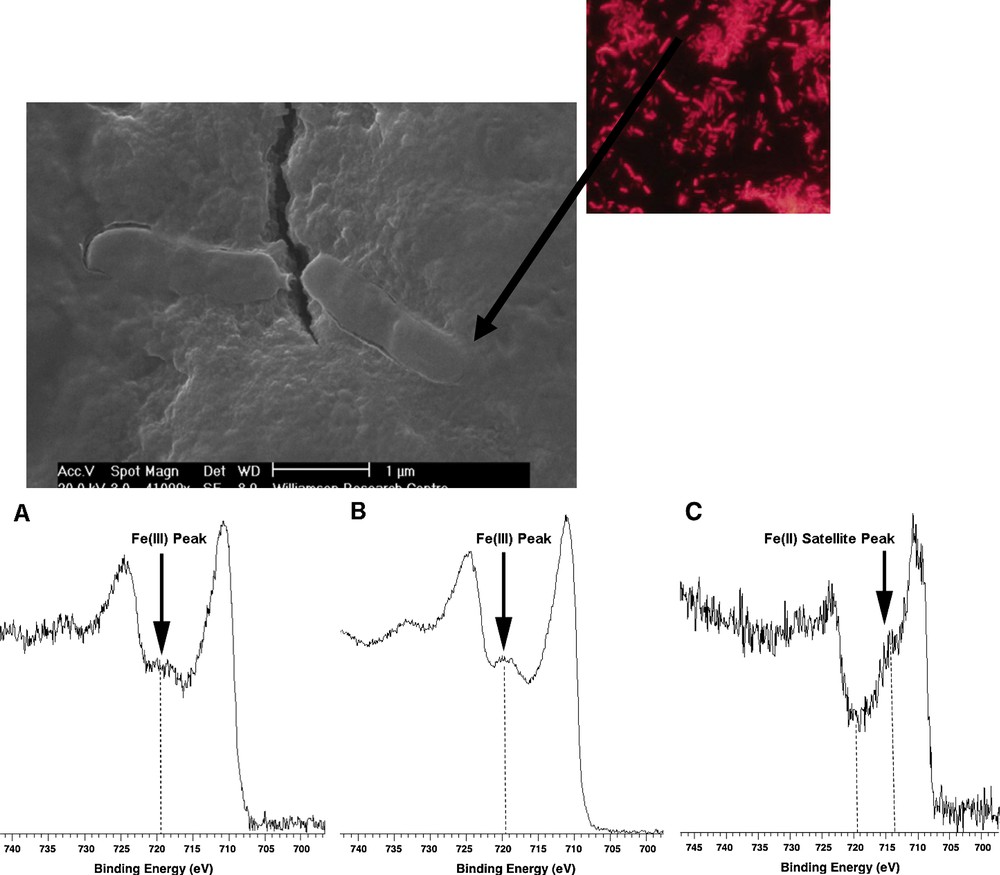
(Top) Fluorescence microscope and Environmental Scanning Electron Microscope (ESEM) images of Geobacter sulfurreducens interacting with a hydrous ferric oxide (HFO) substrate. Three Fe2p XPS spectra (bottom) show HFO starting material (A); the same surface after 55 days incubation with G. sulfurreducens (B); a vivianite standard showing an Fe(II) satellite peak. Spectra (A) and (B) show only Fe(III) peaks and no evidence for Fe(II) at the surface (after Wilkins et al., 2007a, 2007b).
À la partie supérieure, images obtenues au microscope à fluorescence et au microscope électronique à balayage environnemental (MEBE), de Geobacter sulfurreducens en interaction avec un substrat ferrique hydraté (HFO). À la partie inférieure, trois spectres de photoélectrons RX pour Fe2p correspondant (A) au matériau de départ (HFO) ; (B) à la même surface après 55 jours d’incubation avec G. sulfurreducens ; (C) à une vivianite standard montrant un pic satellite Fe (II). Les spectres (A) et (B) ne révèlent à la surface, que des pics Fe(III), mais pas de pic Fe(II).
For higher resolution imaging of the microbe-mineral interface, Transmission Electron Microscopy (TEM) offers near atomic resolution at the price of working in a vacuum with dried samples. Here, detailed characterization of the starting and end products of Fe(III) reduction has confirmed the recalcitrance of crystalline phases such as hematite to reduction by Geobacter sulfurreducens, in stark contrast to highly bioavailable nanoparticulate phases such as ferrihydrite or feroxyhyte which are transformed rapidly to magnetite (Cutting et al., 2009; see Fig. 6). Colorimetric (ferrozine) assays for Fe(II), in combination with XPS analyses confirmed a lack of ferrous iron associated with the more crystalline mineral phases after incubation with the Fe(III)-reducing bacterium (Fig. 7). The bar diagram with these results shows both the much lower bulk rates of reduction of the more crystalline phases and the dramatic effects of introducing an electron shuttle compound (AQDS) into the system. The XPS analyses illustrated in Fig. 7 show the surface Fe(III):Fe(II) data for the crystalline hematite compared with that for the poorly crystalline hydrous ferric oxide (HFO), before and after 144hrs incubation with G. sulfurreducens, and both without and with AQDS. Both starting materials have surfaces (i.e. < 6 nm depth) which are 100% Fe(III), and whereas HFO shows ∼8% reduction after exposure without AQDS and ∼20% reduction with AQDS, the hematite only shows ∼ 2% reduction with AQDS present and none in its absence. A major conclusion from these data is that surface area alone is not the critical factor controlling the rate and extent of Fe(III) reduction in the subsurface. Of course, in natural environments Fe(III)-reducing bacteria will not interact with Fe(III) phases in isolation, and sorbed trace elements can play a role in controlling both the rate and the end-point of microbial reduction (Zachara et al., 2001).

TEM images and selected area electron diffraction (SAED) patterns for synthetic Fe(III) oxyhydroxide starting materials (lepidocrocite, akaganeite, feroxhyte and HFO) and for the same materials following 144 hrs of incubation with G. sulfurreducens in the presence of 10 μM AQDS (after Cutting et al., 2009).
Images obtenues au microscope électronique à transmission (MET) et diagrammes de diffraction électronique sur aires sélectionnées de matériaux de départ synthétiques d’oxy-hydroxydes de Fe (III) (lépidocrocite, akaganéite, feroxhyte et HFO) et pour ces mêmes matériaux, après 144 h d’incubation avec G. Sulfurreducens, en présence de 10 μM d’AQDS (selon Cutting et al., 2009).
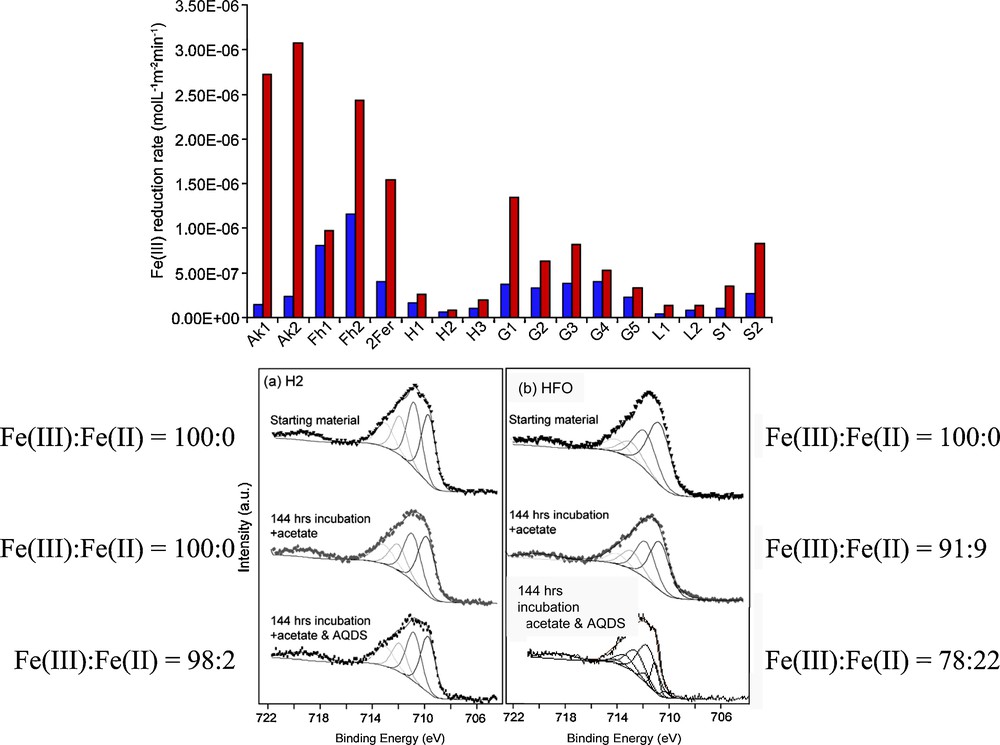
Bar chart (top) showing the rates of Fe(III) reduction by G. sulfurreducens of a series of synthetic Fe(III) minerals in the presence of acetate only (red bars) and acetate plus AQDS (10 μM) (blue bars). Key to figure: Ak- akaganeite; Fh- feroxhyte; Fer- ferrihydrite; H-hematite; G-goethite; L-lepidocrocite; S-schwertmannite. Also shown (bottom) are XPS data used to determine Fe(III):Fe(II) ratios at the surfaces of synthetic hematite (H2) and HFO starting materials and after 144 hrs exposure to G. sulfurreducens without and with (10 μM) AQDS (after Cutting et al., 2009).
À la partie supérieure, histogramme montrant les taux de réduction par G. sulfurreducens d’une série de minéraux Fe(III) synthétiques en présence d’acétate uniquement (barres rouges) et d’acétate + AQDS (10 μM) (barres bleues). Ak-akaganéite ; Fh-feroxhyte ; Fer-ferrihydrite ; H-hématite ; G-goethite ; L-lépidocrocite ; S-schwertmannite. Dans la partie inférieure de la figure, les données de spectroscopie des photoélectrons RX sont utilisées pour déterminer les rapports Fe(III)/Fe(II) à la surface des matériaux de départ hématite synthétique et HFO, et après 144 h d’exposition à G. sulfurreducens, sans et avec (10 μM) d’acétate (selon Cutting et al., 2009).
3 Mineral-organic interactions
At the most fundamental level, the interaction between a mineral surface and a microbe is that between the surface and an organic molecule, and a proper understanding of this interaction requires knowledge of the nature of the surface at a molecular scale. In fact, molecular-scale studies of such interactions are now possible and involve both experimental and computational approaches; one example is our atomic resolution study of interactions of the magnetite (111) surface with three contrasting organic molecules: formic acid, pyridine and carbon tetrachloride (Cutting et al., 2006). This work involved Scanning Tunneling Microscopy (STM) experiments under carefully controlled (UHV) conditions, and is a good illustration of the complexity of surface structure in even a relatively simple mineral. In Fig. 8, three experimental STM images are shown (and labeled as A, A’ and B surfaces); all are atomic resolution images of the magnetite (111) surface. These STM data are interpreted as different terminations of magnetite (111) arising from different level ‘slices’ through the bulk structure, which is also shown in Fig. 8; thus there may be unexpected complexities when considering only a single ‘face’ of a particular mineral.
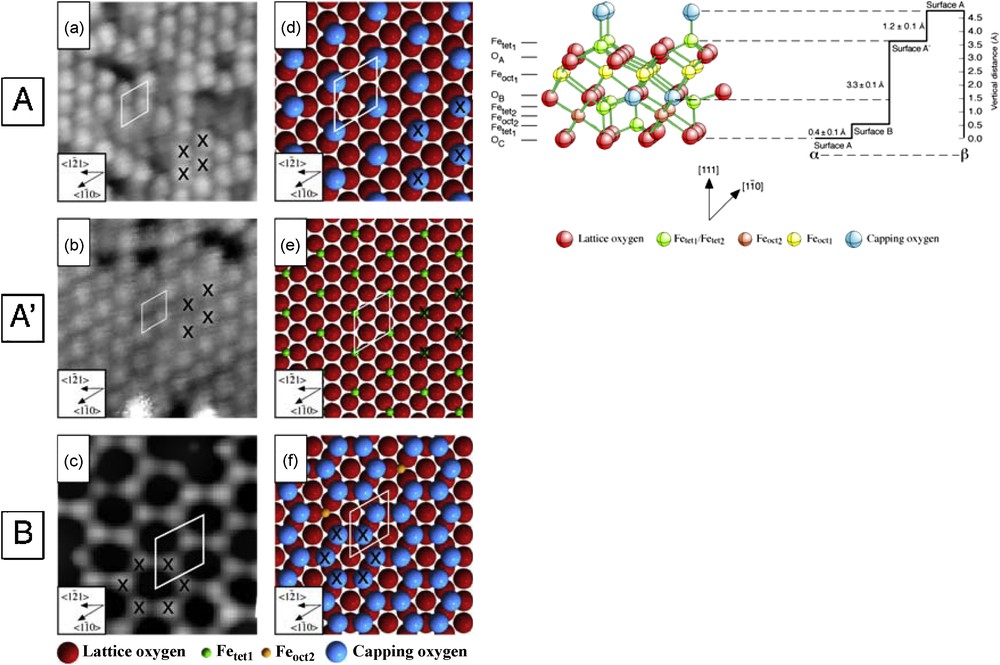
Scanning Tunneling Microscope (STM) images of the (111) surface of magnetite obtained under UHV conditions: (a) 100 × 100 Å2 image of the A surface; (b) 120 × 120 Å2 image of the A’ surface; (c) 16 × 16 Å2 image of the B surface, with corresponding models of the surfaces (d, e and f, respectively) which are interpreted as arising from different level slices through the bulk structure of magnetite, as illustrated on the right hand side of the figure (Redrawn after Cutting et al., 2006).
Images obtenues au microscope à balayage à effet tunnel des surfaces (111) de la magnétite sous conditions UHV : (a) Image 100 × 100 Å2 de la surface A ; (b) Image 120 × 120 Å2 de la surface A’ ; (c) Image 16 × 16 Å2 de la surface B, avec modèles correspondants des surfaces (d, e, f respectivement) interprétées comme des sections, à différents niveaux au travers de la structure brute de la magnétite, ainsi qu’on peut l’observer en haut et à droite de la figure (redessiné d’après Cutting et al., 2006).
As well as probing the ‘molecular’-scale structures of mineral surfaces using methods such as STM, computer modeling techniques may also provide insights, although they need to be carefully compared with experimental data. An example, again from our work (Martin et al., 2009), is shown in Fig. 9. Here, STM images have been generated for three iron (hydr)oxide surfaces using quantum mechanical calculations. The magnetite (111) surface, corresponding to ‘surface B’ mentioned above, shows good agreement with experiment (see Fig. 8); the hematite (0001) and goethite (010) surfaces have, thus far, not been successfully imaged at atomic resolution by STM.

Computer generated STM images for: (a) hematite (0001); (b) goethite (010) and (c) magnetite (111) surfaces and the corresponding molecular models (after Martin et al., 2009).
Images de microscopie à balayage à effet tunnel produites par ordinateur pour les surfaces : (a) de l’hématite (0001) ; (b) de la goethite (010) et (c) de la magnétite (111) et modèles moléculaires correspondants (selon Martin et al., 2009).
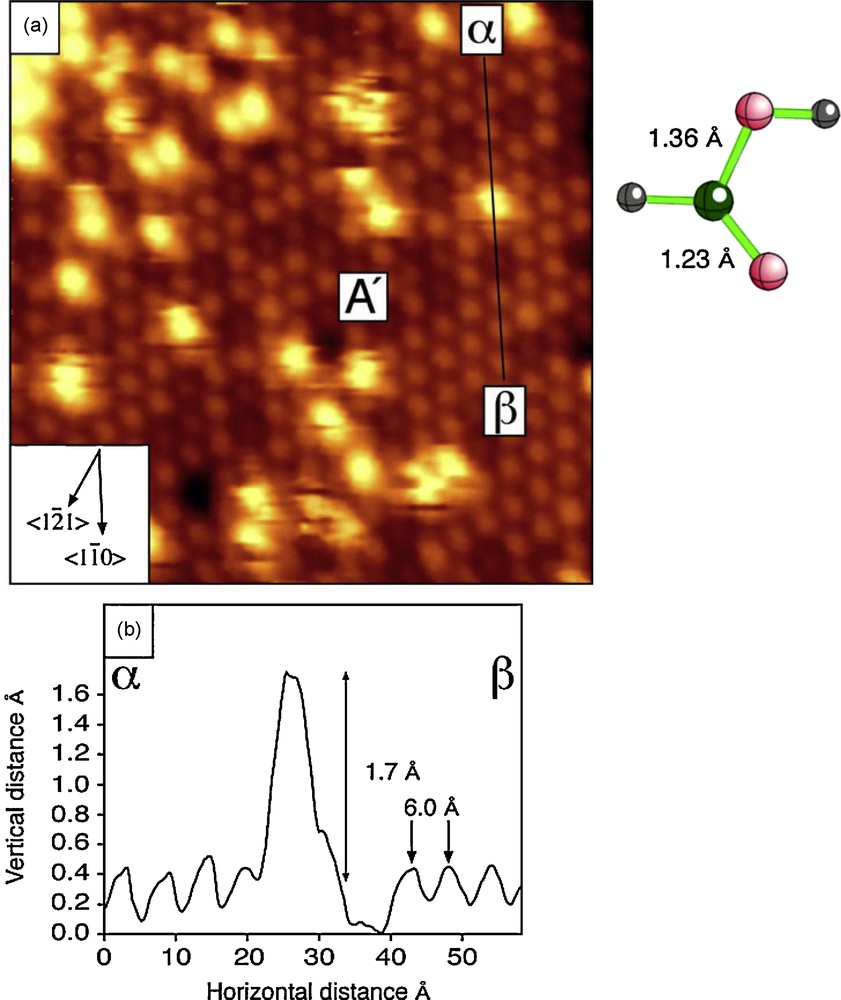
In Fig. 10 an STM image is shown of the magnetite (111) surface after exposure to a small amount of pyridine. Individual molecules of pyridine give rise to the bright features in the image, and the topography determined by the cross section labeled α-β is consistent with interaction of the N atoms of pyridine with tetrahedral site Fe atoms exposed in the A’ surface (see above) and as shown in the molecular model (Fig. 10).

Interaction of magnetite (111) surface with pyridine: STM image (top left); STM line profile (bottom left); the pyridine structure and a model for proposed interaction (right) mechanism (after Cutting et al., 2006).
Interaction de la surface (111) de la magnétite avec la pyridine : image obtenue au microscope à balayage à effet tunnel (en haut à gauche) ; profil linéaire α-β (en bas à gauche) ; structure de la pyridine et modèle de mécanisme d’interaction proposé (à droite) (selon Cutting et al., 2006).
This interaction contrasts with that of the same magnetite surface after interaction with formic acid. Experimental data for this system are shown in Fig. 11 and are consistent with dissociation of the formic acid to protons and formate (HCOO−) and bonding of the formate at the magnetite surface. The three possible models for formate group attachment (bidentate bridging, bidentate non-bridging and monodentate) are illustrated in Fig. 12. Here, again, the arrangement of features in the x-y plane of the STM image, and in the z direction as seen in the line profile (Fig. 11), enables the choice of a bidentate non-bridging configuration as the appropriate model for interaction of the formate molecule.
Interaction of magnetite (111) surface with formic acid: (a) STM image, and (b) line profile(α- β), along with structure of formic acid molecule (after Cutting et al.,2006).
Interaction de la surface (111) de la magnétite avec l’acide formique : (a) image obtenue au microscope à balayage à effet tunnel et (b) profil linéaire (α-β) avec, en regard, la structure de la molécule d’acide formique (selon Cutting et al., 2006).

Models for three possible modes of attachment of the formate molecule at the magnetite (111) surface showing (on left) bidentate bridging (a), bidentate non-bridging (b) and monodentate (c) configurations, and (on right) bidentate non-bridging (proposed model) option seen in plan and cross-sectional view, (after Cutting et al., 2006).
Modèles pour trois modes de liaison possibles de la molécule de formate sur la surface (111) de la magnétite montrant, à gauche, un pontage bidenté (a), un non-pontage bidenté (b), et des configurations monodentées (c) et, à droite, un non-pontage bidenté (modèle proposé) vu en plan et en coupe (selon Cutting et al., 2006).
The third set of experiments involved exposure of the magnetite surface to molecules of carbon tetrachloride. Here, the STM data could only be interpreted with the help of results from other experiments, such as Temperature Programmed Desorption (TPD) studies where, after exposure of the magnetite to CCl4 in a controlled (UHV) environment, gaseous species released from the reacted surface over a range of temperatures (100 to 820 K) are monitored by mass spectrometry. It appears that, at the lowest temperatures, intact CCl4 molecules are sorbed at the surface. Then, at around room temperature, there is dissociation into CCl2 and Cl species, and at successively higher temperatures, removal of surface oxygens from the magnetite to form OCCl2 (phosgene gas), and of Fe to form FeCl2 at the highest temperatures. At around room temperature, both strongly bonded Cl atoms and weakly bonded CCl2 molecules appear to co-exist on the same (A’ type) magnetite surface (Cutting et al., 2006).
What is clear from these studies is that there can be a considerable diversity of behaviour for different organic molecules even on interaction with just one mineral surface. The three molecules discussed above show behaviour ranging from chemisorption of the complete molecule (pyridine), to dissociation and chemisorption (formic acid), and to dissociation giving both strongly bonded atoms and weakly bonded molecules. Interaction of the mineral surface with organic molecules is the first stage in the formation of biofilms, and therefore in mineral-microbe interactions.
4 Biofilms and interfacial processes
In most natural environments, microbial cells actually grow on solid substrates as biofilms; hence the mineral substrate is crucial. A useful definition of the term “biofilm” is that given by Characklis and Marshall, 1990 in stating that microbial cells attach to solids (such as minerals) and “immobilized cells grow, reproduce and produce extracellular polymers which frequently extend from the cell forming a tangled matrix of fibers which provide structure to the assemblage termed a biofilm”. However, traditional microbiological approaches have focused on studying planktonic cell cultures grown under well-characterized, highly mixed growth regimes. The traditional approaches have tended to limit many geomicrobiological studies, because cell growth in association with a biofilm has a major impact on the host cell physiology, and also on the physical and chemical environment associated with the biofilm (Stoodley et al., 2002). It has also long been known that biofilms at a solid-fluid interface can maintain fluid conditions in the region of the mineral surface which are very different relative to those in the associated bulk fluid (e.g., in terms of pH, pO2 etc).
A number of key aspects of the impact of biofilms on geological systems can be illustrated by work in our laboratories (Brydie et al., 2004, 2009), including studies relevant to our theme of systems containing iron. We have conducted experiments with Pseudomonas aeruginosa biofilms at micro-, meso- and macro-scales. At the micro-scale, experiments involved the growth of biofilms in a fracture which was simulated using two silica glass surfaces ∼ 30 μm apart, with the structure and composition of the biofilm at various stages of growth being studied using Confocal Laser Scanning Microscopy (CLSM) imaging (see Fig. 13). In the early stages, imbricate structures were observed associated with the flow of fluid through the simulated fracture. The development of a mature biofilm, however, showed complex columnar structures. Crucially, substantial areas of the “mineral” surface remained free from biofilm material and able to interact with the fluid (see Fig. 13). In subsequent experiments, a Pseudomonas aeruginosa biofilm on a quartz glass substrate was exposed to very low concentrations of iron in solution (100 mg l−1); this led to formation of iron oxyhydroxide precipitates (lepidocrocite) under conditions where precipitation would not have occurred without the presence of such a biofilm.

Synthesis of P. aeruginosa biofilm in a fracture system showing experimental set-up (top left), CLSM image of early stage biofilm (bottom left) and of mature biofilm (top right) with explanatory diagram for the latter below (after Brydie et al., 2009).
Synthèse d’un biofilm de P. aeruginosa biofilm dans un système de fracture montrant la mise en place expérimentale (en haut à gauche), l’image au microscope à balayage laser confocal d’un biofilm au stade précoce (en bas à gauche) et d’un biofilm mature (en haut à droite) avec un diagramme explicatif pour ce dernier (au-dessous) (selon Brydie et al., 2009).
The development of biofilms can also have a significant impact on the flow of fluid through fractures in rocks or through porous media, causing so-called bioclogging. Fig. 14 illustrates the results of experiments involving the flow of fluid through a column packed with pure quartz sand (Brydie et al., 2004). Following inoculation with Pseudomonas and biofilm growth, the hydraulic conductivity of the sand column drops by several orders of magnitude. Also shown in this figure are ESEM images of biofilm grown between the quartz grains in the column at realistic concentrations of nutrients. In related column experiments, Boult et al., 2006 and Hand et al., 2008 have explored the influence of a range of variables (grain size, oxygen availability, organic carbon availability) on bioclogging in porous media, and its influence on the mobility of contaminants such as metals. For example, Boult et al., 2006 show that microbial activity affects the mobility of Cu under the relatively low nutrient fluxes that dominate subsurface systems; microbes and microbial products can immobilize metals such as Cu but may themselves be subject to subsequent mobilization, complicating the pattern of metal storage and release.
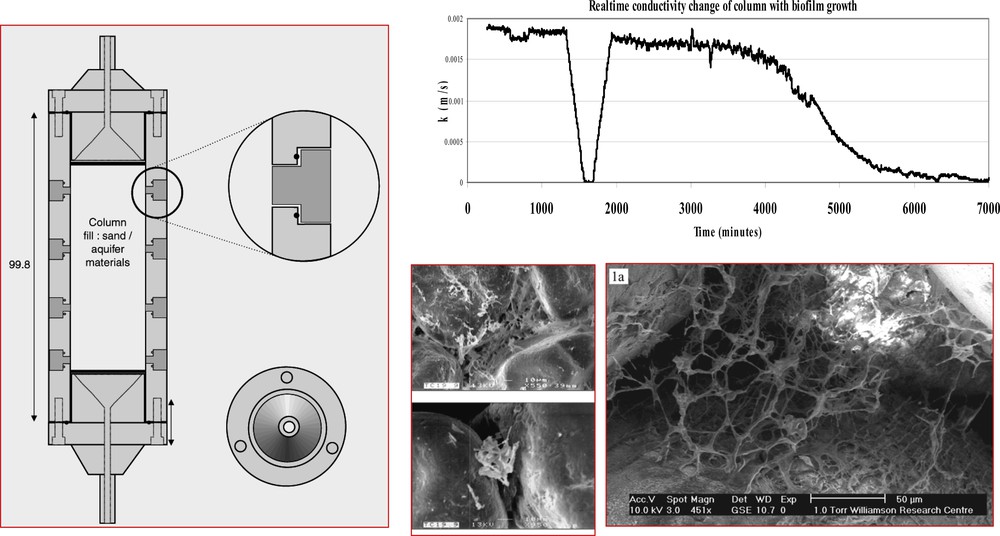
Biofilm fluid flow experiments (meso-scale) showing experimental set-up (on left), hydraulic conductivity experiment data (top right) and ESEM images of material from column experiments, (after Brydie et al., 2004).
Expériences d’écoulement d’un fluide de biofilm (méso-échelle) montrant la mise en place expérimentale, (à gauche), les données expérimentales de conductivité hydraulique (en haut à droit) et les images au microscope électronique à balayage environnemental (selon Brydie et al., 2004).
In a further development of these experiments concerned with fluid flow, and aimed at exploring behaviour at the macro-scale, Brydie et al., 2004 employed a 2.7m radius geotechnical centrifuge in order to simulate 100m field-scale pressure in manageable 0.6m high columns which were spun at 100 gravities. This experimental set-up is shown in Fig. 15, which also shows the results of measurements made on a (quartz) sand-filled column in the centrifuge. Initial hydraulic conductivity (shown by the dashed line at the top of the figure) falls dramatically after biofilm formation. The injection of iron in solution, actually accomplished during the operation of the centrifuge, can be seen to cause a further reduction in hydraulic conductivity. The important point here is that, whereas biofilms may be ephemeral phenomena, they can bring about precipitation of minerals, such as iron hydroxides. Such minerals may have long lasting effects on the chemical and hydraulic properties of a system, reducing fluid flow and, in some cases, trapping toxic trace elements by sorption to their surfaces or incorporation into their structures.
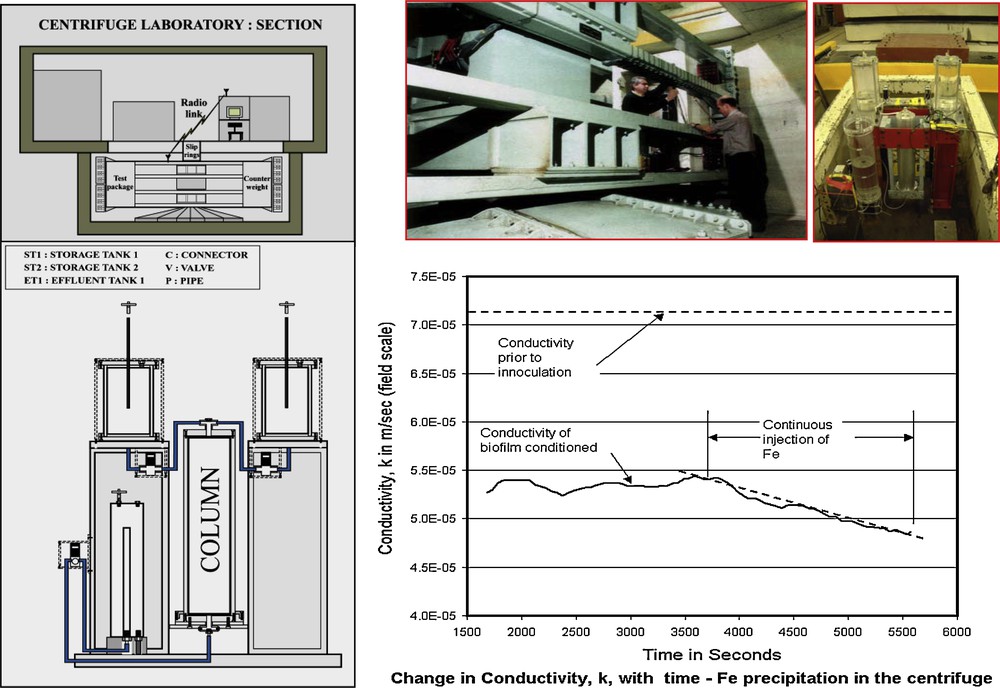
Biofilm fluid flow experiments (macro-scale) using geotechnical centrifuge: a scheme of the centrifuge is shown (top left) with experimental payload shown below and photographs of both shown (top right). Experimental results are shown bottom right with varying hydraulic conductivity over time under various conditions (after Brydie et al., 2004).
Expériences d’écoulement d’un fluide de biofilm (macro-échelle) utilisant une centrifugeuse géotechnique: un schéma de la centrifugeuse est présentée en haut à gauche, avec la charge utile indiquée au-dessous de la photographie des deux, en haut à droite. Les résultats expérimentaux sont fournis en bas à droite, avec une conductivité hydraulique variable en fonction du temps sous des conditions variées (selon Brydie et al., 2004).
5 Concluding remarks
The processes taking place at the mineral-microbe interface were surely amongst the very first associated with the emergence of life on Earth. They have played a key role in the biogeochemical cycling of the elements since that time, and the part played by them in the past, in processes such as the formation of the geologically ancient banded iron formations (currently the dominant world suppliers of iron), is only now being fully appreciated. Recent years have seen rapid growth in the understanding of mineral-microbe interactions, prompted by both advances in methods for studying minerals and their surfaces at the molecular scale, and by developments in microbiology, not least those involving molecular techniques. Only a few examples of the systems already studied just in our laboratories have been discussed in the present article; we have also recently investigated mineral-microbe interaction processes in systems involving a range of other metals including Cr, Hg, Tc, U, Pu and As (see, for example, Burke et al., 2006; Islam et al., 2004; McBeth et al., 2007; Renshaw et al., 2009; Wilkins et al., 2007a, 2007b). There is also much work from other groups including, for example, that published by Fendorf and Kocar, 2009; Fendorf et al., 2010; Icopini et al., 2009 and Jaisi et al., 2009. Such studies are of relevance to the mobility of toxic metals in the environment, to strategies limiting their environmental impact, and to the development of technologies for the bioremediation of contaminated sites. For example, recent studies from our laboratories have focused on the reductive immobilization of high oxidation state metals and radionuclides including Tc(VII) and Cr(VI) (Cutting et al., 2010). Here we have shown that the formation of highly reactive biogenic magnetite nanoparticles can be optimized through the choice of an appropriate Fe(III)-bearing mineral phase for bioreduction by G. sulfurreducens. Techniques used to identify the mechanism of metal immobilization by the magnetite included XPS, XMCD and TEM, showing that metal removal was linked to the reduction of Tc(VII) and Cr(VI) to Tc(IV) and Cr(III) respectively, mediated by biogenic Fe(II). In the case of Cr(III), the reduced metal was incorporated into the spinel structure, which should result in an end product that is relatively resistant to re-oxidation.
Future research in mineral-organic-microbe interactions will surely see benefits to our understanding of biogeochemical cycles at the molecular level, helping in the quantitative prediction of migration of contaminants in the ‘critical zone’. It should also bring a more fundamental understanding of how particular microorganisms function, including those capable of flourishing under extreme conditions of pH, temperature or pressure. The practical applications of such work are relevant to many of the key problems now facing society, including the safe containment of hazardous (including nuclear) wastes, the cleanup of contaminated soils and waters, and development of efficient clean technologies for processing of minerals or recovery of metals and toxic materials from wastes.
The year 2009 was the 200th anniversary of the birth of Charles Darwin and the 150th anniversary of the publication of his book “On the Origin of Species”. Although almost always thought of as a biologist, Darwin was also a geologist (three quarters of the notes produced by him from the famous voyage of HMS Beagle dealt with geological topics). Darwin acknowledged his debt to the founders of geology such as Charles Lyell, and is quoted as saying “I always feel as if my books came half out of Lyell's brain”. He also acknowledged the debt he owed to biologists of earlier generations, including Jean Baptiste Lamark, described by Darwin as a “justly celebrated naturalist” and who, 50 years before him had proposed a full blown theory of evolution. We owe much to the tradition of the great British and French naturalists of the 19th century, and it is appropriate that current developments in ‘environmental mineralogy’ are again bringing together the biological and geological sciences.
Acknowledgements
The authors acknowledge the funding received from the UK Natural Environment Research Council, Engineering and Physical Sciences Research Council, and Biotechnology and Biological Sciences Research Council in support of the work described in this article. The contributions made by numerous colleagues, particularly J. Brydie, C. Corkhill, R. Cutting, V. Hand, M. Wilkins and P. Wincott are also gratefully acknowledged.