

1 Introduction
Many concepts used in inorganic chemistry to highlight the role of physical and chemical processes on crystal structure, crystal growth, size and morphology of synthetic metal oxides may also be used to rationalize the observations made on natural processes by Environmental Mineralogy. In fact, in the laboratory as under natural conditions, similar processes are involved during the formation and/or transformation of crystalline phases from soluble precursors, hydroxylation, complexation, and condensation of metal complexes as well as nucleation, growth or aggregation leading to solid phases (Jolivet, 2000). These transformations may be illustrated with aluminum chemistry. Aluminum oxy(hydroxi)des are important materials or nanomaterials for many applications and uses, for instance in water treatment, as the active phase in adsorption and catalysis, as inert or active charged species in polymers, as refractory material in the ceramic industry, or as abrasives, to name a few (Castel, 1990; Euzen et al., 2002). Aluminum is also an abundant element in Earth's crust, in the form of many mineral phases as pure hydroxides and oxides, and in combination with other elements, particularly with silicon in aluminosilicates such as feldspars, zeolites, clays, and imogolites (Wells, 1991). Aqueous equilibria involving aluminum are also of major importance in biogeochemical cycling of this element (Pédro, 2007).
The structural chemistry of aluminum oxy(hydroxi)des is rich. Aluminum forms different hydroxide phases – Al(OH)3: gibbsite, bayerite, nordstrandite; oxyhydroxides AlOOH: boehmite and diaspore; and a variety of oxides – Al2O3 such as transition aluminas known as γ-alumina exhibiting the defect spinel structure with various degrees of ordering of Al3+ cations in a cubic close packing of oxygen atoms, and α-alumina (corundum), the most thermodynamically stable phase of aluminum oxide, based on a hexagonal closest packing of oxygen atoms. β-aluminas are phases based on a structure closely related to that of spinel, stabilized by incorporation of alkali ions such as Na+ or K+ (Wells, 1991). It is interesting to compare briefly the behavior of Al3+ ions to that of Fe3+ ions (Cornell and Schwertmann, 2003): several forms of Al(OH)3 are well identified stable phases at low temperature, whereas Fe(OH)3 does not exist; the two elements form MOOH isostructural phases:
Here, we present the main aspects of aluminum chemistry in aqueous solution involving condensation phenomena leading to the formation of polycations and nanoparticles, emphasizing the influence of various parameters on particle size and shape. The conditions of formation of different aluminosilicates will be also examined.
2 Hydroxylation and condensation mechanisms in aqueous solution
Like many metal cations in water, Al3+ ions form hexacoordinated aquo complexes [Al(OH2)6]3+. They behave as acid species due to the polarization of coordinated water molecules in forming a series of hydroxylated species [Al(OH)h(OH2)6-h](3-h)+, with h varying in the range 0 to 4 depending on the pH of the solution (Baes and Mesmer, 1976). [Al(OH2)6]3+ complexes, stable up to pH ∼4, are less acidic than the ferric homologue. With the same charge (+3) but larger size (rVIFe3+ = 0.64 Å vs. rVIAl3+ = 0.53 Å (Shannon, 1976), ferric ions are, however, more polarizing than Al3+ ions because iron is more electronegative than aluminum (1.72 vs. 1.47) (Jolivet, 2000).
Hydroxylated complexes exhibit a strong tendency to condense and to form polynuclear entities or solids. Two mechanisms based on substitutions induced by the nucleophilic character of the hydroxo ligand are involved, depending on the nature of the coordination sphere of the cations (Jolivet, 2000). With aquohydroxo complexes [Al(OH)h(OH2)6-h](3-h)+, condensation proceeds by elimination of water molecules and formation of hydroxo bridges (olation mechanism):

Due to the high lability of coordinated water molecules, such a reaction is fast, hardly able to be kinetically monitored, and it always occurs if the concentration of complexes is high enough. With hydroxo or oxohydroxo complexes in which the coordination spheres do not include water molecules, there is no leaving group, and condensation may proceed via a two step mechanism allowing formation of a water molecule as a leaving group and leading to an oxo bridge (oxolation mechanism):

This reaction may stop when the electrophilic power (the partial or effective positive charge) of the cation is too low. This is typically the case for aluminate, a tetrahedral hydroxo complex [Al(OH)4]− existing in alkaline media which does not condense. Even if the hydroxo ligands are highly nucleophilic, the partial charge on aluminum is too low to suffer a nucleophilic attack, because of the ligand-metal charge transfer, and this species usually remains a monomer, even at high concentration. Condensation occurs with other species, for instance silicates [SiO4-h(OH)h](4-h)− in which the electrophilic power of silicon is high enough to allow substitution (Jolivet, 2000).
The hydroxylation rate, h, of the complexes represents their functionality towards condensation and controls the extent of the reaction and the type and structure of condensed species. h is obviously strongly dependent on pH and also on size, formal charge, and electronegativity of the cation. As long as the soluble species are positively (negatively) charged, condensation in solution is always limited and forms polycationic (polyanionic) species. Aluminum hydroxoaquo complexes form a series of structurally well-identified polycations corresponding to the successive steps of condensation of octahedra by edge sharing (Fig. 1a) (Akitt and Elders, 1988; Thompson et al., 1987). Species such as a dimer, a trimer, and a tetramer, with formal h values of 1, 1.33. and 1.76, respectively, are globally characterized by an 27Al NMR resonance around 9 ppm. Their relative amounts vary with hydroxylation rate and globally decrease beyond h = 1 to the benefit of the well-known “ɛ-Al13” polycation ([Al13O4(OH)24(OH2)12]7+) with Keggin's structure (Fig. 1b) (Akitt and Elders, 1988; Johansson, 1963). This polycation, quantitatively formed for h = 2.46, is characterized by the central Al atom in tetrahedral coordination giving an NMR “signature” at 63 ppm in the 27Al NMR spectrum. These observations probably explain why this species only exists with Al3+ and Ga3+, two ions that easily adopt both tetrahedral and octahedral coordinations. Such big polycations seem to be formed by direct addition from trimer and tetramer polycations (Michot et al., 2000). Thermolysis of aluminum chloride hydrolyzed for three days at h = 2.25 and 80 °C, results in the formation of “Al30” polycationic species (Al30O8(OH)56(H2O)24]18+), which have been structurally characterized (Allouche et al., 2000; Rowsell and Nazar, 2000). The structure of this polycationic species, surprinsingly formed by two moities δ-Al13 (an isomer of the ɛ-Keggin's structure) and connected by a ring of four AlO6 octahedra, gives a specific 27Al NMR resonance at 70 ppm, corresponding to the tetrahedral Al ions (Fig. 1c). Clearly, the Al30 polycation results from the transformation during thermolysis of the initially formed Al13 species. Some other aluminum polycations have been obtained but with unusual synthesis conditions or in the presence of aminocarboxylate ligands (Casey et al., 2005; Jordan et al., 1996; Seichter et al., 1998).

Structures and 27Al NMR spectra of aluminum polycations. a, b: from Hernandez (1998); c: from Rowsell and Nazar (2000).
Structures et spectres RMN 27Al des polycations de l’aluminium. a, b : d’après Hernandez (1998) ; c : d’après Rowsell et Nazar (2000).
By increasing the hydroxylation ratio, species corresponding to non-charged complexes h = 3 appear and lead to a solid. In highly alkaline media, the soluble hydroxo complex [Al(OH)4]− is formed and does not condense, as seen before.
3 Formation of solid phases
The precursor of solid phases is always a non-charged complex such as an aquohydroxo, oxohydroxo form, but other ligands may also participate in the coordination sphere of the cation. If the complexing strength of the ligands towards the cation is high, the ligand is incorporated with well-defined crystallographic locations in the solid phase–a so-called basic salt. A low complexing ligand will not be present in the final solid phase but may, however, have an important role in the formation of the solid during transient steps of the precipitation process. The process involved in the formation of the solid, for instance direct alkalinization, thermolysis, or hydrothermal conditions, also have a large influence on the characteristics of the solid.
3.1 Aluminum hydroxides, oxyhydroxides and oxides
Hydroxylation by addition of a base at room temperature to an aluminum chloride or nitrate solution up to pH 6 to 8 forms a transparent amorphous gel quasi-instantaneously. It is interesting to observe that the 27Al NMR spectrum of the gel exhibits a large signal centered at ∼0 ppm relative to the 6-coordination of aluminum ions and any detectable peak at 63 ppm characteristic of the Al13 species. As a rule, this indicates that transient polycations formed during the progressive alkalinization of the medium are rapidly destroyed beyond their acidity range of formation and are not favored as building blocks in the formation of the solid (Bottero et al., 1987; Hernandez, 1998). The metastable gel must evolve, following different mechanisms depending on the acidity of the medium. At 6 ≤ pH ≤ 8, the gel spontaneously transforms into small particles of boehmite, γ-AlOOH (Fig. 2). The solubility of aluminum being very low at this pH (around 10−6 mol.L−1), the transformation proceeds in situ by dehydration and local rearrangement in the solid state, which explains the small size of the crystallites comprising the solid which is not the most stable phase and with a high specific area of around 300 m2g−1. At pH < 6 or pH > 8, the solubility is higher and the transformation of the gel can proceed more easily via a dissolution-crystallization process leading to the hydroxide Al(OH)3, the thermodynamically stable phase at room temperature (Fig. 2). Ageing of this material leads to the formation of bayerite at pH > 8 and gibbsite at pH < 5. These two crystalline phases are based on a different stacking of layers made of Al(OH)6 octahedra sharing three edges and forming hexagons of polyedra (Wells, 1991). In bayerite, the successive layers have the same orientation, while in gibbsite the orientation of each successive layer is rotated by 30°. The particles of bayerite so obtained are strongly aggregated as stacked plates forming cylindrical particles, whereas gibbsite particles do not aggregate and form stable dispersions after two or three weeks of ageing (Fig. 2). Such a difference in behavior is not clearly understood. Although the same layers are formed by dissolution-crystallization, their different stacking as a function of pH may result from different systems of intra- and inter-layer hydrogen bonds. Another possibility may be a stabilization of the particles at pH < 5. Dissolution of the gel occurs under conditions where different species simultaneously exist, so that polycations are present and adsorb on hydroxide particles, allowing their stabilization and preventing their aggregation. Such effects of particle stabilization against aggregation in suspension by adsorption of the Al13 polycation have been observed with iron compounds (goethite and haematite) (Hernandez, 1998). Even if the polycation is destroyed by adsorption, the fragments stay adsorbed and keep their electrostatic surface charge for the stabilization of particles.1 A similar mechanism could likely occur with aluminum hydroxide.
Hydroxylation by thermolysis at 95 °C of aluminum nitrate leads to the formation of boehmite, γ-AlOOH, the thermodynamically stable phase under these conditions, but the morphology and particle size are strongly dependent on the pH of the medium (Chiche et al., 2008; Jolivet et al., 2004; Poisson et al., 1987). Particles are larger than those formed at RT as indicated before, with a specific surface area of around 100 m2.g−1. Electron microscopy and X-ray diffraction showed platelets with different extensions of the lateral faces (Fig. 3) (Chiche et al., 2008).

Electron micrographs of the different morphologies of boehmite obtained by thermolysis of aluminum nitrate at different pH. The surface charge densities are σ = +0.2, +0.05, −0.15 C.m−2, respectively. Particle shapes were deduced from XRD pattern simulation (from Chiche et al., 2008).
Clichés de microscopie électronique des différentes morphologies de boehmite obtenue par thermolyse du nitrate d’aluminium à différents pH. Les densités de charge de surface sont σ = +0,2, +0,05, −0,15 C.m−2 respectivement. Les formes des particules ont été déduites de la simulation des diagrammes de diffraction des rayons X (d’après Chiche et al., 2008).
The change in morphology results from variation in surface energy due to adsorption. Under the conditions used here, the change in pH involves changes in acid-base surface equilibria and in the degree of protonation of oxygenated surface groups. Modeling the influence of proton adsorption on the surface energy for various crystalline faces of boehmite accounts well for these changes in morphology (Jolivet et al., 2004). Adsorption of other species and organic molecules such as polyols and carboxylic acids may also influence particle shape. Prominent effects are obtained when the conformation of the adsorbed molecule has a structural similarity to the surface atomic arrangement (Chiche et al., 2009). Other morphologies of boehmite were obtained by hydrothermal treatment of suspensions of aluminum salts in the presence of surfactants, including: nanofibers from heating of Al(OH)3 in the presence of POE (polyethyleneoxide) at 120 °C (Zhu et al., 2004), nanorods and nanoflakes from aluminum nitrate at pH 5 and 10, respectively, adjusted with hydrazine at 200 °C (Chen et al., 2008), and nanotubes from aluminum chloride in alkaline medium in the presence of CTAB (N-cetyl-N,N,N-trimethylammonium bromide) at 120 °C (Kuang et al., 2003). Interest in the control of the morphology of boehmite particles lies in the production of γ-alumina, γ-Al2O3, which is largely used as a catalyst and a support in the petroleum refining industry (Euzen et al., 2002). The topotactic transformation of boehmite, made by thermal treatment around 500 °C, maintains the size and shape of all these particles (Fig. 4) (Chiche, 2007; Digne et al., 2002). It is also possible to optimize alumina catalysts by the development of specific crystalline faces for a given reaction.

Electron micrographs and X-ray diffraction patterns of boehmite obtained at different pH (Fig. 3) and of corresponding γ-alumina obtained by heating at 500 °C for 2 hours (from Chiche, 2007).
Clichés de microscopie électronique et diagrammes de diffraction des rayons X des différentes morphologies de boehmite obtenue à différents pH (Fig. 3) et des alumines γ correspondantes, obtenues par chauffage à 500 °C pendant deux heures (d’après Chiche, 2007).
In comparison with ferric ions, the behavior of Al3+ ions seems to be marked by a lower polarizing power towards oxygenated ligands, allowing a higher stability of the hydroxo ligand for aluminum compounds. So, aluminum hydroxides are stable phases while Fe(OH)3 does not exist. Oxyhydroxides are formed by the two ions, but under very different conditions: in contrast with goethite, α-FeOOH, which is the most widely obtained ferric phase from solutions, the isostructural α-AlOOH phase, diaspore, is only formed by hydrothermal treatment of Al(OH)3 or γ-Al2O3 in the presence of sodium carbonate at 380 °C under 6 kbars (Dillinseger, 1995); it has been seen that boehmite is easily obtained by precipitation of an aluminum salt, but the isostructural iron phase, lepidocrocite, γ-FeOOH, is only formed from oxidation of ferrous hydroxide. Aluminum oxides are never formed in solution and require heating of oxy(hydro)xides in the solid state, whereas hematite or magnetite is easily precipitated from solutions (Cornell and Schwertmann, 2003; Jolivet et al., 2004, 2006).
3.2 Aluminosilicates
Aluminosilicates form a large family of inorganic compounds (Wells, 1991). They may be considered as derivatives of aluminum complexes by the silicate ligand in solution. Silicates [SiO4-h(OH)h](4-h)− are strong basic, highly nucleophilic species that are able to autocondense to form anionic polysilicates (h < 4) with a large variety of structures, as evidenced by 29Si NMR (Knight, 1988; Knight et al., 1989). Silicic acid, Si(OH)4, as a zero charged species (h = 4), condense indefinitely by oxolation to form amorphous hydrated silica, with a morphology that depends strongly on pH, including transparent polymeric gels made of long chains of SiO4 tetrahedra with few reticulation points and many dangling Si-OH bonds in acidic medium and strongly diffusing gels containing particles made of highly reticulate chains in neutral medium (Iler, 1979; Jolivet, 2000). Silica and silicates may react with aluminum following very different pathways, which will be briefly examined below.
3.2.1 Adsorption of aluminum by amorphous silica
Amorphous hydrated silica reacts with soluble aluminum by adsorption phenomena involving different types of interactions. The polar or charged surface oxygenated groups may induce non-specific electrostatic interactions with molecules or metal complexes in forming, through the solvation layer of particles, the formation of outer sphere surface complexes (physisorption process, Scheme 1a). This type of interaction may have various consequences, including a restricted mobility of soluble species near the surface (as nicely showed by ESR spectroscopy of hexaaquocopper(II) complex as a paramagnetic probe adsorbed onto silica at pH 3 (Bassetti et al., 1979)), electrostatic screening leading to destabilization of sols and flocculation of silica particles by aluminum-based flocculants (Lartiges et al., 1997), and pollutant transport such as molecules and weakly hydrolyzable metals in water.
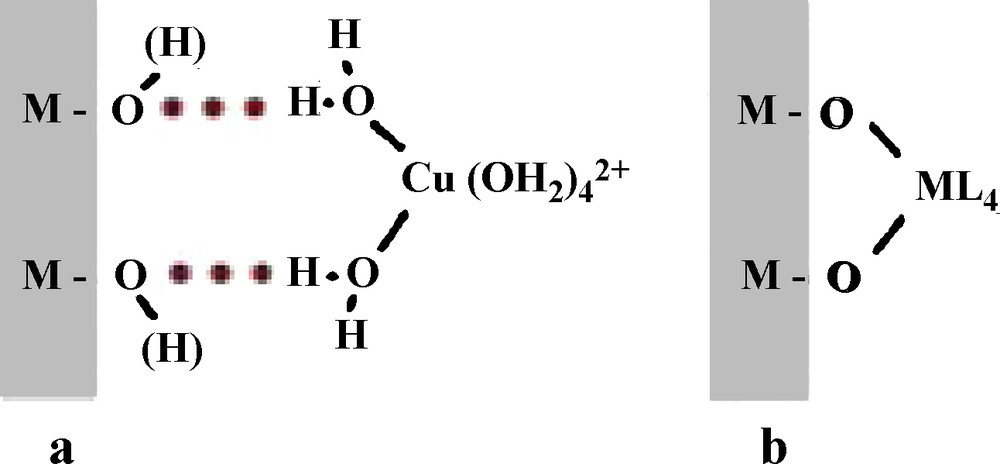
Physisorption (a) and coordination (b) of a metal complex onto an oxide surface.[[fr]]Physisorption (a) et coordination (b) d’un complexe métallique sur une surface d’oxyde.
Adsorption may also involve specific interactions in a chemisorption process. Polar or charged surface oxygenated groups act as hydrolyzing ligand towards hydroxylable cations in solution, leading to the coordination of cations onto the surface in forming inner sphere surface complexes (Scheme 1b). The role of the surface is well evidenced in this process because it occurs at a pH lower than the pH corresponding to hydroxylation of the cation alone in solution (James and Healy, 1972). This behavior is observed with many metal oxide particles, but amorphous silica with a high porosity and a high surface reactivity due to many strongly nucleophilic dangling silanol bonds, may adsorb and also incorporate Al3+ ions into its framework because of the ability of the cation to adopt fourfold coordination. This was observed with colloidal silica (Lartiges et al., 1997) and also with biogenic silica from diatom frustules (Gehlen et al., 2002; Koening et al., 2007). With silica from post-mortem diatom frustules (after removal of the organic matter) and incubation with very dilute aluminum in seawater at pH ∼8, incorporated aluminum exhibits essentially tedrahedral coordination and causes an unexpected slight decrease in the pore-size distribution, suggesting the formation of a surface aluminosilicate layer (Koening et al., 2007). In contrast, with living diatoms, Al is exclusively incorporated in tetrahedral coordination inside the silica framework by a biosynthetic mechanism, leading to a decrease of solubility of silica in marine environment (Gehlen et al., 2002). The incorporation of aluminum into silica stabilizes it against hydrolysis and dissolution likely because of a reinforcing of the silica network by elimination of the reactive dangling bonds and by filling and flattening the silica surface.
3.2.2 Crystalline aluminosilicates: zeolites, clays, imogolite
Silicates [SiO4-h(OH)h](4-h)− can behave as strong ligands towards aluminum in solution, as is the case for phosphates for instance, but silicates have a special role because they polymerize in contrast to phosphates which always occur as monomers in solution. Such a difference may explain the diversity of the crystal chemistry of silicates, in comparison to that of phosphates. Even if the crystal chemistry of aluminosilicates appears very complicated, and regardless of specific conditions of their formation, one may observe that acidity seems the most important parameter governing the formation of the main types of crystalline aluminosilicates. This is the case because acidity controls the relative reactivity of aluminum and silica towards condensation. In a basic medium (pH around 12), silicates, which condense and simultaneously complex tetrahedral aluminate, drive the formation of zeolites made of a three dimensional framework of AlO4 and SiO4 tetrahedra with no Al-O-Al bonds. In a slightly basic to neutral medium (pH 9–5), both silicates and octahedral aluminum species condense, but the strong complexing ability of silicates towards gibbsite-like layers orients their own condensation resulting in the formation of adjacent polysilicate layers. Clays, which consist of different types of sandwich structures may also form. In a slightly acid solution (pH in the range 5–3), aluminum still forms similar gibbsite-like layers rather than clays, but the tendency of silicic acid to condense is low (see the structure of acidic silica gels), and it is coordinated only as a monomeric ligand involving bonding to a gibbsite layer by three OH groups per tetrahedral. This bonding results in the curvature of layers and the formation of tubes of imogolite. A more detailed insight is given below.
In a strongly basic medium, small polysilicate units may complex tetrahedral aluminate [Al(OH)4]−. This behavior results in a complex chemistry of aluminosilicic polyanions which have been structurally well-characterized by 29Si and 27Al NMR (Fig. 5) (Mortlock et al., 1991a, 1991b, 1991c; Swaddle, 1994). These mixed species are highly labile and never contain Al-O-Al bonds because aluminate cannot auto-condense, and polyanionic species result from the ability of silicate to condense. Such aluminosilicates species are believed to be the building blocks for crystallization of zeolites. These very numerous compounds are made of a three-dimensionnal largely open anionic framework of SiO4 and AlO4 tetrahedra, forming cavities and channels of variable size and shape containing water molecules and counter cations (Fig. 6) (Barrer, 1982; Breck, 1974). There are many natural compounds as well as many synthetic phases mainly obtained by (hydro)thermal treatment of concentrated aluminum hydroxide or aluminate mixed with silica or silicates in a strongly alkaline medium (sodium or tetraalkylammonium hydroxide) in the presence of a template such as surfactant (e.g., amine or crown-ether). Under these conditions, the thermal treatment of solutions allows formation of zero-charged precursors (Jolivet, 2000). The main parameters controlling the structure are the Si/Al ratio, pH, the nature of cations (e.g., Na+, NR4+) and of the surfactant, temperature, pressure…
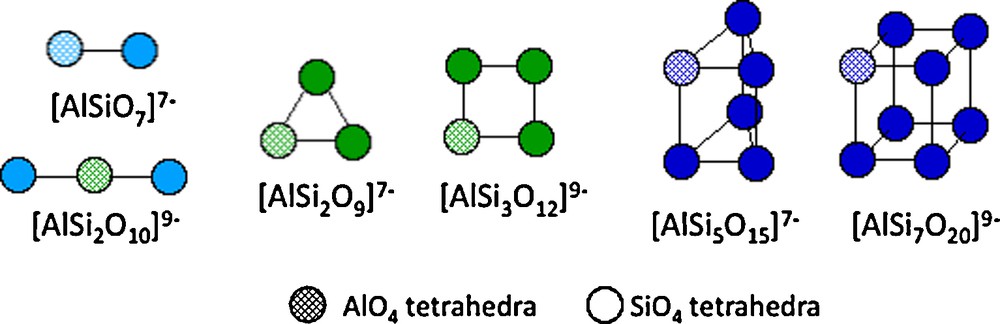
Some aluminosilicic polyanions formed at pH > 12 in the presence of Na+ or K+. The polyhedral species are formed in the presence of tetralkyl NR4+ cations (from Swaddle et al., 1994).
Polyanions aluminosiliciques formés à pH > 12 en présence d’ions Na+ ou K+. Les espèces polyédriques sont formées en présence de cations tétralkyl ammonium NR4+ (d’après Swaddle et al., 1994).
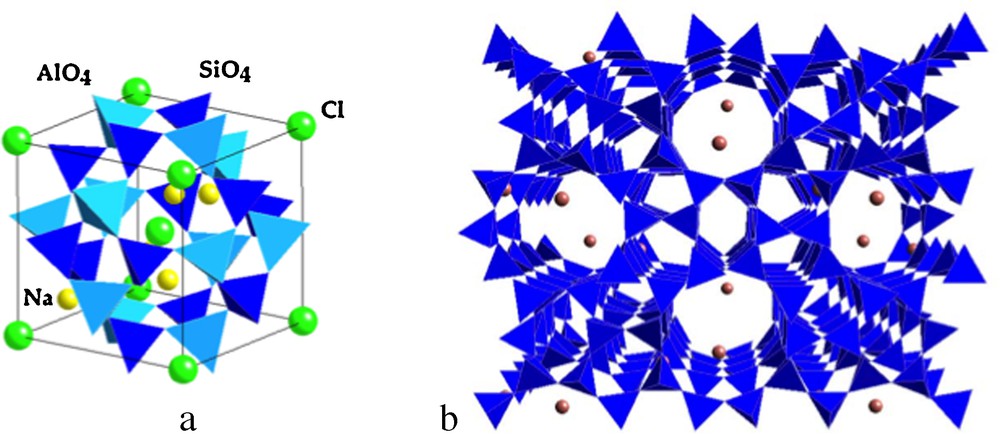
Representation of (a) the sodalite cavity (Na4(AlO2)3(SiO2)3Cl), a basic structural unit of natural zeolites and (b) view of the ZSM-5 synthetic zeolite (Zeolite Socony Mobil).
Représentation de (a) la cavité sodalite (Na4(AlO2)3(SiO2)3Cl), une unité structurale de base des zéolites naturelles et (b) vue de la zéolite synthétique ZSM-5 (Zeolite Socony Mobil).
Clays form a large family of aluminosilicates characterized by different two-dimensionnal structures (phyllosilicates), basically constituted by assembling layers of AlO6 octahedra (O) and layers of SiO4 tetrahedra (T). There are many stacking sequences of such layers, for example T-O T-O in kaolinite Si2O2Al2(OH)4 and T-O-T T-O-T in pyrophyllite Si4O10Al2(OH)2. The chemical composition of the layers is highly variable because of many substitutions inside each layers, for example Al3+ or Fe3+ to Si4+ in T layers as in muscovite or mica and Al3+ to Si4+ in T layers and Mg2+ to Al3+ in O layers as in montmorillonite. These substitutions induce an excess negative charge in the layers, which is compensated by intercalation of cations (e.g., K+, Ca2+) and possibly water molecules through the interlayer space, as for smectites (Fig. 7). Clays may also contain other substituting cations depending on the elements present during their formation (Berry and Mason, 1972; Caillère et al., 1982).
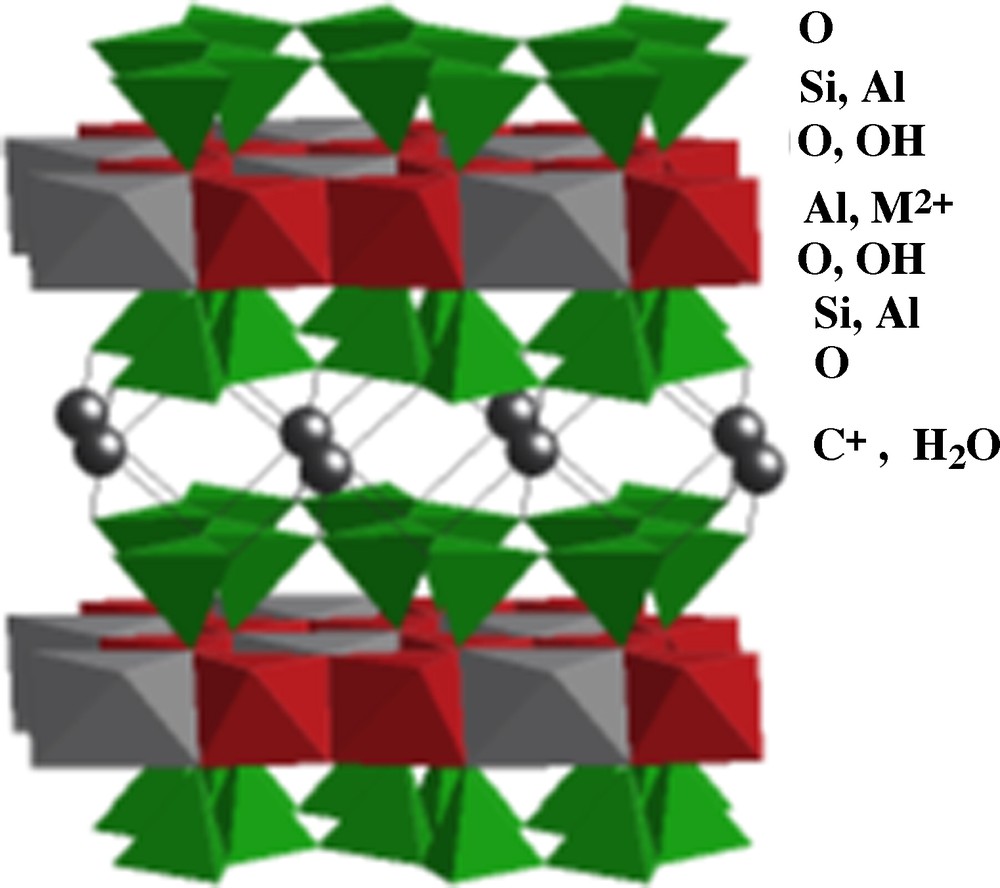
Idealized structure of clay montmorillonite, [Si4O10Al2−xM2+x(OH)2] Cx nH2O.
Structure idéalisée de l’argile montmorillonite, [Si4O10Al2−xM2+x(OH)2] Cx nH2O.
In the laboratory, the synthesis of smectites may involve a variety of precipitation conditions at low temperature (< 100 °C) or hydrothermal conditions (Kloprogge et al., 1999). Coprecipitation of Al3+, M2+ and Si4+ is in general carried out in presence of Ca(OH)2, NaOH or KOH and with a pH in the range 5 to 9. The general requirements for synthesis are a basic to neutral pH, a dilute medium, especially for Si (concentration in the range 10−4–10−3 mol.l−1), and slow precipitation (Decarreau, 1980; Harder, 1972, 1977) Under these conditions, one may suggest a simple possible mechanism of formation in which the acidity conditions lead first to the formation by olation of brucite-type (Mg(OH)2) or gibbsite-type (Al(OH)3) mixed layers, including the cations in an O-type layer. Due to their very low concentration, the auto-condensation of silicon in the form of Si(OH)4 or SiO(OH)− species is slow, but these highly nucleophilic monomeric species may strongly complex the cations by substitution on each face of the layers and then polymerize to form connected rings of tetrahedra sharing corners comprising the T-layers. Such a possibility explains how the Al-based layers result in the bi-dimensionality of clay mineral structures.
Another interesting type of aluminosilicates is imogolite. It is a tubular nano-object of ∼2 nm in diameter and several micrometers in length associated as bundles of ∼30 nm in diameter, with the global composition Al2SiO7H4 (Levard, 2008). As is the case for many other natural aluminosilicates, imogolite results from alteration of volcanic glasses and ashes. Imogolite is structurally described by a gibbsite-type layer of AlO6 octahedra wrapped up to form a cylinder, with isolated SiO4 tetrahedra grafted by three corners on the internal surface at the upright of vacancies in the gibbsite layer (Fig. 8) (Cradwick et al., 1972).
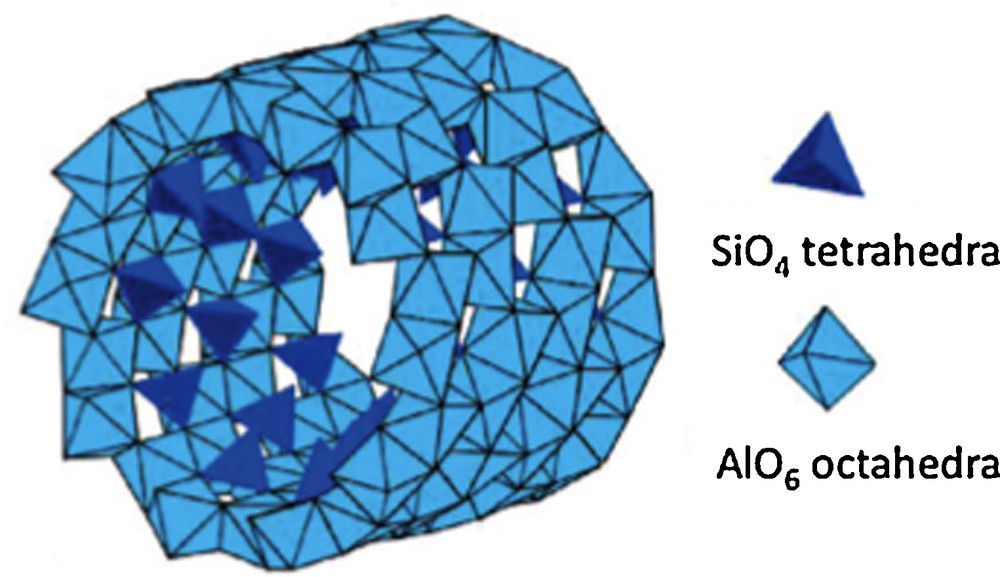
Structure of imogolite showing SiO4 tetrahedra bonded onto the internal face of the wrapped up gibbsite layer (from Levard, 2008).
Structure de l’imogolite montrant les tétraèdres SiO4 liés sur la face interne de la couche de gibbsite enroulée (d’après Levard, 2008).
Imogolite is classically synthesized by heating dilute solutions of the hydroxylated Al3+ complex (OH/Al = 2, millimolar Al concentrations) for five days at 95 °C in presence of silica (Al/Si = 2) in a slightly acidic medium (pH in the range 3.5–4.5) (Farmer et al., 1977). These conditions are somewhat similar to those involved in the synthesis of clays, although the solution is more acid, the Si/Al ratio is lower, and the dilution is greater. These factors favor the formation of aluminosilicic complexes by avoiding the auto-condensation of silicic species. Recently, it was shown that imogolite can be obtained using more concentrated media (decimolar concentrations) which increases the yield of the reaction; however, under these conditions the growth of imogolite was notably delayed during up to one month of heating at 95 °C (Levard et al., 2009). This experiment, carried out with aluminum perchlorate and tetraethylorthosilicate, showed, through 29Si and 27Al NMR observations that the formation of imogolite is limited by the decondensation rate of polysilicic species which was concomitantly formed. It is also likely that the complexation of aluminum by monomeric SiO4 tetrahedra, inhibits boehmite formation, as is also the case for clays, and results in the curvature of the aluminate layer. This last effect was nicely demonstrated using germanate analogues of imogolite (Levard et al., 2010). With a better electronic contrast between Al and Ge than between Al and Si, the use of X-ray spectroscopy and scattering and NMR spectroscopy showed that the curvature of gibbsite-like fragments was driven by the coordination of GeO4 tetrahedra, and the maximum in curvature was obtained when all vacancies in the gibbsite layer were coordinated to tetrahedra, due to a shorter O-O distance in tetrahedra than in the vacancies. These studies have also shown that imogolite precursors were curved, rooftile-shaped fragments of around 5 nm in size, which assemble edge to edge to form short cylindrical sections. Finally, imogolite nanotubes result from both direct asemblage of these sections and by Ostwald ripening.
4 Conclusion
This short review devoted to aluminum chemistry emphasizes the role of many parameters, particularly the acidity, in the formation of solids from solutions. The aim was to show that the knowledge of basic mechanisms of inorganic chemistry in solution allows one to understand the formation of various materials in the laboratory as well as in natural environments. Of course, in a general way, it is also necessary to take into account in many cases specific factors such as biologic or microbial mediation as seen for the interaction of aluminum with diatoms, and also some photosynthetic processes that induce possible photoredox reactions.
1 Stable aqueous dispersions of hematite α-Fe2O3 nanoparticles were obtained at pH 2, with a zeta potential of 32 mV. When the pH of the suspension was increased up to 5 by addition of a base, the zeta potential was lowered to around 10 mV and particle aggregation occurred and flocculation was quasi complete after two days. If the pH was increased up to 5 in the presence of Al13 polycations (pH 5 is the optimum for the stability of Al13 in solution), the zeta potential of particles remained near 30 mV and the stability of the suspension was maintained thanks to the adsorption of polycations, even though its structure was destroyed (Hernandez, 1998).

Solubility diagram of aluminum and electron micrographs and structures of the phases obtained at RT from the ageing of aluminous gel (from Hernandez, 1998).
Diagramme de solubilité de l’aluminium, clichés de microscopie électronique et structures des phases obtenues à température ambiante par vieillissement du gel aluminique (d’après Hernandez, 1998).