

1 Introduction
The chemical and physical states of soils and sediments are the legacy of a host of past and present processes, acting both independently and in synergy one with another. The oxidation state of iron (Fe) in the crystal structure of expandable clay minerals is one such factor, being well known for its effect on the surface and colloidal chemistry of the clay (Gates et al., 1993; Stucki, 2006; Stucki et al., 1984b, 1988) which, in turn, influences the behavior of the overall soil matrix and the fate of environmental contaminants (Hofstetter et al., 2003, 2006; Neumann et al., 2008, 2009; Shen et al., 1992; Tor et al., 2000; Xu et al., 2001). The respiration of soil-borne microorganisms creates a dynamic reduction-oxidation (redox) system in which electrons are shuttled between the organisms and their surroundings, including soil organic matter and clay minerals (Stucki and Kostka, 2006). When water either fills soil pores or forms films around mineral particles, local anaerobic conditions are quickly attained and heterotrophic microorganisms seek alternative electron acceptors such as structural Fe(III) in the soil clay minerals, converting it to Fe(II) and thereby invoking a variety of chemical and physical consequences. Upon evaporation or drainage of the water, aerobic conditions return and reoxidation occurs. This cycle is and has been repeated frequently over the history of the Earth, but only a few studies have documented its potential ramifications with respect to soil formation and transformation.
While studies of the effects of redox cycles on clay properties have been more limited than those from the first or a single reduction event, they have revealed that the reoxidized state of the clay may be different from the original oxidized state, depending on the property being investigated and the nature of the reductant. In other words, redox cycles likely invoke an evolution of the character and performance of soils and sediments over time, but the extent of that evolution differs depending on the extent of reduction and whether reduction was invoked by synthetic inorganic reductants or mediated by microorganisms. The purpose of this manuscript was to review the present state of knowledge regarding the effects of redox cycles on the properties and behavior of soils and sediments. Reviewed below are comparisons between the effects of redox cycles when reduction is achieved by either dithionite (a synthetic inorganic reductant) or by naturally occurring microorganisms.
2 Results and discussion
2.1 Color
One of the properties of Fe-rich smectites that is greatly affected by redox reactions is the color. In the unaltered state, ferruginous smectite and nontronite are yellow in aqueous suspension (Fig. 1). Upon reduction, the color changes from yellow to green, then to blue-green, and finally to blue-grey as the extent of reduction proceeds from 0 to about 96%. These color changes appear because of changes in intra- and intervalence electron transfer transitions involving Fe(II), Fe(III), and O → Fe(III) charge transfer transitions (Banin and Lahav, 1968; Lahav and Banin, 1968; Sherman and Vergo, 1988). The differences in color are striking (Fig. 1) and produce rather significant shifts in the UV-Visible spectra (Fig. 2). The unaltered (oxidized) sample, for example, has a high background at lower wavelengths due to an O → Fe(III) charge transfer transition that occurs at about 260 nm, which is greatly diminished in the reduced and reoxidized samples (Fig. 2). A primary contributor to the color is an intervalence electron charge transfer transition (IVCT) that occurs between Fe(II) and Fe(III) in the octahedral sheet of the reduced clay, and appears in the UV-Visible spectrum at about 700 to 750 nm as reported and discussed by Lear and Stucki (1987). The color reversibility in ferruginous smectite with redox cycles can be quantified by comparing the IVCT band in the UV-Visible spectra (Fig. 2) at a wavelength of about 720 nm for ferruginous smectite, the intensity of which varies with reduction level as described above. The spectra for the unaltered and reoxidized samples (Fig. 2) are very different, thus providing spectroscopic documentation of the visual observations that the reoxidized color differs somewhat from the unaltered color, even though both have a yellow hue. To understand these differences from a mechanistic perspective, a more complete analysis than has been carried out in the past of changes in particle size, Fe site coordination, Fe clustering, and other factors controlling the light scattering and chromophoric absorption phenomena is necessary.

Illustration of color change from yellow to blue-green in Fe-rich smectite upon reduction (sample NG-1 (Hohen Hagen, Germany) from the Source Clays Repository of The Clay Minerals Society).
Illustration du changement de couleur de jaune à bleu-vert par réduction, dans une smectite riche en fer (échantillon NG-1 (Hohen Hagen, Allemagne) en provenance de la collection de référence des argiles de la Clay Minerals Society).

Diffuse-reflectance spectra (converted to Absorbance) of unaltered, reduced, and reduced-reoxidized Fe-rich smectite (sample SWa-1 from the Source Clays Repository of The Clay Minerals Society), using dithionite as the reducing agent (unpublished results of Lubos Jankovič).
Spectres de réflectance diffuse (convertie en absorbance) d’une smectite riche en fer, inaltérée, réduite et réduite-réoxydée (échantillon SWa-1 en provenance de la collection de référence des argiles de la Clay Minerals Society), avec utilisation de dithionite comme agent réducteur (résultats de Jankovič, non publiés).
These changes in IVCT were followed continuously by UV-Visible spectroscopy by Komadel et al. (1990) during the course of reduction of Garfield nontronite (Fig. 3A). The intensity of the IVCT band initially increases in an approximately linear fashion as the number of Fe(II)-O-Fe(III) groups increases within the octahedral sheet, indicating that at this stage of reduction (Phase I, Fig. 3A) the entering electrons preferentially avoid reducing the Fe(III) ions that are already paired with Fe(II). This is probably due to the structural instability that would be produced by an uncompensated local excess of negative charge. The color change in this phase is from yellow to green. As the level of reduction approaches about 50%, however, this condition cannot persist and the Fe(III) paired with Fe(II) begins to be reduced also, thus converting Fe(III)-O-Fe(II) groups to Fe(II)-O-Fe(II). As this occurs, the intensity of the IVCT band decreases (Phase II, Fig. 3A) and the color becomes blue-green. This color is commonly observed in both dithionite- and bacteria-reduced smectites (Fig. 1).

Change in intensity of the IVCT band of Fe-rich smectite (sample SWa-1) during (A) dithionite reduction (Phases I and II) and reoxidation by O2 (Phases III and IV) (adapted from Komadel et al., 1990; or (B) bacterial reduction followed by O2 reoxidation.
Changement d’intensité de la bande de transfert de charge intervalence d’une smectite riche en fer (échantillon SWa-1) pendant la réduction par dithionite (A) (Phases I et II) et la réoxydation par O2 (Phases III et IV) adapté d’après de Komadel et al. (1990) ou par réduction bactérienne (B), suivie de réoxydation par O2.
As Fe(III) reduction continues to maximum levels (∼3.9 mmol Fe(II)/g clay or 96%), which is achievable only when dithionite is used as the reductant (bacterial reduction is limited to not more than about 1 mmol Fe(II)/g clay, which is about 20 to 30% reduction in nontronite) in an open system under an inert-atmosphere purge (Komadel et al., 1990), the color changes to blue-grey or grey (Phase II, Fig. 3A) and virtually all of the Fe is present as Fe(II)-O-Fe(II). Reoxidation of this maximally reduced smectite yields a sharp reversal in the color back to blue-green then green during Phase III (Fig. 3A), and finally to a brownish yellow (Phase IV, Fig. 3A). The color of the reoxidized sample, although yellow, appears somewhat different from the original, unaltered sample, indicating the process may not be completely reversible.
Using this same approach to follow the progress of bacterial reduction, the intensity of the IVCT band reached a maximum and remained approximately level until reoxidation was invoked, at which point the intensity decreased (Fig. 3B). The bacteria appear, therefore, to lack the reduction potential to form Fe(II)-O-Fe(II) entities in the clay structure.
2.2 Layer charge and cation exchange
The potential reversibility of the effects of Fe oxidation state on the layer charge or cation exchange capacity (CEC) of clays was first reported by Stucki and Roth (1977), who found that the previously proposed (Roth et al., 1969), theoretical 1:1 relationship between layer charge
Q = Fe(II) + 95.91 meq/100 g (1)
was invalid (Fig. 4). Instead, the relationship was curvilinear, the CEC increased at a much smaller rate, and reoxidation only partially reversed the CEC values. Stucki et al. (1984a) analyzed four different smectites and also found curvilinear relationships. These studies were limited in the extent of reduction achieved in the samples, however, because reduction was performed in a closed system, thus shutting down the reaction due to accumulation of gaseous reaction products (Komadel et al., 1990).

Effect of structural Fe(II) content on the cation exchange capacity (CEC) of dithionite-reduced and O2-reoxidized Garfield nontronite (sample API 33a from Wards Natural Science Establishment). Reduced 1, from Stucki and Roth (1977); Reduced 2, from Stucki et al. (1984a).
Effect de la teneur en Fe(II) structural sur la capacité d’échange cationique (CEC) d’une nontronite de Garfield (échantillon API 33a en provenance de l’Établissement de Sciences Naturelles Wards), réduite par dithionite et réoxydée par O2. Réduction 1 (Stucki et Roth, 1977) ; Réduction 2 (Stucki et al., 1984a).
Further studies of these relationships were carried out by Lear and Stucki (1985), who quantified the extent of charge compensation occurring by dehydroxylation of the clay structure during or after Fe reduction in aqueous solution. The corresponding reduction mechanism that they proposed was:
m Fe(III) + m e− = m Fe(II) (2)
2n (OH−)x = n (O2−)x + n H2O (3)
n (O2−)x + n (H+)s = n (OH−)x (4)
and the observed relationship between m and n is:
n = 0.32 m (5)
This adjustment still, however, leaves the Fe(II) vs layer charge relationship linear, so it fails to describe fully the observed relationship between layer charge and Fe(II) content, which is curvilinear (Fig. 4). Stucki and Lear (1989) addressed this question and proposed a “self-reduction” process by which excess electrons in the clay structure, such as from electron-rich tetrahedral sites (where Fe(III) or Al has substituted for Si), could be captured by the octahedral Fe(III) once the clay is “activated” by initial stages of reduction. They represented this process by the expression:
m (Fe3+)x + a(Z−q)x + (m−a) e− = m (Fe2+)x + a(Z1−q)x (6)
where the identity of Z−q is speculated to be an electron donor source somewhere within the clay crystal which gives up an electron. The electron, in turn, finds a pathway to a structural Fe site and brings about reduction. The net result of this aspect of the reaction is an increase in Fe(II) content with no increase in layer charge because, in effect, self-reduction of the clay has occurred. They speculated that this unknown electron donor (Z−q) could involve O− or O22− centers as discussed by Coyne et al. (1989) and Freund et al. (1989). Drits and Manceau (2000) proposed that structural protonation may account for the observed relationship. No further work has been performed on this topic, however.
Upon reoxidation, Lear and Stucki (1985) reported possible scenarios involving re-hydroxylation of the structure, but Komadel et al. (1995) found that the structural hydroxyl content was only partially restored upon reoxidation, indicating that some of the structural alterations during reduction were irreversible. Some of the irreversibility may occur because of the collapsing of superimposed smectite layers provoked by the reduction reaction. Collapsed smectite interlayers decrease the specific surface area (Lear and Stucki, 1989) as well as the basal spacing of the clay mineral (Wu et al., 1988). As a result of this process, interlayer cations become trapped or fixed (Fig. 5) between the layers (Khaled and Stucki, 1991). The extent of cation fixation depends inversely on the hydration energy of the cation (Fig. 6). Shen and Stucki (1994) investigated the fixation of K+ in the clay interlayers after numerous reduction-reoxidation (redox) cycles (Fig. 7). The layer charge, amount of interlayer K+, and Fe(II) content of the smectite were measured in the oxidized (or reoxidized) and reduced (or re-reduced) state of the smectite. The trend was that the amount of fixed K+ and the amount of Fe(II) remaining after reoxidation steadily increased with increasing number of redox cycles. The collapsing of clay interlayers prevented the K+ ions from being exchanged or removed even though the suspension was washed with a 1 M NaCl solution and exposed to an abundance of oxygen. The collapsed layers also prevented the oxygen from accessing all of the structural Fe(II), thereby allowing some of it to remain even though oxygen in solution was abundant. These observations clearly demonstrate that the reduction reaction is at least partially irreversible (Fig. 7; (Shen and Stucki, 1994)) and the redox cycles actually contribute to a transformation of the smectite into a more illite-like structure, becoming enriched in K+ and Fe(II) with limited expandability.

Cation fixation in ferruginous smectite (sample SWa-1) as affected by reduction of structural Fe (adapted from Khaled and Stucki, 1991). Ox = unaltered (oxidized) sample; Red = reduced with dithionite to 81.3% of total Fe.
Fixation de cations dans une smectite ferrugineuse (échantillon SWa-1), affectée par réduction du Fe structural (adapté d’après Khaled et Stucki, 1991). Ox = échantillon non altéré (oxydé) ; Red = réduit par action de dithionite à 81,3 % du Fe total.

Relation between cation hydration energy and fixation in ferruginous smectite (sample SWa-1) upon structural Fe reduction (adapted from Khaled and Stucki, 1991).
Relation entre énergie d’hydratation cationique et fixation de cations dans une smectite ferrugineuse (échantillon SWa-1) par réduction du Fe structural (adapté d’après Khaled et Stucki, 1991).

Effects of multiple redox cycles on the structural Fe(II) content and interlayer K+ in ferruginous smectite (sample SWa-1) (adapted from Shen and Stucki, 1994).
Effets de multiples cycles redox sur la teneur en Fe(II) structural et en K+ interfoliaire dans une smectite ferrugineuse (échantillon SWa-1) (adapté d’après Shen et Stucki, 1994).
2.3 Structural transformations
Changes in smectite structure during redox cycles have been studied by infrared, Mössbauer, and EXAFS spectroscopy and magnetic susceptibility. Some of the findings from these studies are now summarized.
Structural OH groups bonded to octahedral cations are sensitive probes of local structural changes due to redox reactions involving the octahedral Fe. Shifts are observed in O-H stretching, Si-O stretching, and M-O-H bending modes. The focus of the present discussion will be on the O-H stretching band, which is representative of changes that occur in the other vibrational modes. The peak for the O-H stretching mode in Fe-rich smectites occurs at 3570 cm−1 and is best studied when the sample is dehydrated sufficiently to remove overlap with the adsorbed water bands. The energy of this vibrational mode shifts downward by about 20 to 30 cm−1 and its intensity decreases as Fe(II) content increases (Fig. 8) (Fialips et al., 2002a, 2002b; Lee et al., 2006; Stucki and Roth, 1976; Roth and Tullock, 1973; Yan and Stucki, 1999, 2000). Changes in intensity are consistent with increasing structural dehydroxylation with increasing Fe(II) content, except two other interesting structural changes occur as reduction progresses. The first is the appearance of a band for H2O that survives the dehydration treatment, indicating that in the reduced state a strongly bound form of H2O is created, probably by the protonation of structural OH groups. So the loss of intensity in the O-H stretching band may be the result of dehydroxylation or it may be due to protonation. The extent to which this H2O band is removed upon reoxidation depends on the initial level of reduction. The greater the initial reduction level, the greater the tendency for this H2O band to remain. In other words, the reversibility of the hydration or protonation decreases with increasing reduction.

FTIR spectra of the O-H stretching region of Garfield nontronite (sample API 33a) reduced by dithionite to increasing levels (adapted from Fialips et al., 2002a, 2002b).
Spectres infra-rouges à transformée de Fourier dans la région des élongations O-H d’une nontronite de Garfield (échantillon API 33a), réduite par dithionite à des niveaux croissants (adapté d’après Fialips et al., 2002a, 2002b).
The second structural change is the creation of a weak, pleochroic shoulder on the high-energy side (3622 cm−1) of the main O-H stretching band. This band is consistent with the formation of a trioctahedral Fe(II)-Fe(II)-Fe(II) environment for OH in which the O-H vector is oriented perpendicular to the clay layer, as in biotite. This infrared evidence for the formation of trioctahedral domains was confirmed by EXAFS measurements which were interpreted to show that, as the reduction level increases, the Fe migrates from cis to trans sites, leaving behind structural voids where cis sites had previously been (Manceau et al., 2000a, 2000b) (compare panels A and D in Fig. 9).

Illustration of changes in structure and in the distribution of Fe in the octahedral sheet of Garfield nontronite: (A) unaltered (oxidized); (B) partially reduced by dithionite; (C) partially reduced by bacteria; (D) fully reduced by dithionite (adapted from Ribeiro et al., 2009).
Illustration des changements de structure et de distribution du Fe dans le feuillet octaédnique d’une nontronite de Garfield : (A) non altérée (oxydée), (B) partiellement réduite par dithionite, (C) partiellement réduite par des bactéries, (D) complètement réduite par dithionite (adapté d’après Ribeiro et al., 2009).
Interpretation of the downward shift in wavenumber requires some explanation. The meaning of this shift is that the bond energy between O and H in the OH group becomes weaker as Fe(II) content increases. Adding an electron to the Fe should actually increase the bond order (strength) in the adjacent O-H bond, so how does reduction of Fe bring about the observed downward shift in bond energy? The answer lies in the fact that the orientation of the O-H vector in a [Fe(III)-Fe(III)-vacancy], cis-vacant, dioctahedral sheet is tilted toward the vacant octahedral site to a position that is symmetrically intermediate between two apical oxygens from the tetrahedral sheet. Converting one Fe(III) to Fe(II) preserves the tilting of this vector, but deflects the H away from the intermediate position, moving it closer toward one or the other of the apical oxygens. This closer proximity allows a mutual attraction between the apical oxygen and H, thus weakening the bond between O and H in the structural hydroxyl group.
Restoration of the infrared spectrum upon reoxidation is incomplete regardless of the extent of reduction (Fig. 10), even though the deviations between the spectra from the unaltered and reduced-reoxidized samples increase with increasing levels of reduction. If the level of reduction is small prior to reoxidation, the O-H stretching band of the reoxidized sample is almost completely restored to its original position and intensity, although slight differences are noted including the loss of the low-energy shoulder at about 3533 cm−1. Complete reduction (∼96%) prior to reoxidation, on the other hand, yields an O-H stretching band in the reoxidized sample that is still shifted to lower wavenumber and much less intense than in the unaltered sample. The pleochroic band in the highly reduced sample also disappears after reoxidation and some of the H2O band is still visible.
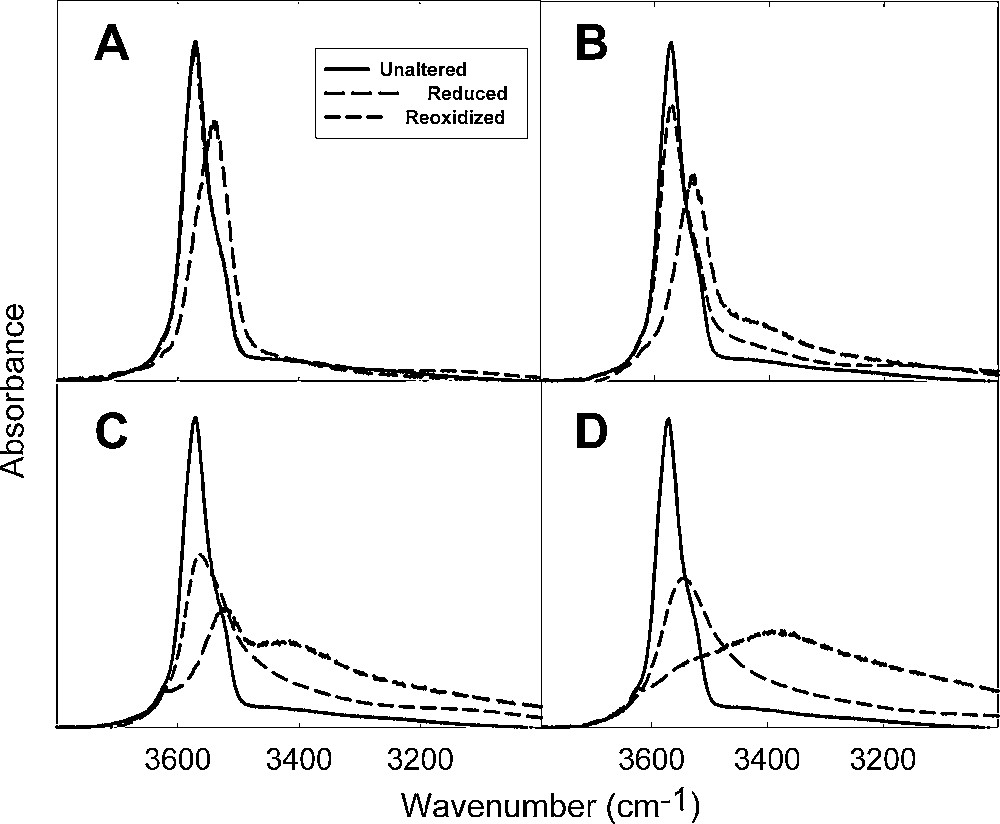
FTIR spectra of the O-H stretching region of Garfield nontronite (sample API 33a) reduced by dithionite to increasing levels, then reoxidized by O2 (adapted from Fialips et al., 2002a).
Spectres infra-rouges à transformée de Fourier de la région des élongations O-H d’une nontronite de Garfield (échantillon, API 33a), réduite par dithionite à des niveaux croissants, puis réoxydée par O2 (adapté d’après Fialips et al., 2002a).
The infrared spectra of samples reduced by bacteria exhibit shifts similar to those observed after short dithionite treatments in which the level of reduction is also rather small (< 1 mmol Fe/g), and the observed shifts seem to be largely reversible (Fig. 11) (Lee et al., 2006). In fact, bacterial reduction appears to be a very mild form of reduction that is rather readily reversed and may involve a different reduction mechanism than dithionite (see below). Based on this evidence, smectite mineral transformations in natural soils and sediments due to bacteria-mediated redox cycles may be advanced in only very small increments. More work is needed in this regard, however, because studies have also shown that under some conditions bacterial reduction causes extensive dissolution of the smectite (Kim et al., 2004; Jaisi et al., 2005, 2007a, 2007b, 2007c, 2008), including a role for organic acids (Kostka et al., 1999).

Comparisons of FTIR spectra of the O-H stretching region of ferruginous smectite (sample SWa-1) reduced to similar levels by bacteria, then reoxidized by O2 (adapted from Lee et al., 2006).
Comparaison des spectres infra-rouges à transformée de Fourier de la région des élongations O-H d’une smectite ferrugineuse (échantillon SWa-1), réduite à des niveaux similaires par des bactéries, puis réoxydée par O2 (adapté d’après Lee et al., 2006).
Variable-temperature (298 to 4 K) Mössbauer spectroscopy provides rich, complementary information about the oxidation state and local environment of Fe in redox-treated smectites. It has the advantage over infrared because it probes the Fe directly rather than indirectly. The unaltered sample is dominated at all temperatures by a central doublet due to octahedral Fe(III) (Fig. 12). The spectrum of this sample at 4 K differs from 77 K and 298 K only by the appearance of a small sextet due to goethite (Murad, 1987), which is removed by reduction treatments with either dithionite or bacteria. After complete reduction (∼3.9 mmol Fe(II)/g clay or 96%) by dithionite, the spectrum at 77 K (which is virtually unchanged from 298 K) is dominated by a doublet that is typical of high-spin octahedral Fe(II) (Fig. 13A) with only a minor doublet for structural Fe(III). Reoxidation of this completely reduced sample restores the spectrum at 77 K to a central Fe(III) doublet (Fig. 13B), which is similar to that of the unaltered sample except it is broadened and more split due to a greater quadrupole splitting, indicating the Fe is in a more distorted environment. A small amount of the Fe failed to be reoxidized, as indicated by the small peak at about 2.5 mm/s, which represents the right-half of a Fe(II) doublet.

Mössbauer spectra of unaltered Garfield nontronite (sample API 33a) at 4 K (adapted from Ribeiro et al., 2009).
Spectres Mössbauer d’une nontronite de Garfield non altérée (échantillon API 33a) à 4 K (adapté d’après Ribeiro et al., 2009).

Mössbauer spectra of fully reduced (∼3.9 mmol Fe(II)/g clay or 96%) Garfield nontronite (sample API 33a) at 77 K before (A) and after (B) reoxidation with O2 (adapted from Ribeiro et al., 2009). The spectrum at 77 K is also representative of that observed at 298 K.
Spectres Mössbauer d’une nontronite de Garfield (échantillon API 33a) totalement réduite (∼3,9 mmol Fe(II)/g d’argile ou 96 %) à 77 K avant (A) et après (B) réoxydation par O2 (adapté d’après Ribeiro et al., 2009). Le spectre à 77 K est aussi représentatif de ce qui est observé à 298 K.
Lowering the temperature to 4 K, however, changes the spectrum completely (Fig. 14A) compared to 77 K. At 4 K, magnetic exchange interactions among the Fe ions in the clay mineral lattice manifest themselves. In the fully reduced state the spectrum is dominated by complex features that are divided into two general resonance regions; one at about 0 mm/s and the other at about 4 mm/s. Full interpretation of this spectrum by curve fitting and the assigning of hyperfine parameters has yet to be achieved, but its overall features are regarded as representative of a fully reduced nontronite. When the fully reduced sample is reoxidized by O2 (Fig. 14B), the 4 K spectrum also differs radically from that at 77 K. Instead of a broadened central doublet for Fe(III), the pattern consists of six peaks. These peaks are evidence for magnetic order among Fe(III) ions in the octahedral sheet of the reoxidized clay. This magnetic ordering was absent from the unaltered sample (Fig. 12) and was not visible in the fully reduced, reoxidized sample at 77 K. The magnetic ordering temperature is, therefore, above 4 K and below 77 K, whereas in the unaltered sample it is below 4 K. The appearance of magnetic ordering is strong indication that the bonding relationships among the Fe(III) ions in the structure have changed irreversibly during the redox reactions and the process is far from reversible.

Mössbauer spectra of fully reduced (∼3.9 mmol Fe(II)/g clay or 96%) Garfield nontronite (sample API 33a) at 4 K before (A) and after (B) reoxidation with O2 (adapted from Ribeiro et al., 2009).
Spectres Mössbauer d’une nontronite de Garfield (échantillon API 33a) totalement réduite (∼3,9 mmol Fe(II)/g d’argile ou 96 %), à 4 K avant (A) et après (B) réoxydation par O2 (adapté d’après Ribeiro et al., 2009).
Reduction of the clay to a lesser extent uncovers significant differences in the distribution of Fe(II) and Fe(III) in the octahedral sheet, depending on whether reduction is brought about by bacteria or by dithionite. Samples of Garfield nontronite reduced by bacteria or by dithionite to about the same level (approximately 1 mmol Fe(II)/g clay, or ∼25% of total Fe) exhibit similar spectra at 77 K, featuring doublets for both Fe(II) and Fe(III), and no important distinctions can be made based on the method of reduction (Fig. 15A and B).

Mössbauer spectra at 77 K of partially reduced (∼1 mmol Fe(II)/g clay) Garfield nontronite (sample API 33a) using (A) dithionite or (B) bacteria (adapted from Ribeiro et al., 2009).
Spectres Mössbauer à 77 K d’une nontronite de Garfield (échantillon API 33a) partiellement réduite (∼1 mmol Fe (II)/g d’argile) par utilisation de dithionite (A) ou de bactéries (B) (adapté d’après Ribeiro et al., 2009).
At 4 K, however, the spectra are different depending on the method of reduction (Fig. 16A and B). The dithionite-reduced sample produces a central doublet that is broadened widely at the bottom, indicating that magnetic ordering is beginning, but is incomplete and poorly developed. Ribeiro et al. (2009) explained that this condition probably occurs because the domains of pure Fe(III) or pure Fe(II) are small and the system is dominated more by Fe(II)-Fe(III) clusters or domains (panel B in Fig. 9). The bacteria-reduced samples, on the other hand, yielded spectra with a combination of features resembling the all-Fe(II) domain given in Fig. 14A and a magnetically ordered, all-Fe(III) domain with sextets. Ribeiro et al. (2009) further suggested that, in this case, the octahedral sheet comprises clusters or domains that are all Fe(II) and other clusters or domains that are all Fe(III) (panel C in Fig. 9). The creation of such relatively more homogeneous domains of each oxidation state would occur if reduction proceeded from the edges of the clay layers, with the Fe(II) domain forming first at the edge, then moving as a front toward the center, thus forming an all-Fe(II) domain toward the outside of the layer. The Fe(III) domain remains toward the inside of the layer, away from the layer edges. Reoxidation largely restores the spectrum to its unaltered appearance in both the dithionite- and bacteria-reduced samples (Fig. 17A and B), indicating that the process was mostly reversible.

Mössbauer spectra at 4 K of partially reduced (∼1 mmol Fe(II)/g clay) Garfield nontronite (sample API 33a) using (A) dithionite or (B) bacteria (adapted from Ribeiro et al., 2009).
Spectres Mössbauer à 4 K d’une nontronite de Garfield (échantillon API 33a) partiellement réduite, par utilisation de dithionite (A) ou de bactéries (B) (adapté d’après Ribeiro et al., 2009).

Mössbauer spectra at 4 K of partially reduced (∼1 mmol Fe(II)/g clay), then reoxidized Garfield nontronite (sample API 33a) initially reduced by (A) dithionite or (B) bacteria (adapted from Ribeiro et al., 2009).
Spectres Mössbauer à 4 K d’une nontronite de Garfield (échantillon API 33a) partiellement réduite (∼1 mmol Fe(II)g d’argile), puis réoxydée, initialement réduite par utilisation de dithionite (A) ou de bactéries (B) (adapté d’après Ribeiro et al., 2009).
3 Summary and conclusions
Redox reactions of Fe in clay minerals affect many of the chemical and physical properties of the clay, including color, layer charge, cation exchange capacity, cation fixation capacity, specific surface area, swelling, hydration energy, reactivity with organic species, and crystal structure. Evidence from UV-visible, infrared, and Mössbauer spectroscopy indicates that the reversibility of these changes in properties decreases as the extent of reduction increases. Over many redox cycles, the residual Fe(II) and fixed K+ contents of smectite increase incrementally, suggesting that redox reactions may play a direct role in the in situ transformation of smectite to illite. The greatest level of redox reversibility is observed when bacteria are used to reduce the structural Fe or if the level of reduction by dithionite is limited. Dithionite is capable of reducing virtually all of the Fe in Fe-rich clays, but such levels of reduction invoke irreversible changes in the Fe distribution within the clay structure. Some studies have also suggested extensive mineral alteration even by bacteria. In nature, redox reactions undoubtedly play an important role in promoting transformation in the properties and behavior of soils and sediments.
Acknowledgments
The author is grateful to many students and colleagues who have contributed research results and creative intellectual interpretations leading to a greater understanding of redox processes in clays and soils.