


 CC-BY 4.0
CC-BY 4.0
1. Introduction
Understanding the plumbing systems feeding volcanic eruptions is critical for providing an accurate interpretation of the different signals (seismicity, surface deformation, gas emissions) generated during periods of volcanic reactivation or eruption. Having accurate constraints on magma temperature, storage depths and on the amount of volatiles (H2O, CO2, S, F, Cl … etc.) dissolved in the magma allow to define more realistic scenarios for simulating the ascent and potentially predict the style of the upcoming event. In systems where different types of magmas (i.e., mafic to felsic) are emitted, an important question is to understand under which conditions basaltic melts evolve towards felsic compositions, and in particular whether this process occurs in a single or in multiple reservoirs located at different depths. This is crucial for understanding the final storage level of more evolved compositions and the structure of the plumbing system beneath a volcano, as the generation level of the felsic melts may not correspond to that of their pre-eruptive storage. In this contribution, we address this issue by unravelling the parental relationships between recent erupted basanites and phonolites at Mayotte (North Mozambique channel) using an experimental approach.
Between May 2018 and the end of 2021, Mayotte island has experienced a major submarine volcanic eruption characterized by the emission of more than 6.55 km3 of magma [Feuillet 2019, ReVoSiMa 2022]. Prior to this event, the island has not experienced historical episodes of reactivation, and the latest known volcanic event occurred some 7000 yrs ago on Petite Terre [Zinke et al. 2003]. The 2018–2021 eruption occurred along a WNW–ESE trending submarine ridge on the east flank of the island (Figure 1).
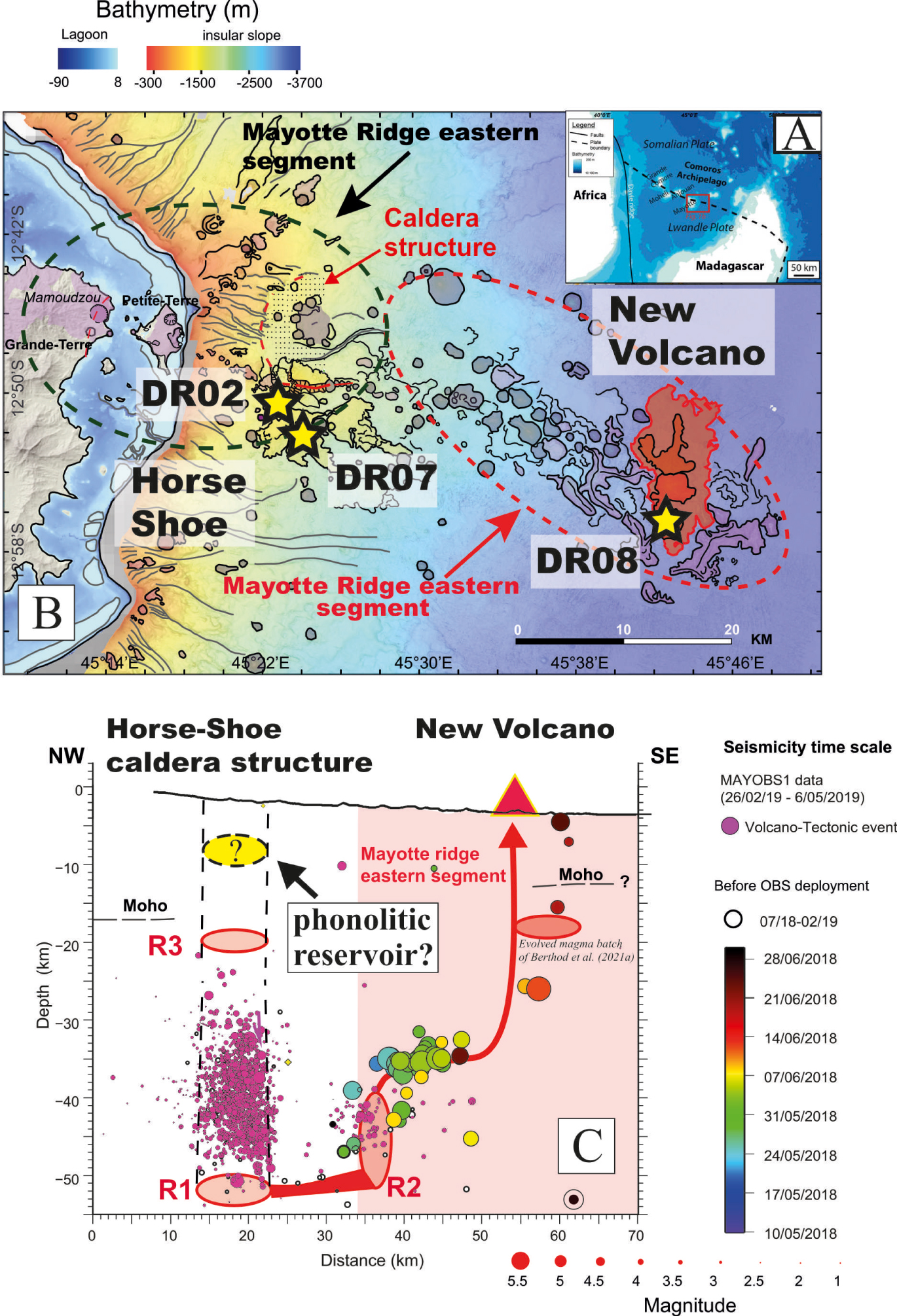
(A) Location of Comoros archipelago in the Mozambique channel. (B) Geological map of the active submarine volcanic ridge at Mayotte (modified from Feuillet et al. [2021]) showing the locations of the new volcano, and the Horse-shoe zone having active submarine degassing. The location of the dredged samples considered in this study is marked by yellow stars (modified from Berthod et al. [2021b]). (C) Depth distribution of the two well identified seismic swarms over the submarine ridge of Mayotte (modified from Feuillet et al. [2021]). The three seismically active zones R1, R2, R3 identified during this eruption, and the petrologically inferred reservoir located below the volcano and intercepted by the rising magma, are also shown [Feuillet et al. 2021, Foix et al. 2021 and Berthod et al. 2021b]. The emptying of a potential shallow phonolitic reservoir, as inferred in this work, could have produced the Horseshoe structure (see text for details). Masquer
(A) Location of Comoros archipelago in the Mozambique channel. (B) Geological map of the active submarine volcanic ridge at Mayotte (modified from Feuillet et al. [2021]) showing the locations of the new volcano, and the Horse-shoe zone having active submarine ... Lire la suite
Two main seismic swarms are still active below the ridge: a proximal and a distal cluster [Bertil et al. 2021, Cesca et al. 2020, Feuillet et al. 2021, Lavayssière et al. 2022, Lemoine et al. 2020, Saurelet al. 2021]. The distal cluster is located 30 km far from the eastern coast of Petite Terre whereas the proximal one is much closer (5–10 km from Petite Terre). Feuillet et al. [2021] inferred that the distal swarm was promoted by the drainage and consecutive stress changes around a ∼40 km deep magmatic reservoir (R2). Relocations of seismic events recorded by seismic stations onshore within the first weeks of the crisis show that a dike propagated from this reservoir R2 to transport the magma toward the seafloor at the eruption site [Feuillet et al. 2021]. Following the eruption, the proximal cluster was activated likely following the drainage of deeper (55 km) reservoir R1 and the consecutive collapse of a deep caldera structure above [Feuillet et al. 2021]. The proximal cluster is located below an old caldera structure where sits the Horseshoe zone [Figures 1B–C, Feuillet et al. 2021]. Active degassing is observed in this zone, giving rise to submarine plumes but without emission of fresh lava [Feuillet et al. 2021]. Ground deformation modeling revealed the drainage of about 5 km3 of a deep -seated reservoir [Lemoine et al. 2020], inferred to be R2 [Feuillet et al. 2021].
Recent passive tomography data [Foix et al. 2021], along with the detection of an important number of very low frequency events at ∼20 km [VLF; Laurent et al. 2021], suggest the presence of a third reservoir (R3; Figure 1C) above the deep caldera structure. The reactivation of this reservoir could be at the origin of the gas-emissions observed on the horseshoe region [Foix et al. 2021].
Geothermobarometry calculations broadly support the above seismic depths for the deep reservoirs (volume ⩾ 10 km3; depths ⩾ 37 km), in which the hydrous (2.3 wt% H2O dissolved in melt, H2Omelt) evolved basanite (generated by ∼50% of crystallization of a more primitive magma; Berthod et al. [2021a]) was sourced. Berthod et al. [2021a] further proposed that during its uprise, the basanite intercepted a shallower pre-existing reservoir at 17 ± 6.5 km below the new volcano (Figure 1C), in which a more evolved magma originated during recent magmatic episodes was residing.
The appearance of the dense seismic swarm beneath the Horseshoe region (Figure 1C) and the discovery of intensive submarine gas plumes, prompted the community for sampling this zone located 15 km away from Petite Terre. The study of samples dredged in this area revealed the presence of a bimodal compositional distribution of the emitted magmas in contrast to the new volcano, where basanite flows predominate. Within the Horseshoe region, several phonolitic bodies occur in abundance in addition to basanites, outcropping on the caldera borders and flowing downwards (Figure 1B; Berthod et al. [2021a, b]). The possible genetic link between the submarine basanites and phonolites was explored in Berthod et al. [2021b] through fractional crystallization modelling of major and trace elements along with Rhyolite-MELTs simulations [Ghiorso and Gualda 2015]. These modelling results show that the production of phonolite liquids from a hydrous (2.3 wt% H2Omelt) basanite requires 80% of fractional crystallization of a mineral assemblage consisting of clinopyroxene (Cpx), olivine (Ol), magnetite (Mt), apatite (Ap), ilmenite (Ilm) and anorthoclase alkali feldspar (Afs) at ⩽1000 °C, P⩾600 MPa and fO2 ∼ FMQ-1. Cpx–Opx barometry performed on mantle xenoliths carried by both phonolitic and basanitic samples from the Horseshoe region, yields similar storage conditions (15–20 km; Figure 1C). Based on these lines of evidence, these authors conclude that crystallisation of evolved basanitic melts at low oxygen fugacity produced the erupted Fe-rich phonolitic melts at Moho levels [Dofal et al. 2021].
It is worth noting that most of the phonolitic magmas known worldwide evolve at relatively shallow conditions [P⩽200 MPa or ⩽6–8 km; Andújar et al. 2008, 2010, 2013, Berndt et al. 2001, Harms et al. 2004, Moussallam et al. 2013, Scaillet et al. 2008]. However, rare are the cases concerning mantle-xenolith bearing phonolites [e.g. Berthod et al. 2021b, Dautria et al. 1983]. At first sight, the presence of mantle fragments within phonolitic magmas, along with thermobarometric calculations, hint at a rapid ascent of these magmas from deep mantle levels. Yet, examples of mafic to intermediate magmas containing significant amounts of different types of mantle xenoliths are manifold in alkaline series [e.g., La Palma, Klügel et al. 1999, 2022, Lanzarote, Neumann et al. 1995, La Garrotxa, Spain, Pedrazzi et al. 2022]. Since the products and mineralogy of evolved phonolites often show evidence of mixing-mingling with more mafic melts [i.e. mixed pumices, presence of inverse zonations in feldspars, olivine; Andújar et al. 2013, Andújar and Scaillet 2012, Berthod et al. 2021b, Wolff 1985; among others], the injection of mantle-bearing mafic magmas into phonolitic reservoirs could also account for the occurrence of these mantle fragments into the phonolites.
Another point worth of note is the use of thermobarometric equations [e.g., Putirka 2008] along with thermodynamic algorithms like MELTS or Rhyolite-MELTS [Gualda et al. 2012] for retrieving the ponding conditions and/or modelling the crystallization behavior of felsic alkaline magmas. These thermobarometric calibrations have important associated uncertainties on intensive parameters, in particular P [e.g. ±50 °C for T and ±150–400 MPa for P or 5–12 km in depth; Berthod et al. 2021a, b, Putirka 2008, 2016, Ubide et al. 2019] which limit their practical use in volcanological contexts, notably for inferring precisely storage conditions or comparing them to geophysical data. In particular, recent works have shown that amphibole- and Cpx- based geobarometers either do not capture real pressure [Erdmann et al. 2014] or overestimate considerably it [Hammer et al. 2016], in particular when applied to alkaline magmas [Hammer et al. 2016].
Similarly, while MELTs algorithm can faithfully simulate the evolution of anhydrous mafic melts at low pressures, the application of this model to the study of hydrous mafic melts is still limited, in large part because it cannot predict the crystallization of important hydrous phases such as amphibole or biotite. Significant differences in the crystallization temperatures and stability fields of phases like Cpx or Plagioclase in hydrous systems have also been reported [Freise et al. 2009, MacDonald et al. 2021].
Whereas the above approaches remain valuable for a first order evaluation of the ponding and evolution conditions of crystal-bearing magmas, the uncertainties and lack of calibration issues outlined above indicate that these methods are not standalone tools, and still need to be confronted to experimental results whenever possible. Accordingly, here we report experimental data gained on mafic magmas from Mayotte in order to put constraints on the generation conditions of phonolitic melts in this area.
2. Geological setting
Mayotte is the easternmost and oldest island of the Comoros archipelago, composed of a main volcanic island (Grande Terre) and a volcanic islet (Petite Terre) located 4 km away to the east (Figure 1B). Mayotte was constructed in three main different phases of volcanism: the first stage corresponds to the shield-building phase, between 20 and 3.8 Ma, followed by a fissure-erupted post erosional phase (3.8–2.5 Ma) and a final episode [2.4–1.5 Ma; Späth et al. 1996]. In detail, the volcanic activity of Grande Terre migrated from the southern part of the island during early stages (10.6–1.9 Ma) towards the north (5–0.75 Ma) and north-east (0.75–Present), with periods of quiescence in between [Debeuf 2004, Nehliget al. 2013]. This period of activity may also concern offshore WNW–ESE volcanic chains extending from Petite-Terre down to the newly discovered Fani Maore offshore volcano, Feuillet et al. [2021].
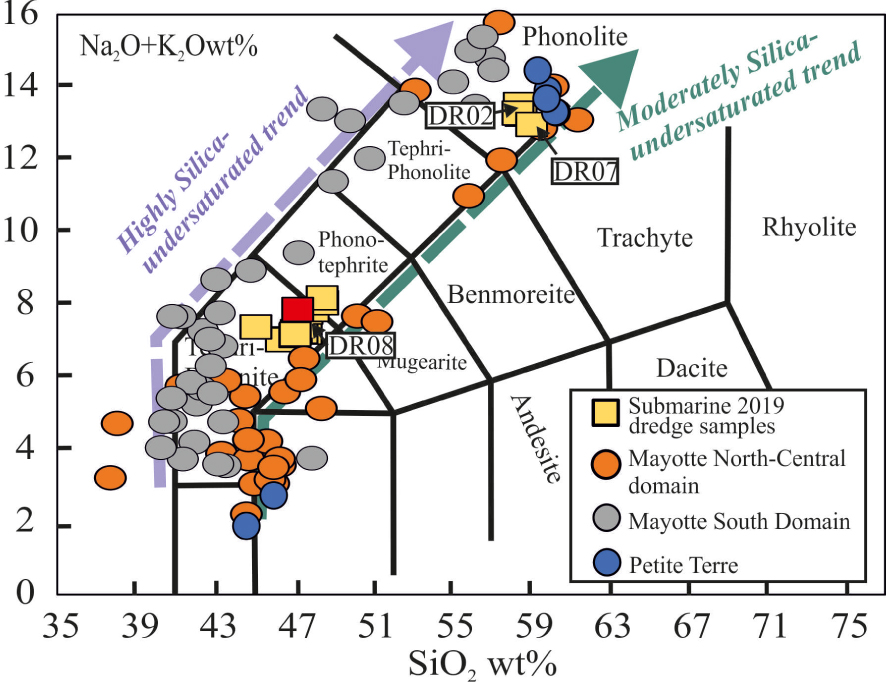
Total alkali (Na2O + K2O) versus SiO2 diagram [TAS after Le Bas and Streckeisen 1991] showing the two different liquid lines of descent of Mayotte magmas: highly silica-undersaturated and moderately silica-undersaturated trends. The products dredged on the active submarine ridge and those from Petite Terre are also shown. Compositional data are from Pelleter et al. [2014] and Berthod et al. [2021a, b].
The compositions of magmas erupted on Mayotte islands and along the 60 km of submarine ridge clearly define two distinct magmatic lineages. Whereas magmas from the southern Mayotte region are alkali-rich and highly silica undersaturated, those from north central-east sectors and the most recent Holocene products (including the 2018–2021 submarine eruption) are also silica-undersaturated but show a moderate alkali enrichment (Figure 2). Compositional differences between these two series are readily explained by the mineralogical heterogeneity of their source regions [Späth et al. 1996]. In both cases, crystal-fractionation has been proposed to be the main mechanism controlling magma evolution [Berthod et al. 2021a, b, Pelleter et al. 2014, Späth et al. 1996]. Despite their different sources, the magmas from these two series are dominated by the same mineralogy sensu latto. Ol+Cpx operate at the very initial stages of the fractionation process, being joined, and progressively replaced by, Fe–Ti oxides, feldspar and amphibole, as well as accessory amounts of apatite and titanite with increasing degrees of differentiation [Debeuf 2004, Pelleter et al. 2014, Späth et al. 1996].
3. Experimental work
3.1. Preparation of the starting material
The starting material used for our experiments is the sample DR08 that was dredged during the early phases of the 2018–2021 eruption by the MAYOBS-2 oceanographic campaign [Figure 1B; Table 1; Jorry 2019]. This sample has been petrologically characterized by Berthod et al. [2021a]; according to these authors (see their Tables 2–4), it is an evolved glassy popping-rock basanite (Mg ∼ 4.5 wt%; Table 1) with normative olivine content being ⩾10%, moderately vesicular (∼31%) with 5–9% (calculated on a bubble-free basis) of olivine (∼Fo72 ± 2 mol%), and trace amounts of Ti-magnetite and apatite.
Several pieces of this glassy rock were first finely ground in an agate mortar, put in a Pt crucible at 1400 °C and melted twice, with grinding in between, during 5 h in open atmosphere. Electron microprobe analyses (EMPA) of the starting glass show a homogeneous composition with no significant Na or Fe loss compared to data from Berthod et al. [2021a, Table 1]. The resulting dry glass was then ground to obtain the powder that was used as starting material for the phase equilibrium experiments and stored in an oven at 120 °C.
3.2. Experimental equipment and strategy
In total, six experiments were performed (Table 2) at the ISTO experimental laboratory, using the same experimental apparatus and procedure of Andújar et al. [2013], Moussallam et al. [2013], Scaillet et al. [1992], which are briefly summarized below. Experiments were carried out in an Internally Heated Pressure Vessel (IHPV) operating vertically, loaded with Ar–H2 mixtures at room temperature to achieve the desired fO2 conditions (see below). Total pressure was recorded by a transducer calibrated against a Heise Bourdon gauge with an uncertainty of ±20 bars. A double-winding kanthal furnace was used as this allows to achieve near-isothermal conditions (gradient <2–3 °C/cm) along a 3 cm long hot spot. Temperature was measured using K-type thermocouples with an accuracy of ±5 °C. A rapid-quench technique was systematically used, imparting isobaric cooling rates of >100 °C/s [e.g., Andújar et al. 2013, 2015, Scaillet et al. 2008]. In all runs reported here, the drop quench was successful as indicated by the rise in total pressure upon the falling of the sample holder into the cold (bottom) part of the vessel.
Experiments were mainly conducted at T–P conditions covering those determined for similar phonolites worldwide [e.g., Andújar et al. 2008, 2010, 2013, Andújar and Scaillet 2012, Giehl et al. 2013, Moussallam et al. 2013, Scaillet et al. 2008]. Temperature was set at 925 and 950 °C whereas explored pressures were 100, 150, 200 and 400 MPa. Based on the iron-rich character of the Mayotte phonolites [Berthod et al. 2021b], the initial fO2 was set at ⩽ NNO buffer (where NNO refers to the Ni–NiO buffer) as Fe enrichment in magmas is favoured by low fO2 [e.g., Giehl et al. 2013, Moussallam et al. 2013, Toplis and Carroll 1995].
3.3. Capsule preparation
We used Au gold capsules (1.5 cm long, 2.5 mm inner diameter, 0.2 mm wall thickness) since this metal minimizes the Fe-loss towards the capsule walls under reducing conditions. Distilled H2O was first loaded, then silver oxalate as the source of CO2 for H2O-undersaturated runs, and then the glass powder. Capsules were weighed and then welded using an arc-electric welder. After welding, capsules were re-weighed and if no significant weight loss occurred (considered to occur for a difference >0.0004 g (or ⩽0.1% of the final weight of the capsule)), they were left in an oven for a few hours at 100 °C, to ensure homogeneous volatiles distribution. Both the amount of H2O + CO2 and fluid/silicate ratio were maintained constant (3 ± 0.5 mg of H2O + CO2, and 30 mg silicate). At a given T–P conditions, various starting H2O–CO2 mixtures were explored: XH2Oin, defined as H2O∕(H2O + CO2) (in moles), varied in the range 1–0.08 (Table 2).
100 wt% anhydrous composition of the starting glass DR08, dredge bulk-rock compositions for submarine basanite DR08 and phonolites [Berthod et al. 2021a, b]; North-central onland phonolites
| DR08 | DR08 Starting Glass | sd | Bulk-rock submarine basanite Berthod et al. [2021a, b] | Bulk-rock submarine phonolites | North central onland-phonolites [Pelleter et al. 2014, Späth et al. 1996] | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR0801-ALF | DR080102 | DR020202 | DR020401 | DR060204 | DR070201 | DR0701 | M29 (aphyric) | M31 | M76 | Ma-5 | Ma65 | |||
| n | 15 | |||||||||||||
| SiO2 | 47.45 | 0.33 | 47.65 | 47.44 | 58.54 | 58.50 | 58.59 | 58.43 | 59.12 | 61.47 | 57.60 | 59.65 | 60.10 | 58.65 |
| TiO2 | 2.87 | 0.13 | 2.90 | 2.92 | 0.37 | 0.36 | 0.36 | 0.35 | 0.36 | 0.35 | 0.87 | 0.56 | 0.58 | 0.81 |
| Al2O3 | 15.35 | 0.16 | 15.69 | 15.53 | 18.51 | 18.47 | 18.65 | 18.67 | 18.37 | 19.79 | 20.24 | 19.95 | 19.91 | 19.14 |
| FeO* | 13.61 | 0.35 | 13.74 | 14.00 | 6.47 | 6.44 | 6.50 | 6.68 | 6.48 | 3.15 | 4.75 | 4.01 | 3.77 | 5.12 |
| MnO | 0.19 | 0.04 | 0.24 | 0.25 | 0.26 | 0.26 | 0.27 | 0.27 | 0.27 | 0.12 | 0.14 | 0.13 | 0.14 | 0.17 |
| MgO | 4.57 | 0.10 | 4.43 | 4.46 | 0.62 | 0.63 | 0.46 | 0.40 | 0.47 | 0.49 | 1.02 | 0.46 | 0.45 | 0.79 |
| CaO | 6.61 | 0.06 | 6.53 | 6.53 | 1.57 | 1.55 | 1.63 | 1.62 | 1.65 | 1.45 | 3.16 | 2.26 | 1.97 | 3.03 |
| Na2O | 4.79 | 0.06 | 4.48 | 4.62 | 7.97 | 8.05 | 7.64 | 7.67 | 7.48 | 7.38 | 7.36 | 7.66 | 8.07 | 7.67 |
| K2O | 2.74 | 0.11 | 2.75 | 2.63 | 5.39 | 5.47 | 5.61 | 5.60 | 5.48 | 5.70 | 4.62 | 5.20 | 4.91 | 4.40 |
| P2O5 | 1.81 | 0.05 | 1.58 | 1.60 | 0.31 | 0.28 | 0.31 | 0.31 | 0.32 | 0.09 | 0.25 | 0.11 | 0.08 | 0.22 |
| Sum | 100.00 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | 100 | |
| P.I. | 0.71 | 0.66 | 0.68 | 1.03 | 1.04 | 1.01 | 1.01 | 1.00 | 0.93 | 0.85 | 0.92 | 0.94 | 0.91 | |
| Normative Nepheline (wt%) | 12.6 | 13.2 | 11.5 | 11.8 | 9.4 | 7.4 | 10.8 | 10.4 | 10.7 | 9.4 | ||||
n: Number of analysis.
sd: Standard deviation.
P.I: Peralkaline index calculated as the (Na+K)/Al in moles.
Experimental run conditions and phase proportions (wt%)
| Charge | T (°C) | P | XH2Oin | XH2Ofin | H2O (wt%) | CO2 (ppm) | fH2O | logfO2 | ΔNNO | ΔQFM | Ol | Mt | Ilm | Ap | Cpx | Pl | Kr | Bt | Observations | % crystals | R2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 950 °C/400 MPa; t = 73h | |||||||||||||||||||||
| DR08-140 | 950 | 400 | 1.00 | 1.00 | 6.78 | 0 | 3880 | −10.7 | 0.3 | 0.87 | 7.7 | 3.8 | 12.4 | 5.2 | 29.1 | 0.61 | |||||
| DR08-141 | 950 | 400 | 0.72 | 0.52 | 4.78 | 4593 | 2015 | −11.3 | −0.3 | 0.30 | 6.15 | 0.1 | 3.3 | 25.8 | 2.5 | 37.85 | 0.42 | ||||
| DR08-142 | 950 | 400 | 0.49 | 0.27 | 3.40 | 5483 | 1062 | −11.9 | −0.8 | −0.25 | 2.9 | 5.6 | 0.37 | 4.7 | 20.6 | 2.1 | 36.27 | 0.23 | |||
| DR08-143 | 950 | 400 | 0.26 | 0.10 | 1.97 | 5974 | 381 | −12.8 | −1.7 | −1.15 | 14.05 | 0.94 | 3.4 | 3.86 | 34.07 | 9 | 0.7 | 66.02 | 0.73 | ||
| DR08-144 | 950 | 400 | 0.14 | 0.04 | 1.29 | 6108 | 172 | −13.5 | −2.4 | −1.83 | X | X | X | X | X | X | X | ||||
| DR08-145 | 950 | 400 | 0.09 | 0.03 | 0.96 | 6154 | 100 | −13.9 | −2.9 | −2.31 | X | X | X | X | X | X | X | X | |||
| 925 °C/400 MPa; t = 71.5 h | |||||||||||||||||||||
| DR08-152 | 925 | 400 | 1.00 | 1.00 | 6.69 | 0 | 3779 | −11.2 | 0.2 | 0.81 | 7.6 | 0.2 | 4.01 | 15.6 | 7.8 | 35.21 | 0.38 | ||||
| DR08-154 | 925 | 400 | 0.50 | 0.31 | 3.59 | 5402 | 1173 | −12.2 | −0.8 | −0.21 | 5.6 | 0.51 | 3.5 | 14.4 | 25.7 | 7.8 | 57.51 | 0.75 | |||
| DR08-155 | 925 | 400 | 0.29 | 0.16 | 2.51 | 5850 | 599 | −12.8 | −1.4 | −0.79 | X | X | X | X | X | X | X | ||||
| DR08-156 | 925 | 400 | 0.18 | 0.09 | 1.86 | 6030 | 340 | −13.3 | −1.9 | −1.29 | X | X | X | X | X | X | X | ||||
| DR08-157 | 925 | 400 | 0.08 | 0.03 | 1.09 | 6170 | 127 | −14.2 | −2.7 | −2.14 | X | X | X | X | X | X | X | X | |||
| DR08-950 °C, 200 MPa, t = 68 h | |||||||||||||||||||||
| DR08-56 | 950 | 200 | 0.90 | 0.85 | 4.65 | 249 | 1527 | −11.6 | −0.5 | 0.81 | 2.3 | 8.8 | 3.2 | 20 | 2.6 | 36.9 | 1.2 | ||||
| DR08-58 | 950 | 200 | 0.51 | 0.34 | 2.73 | 908 | 613 | −12.3 | −1.3 | 1.03 | 3.01 | 7.1 | 3.6 | 25 | 2.1 | 40.81 | 0.93 | ||||
| DR08-925 °C/200 MPa, 69.5 h | |||||||||||||||||||||
| DR08-129 | 925 | 200 | 1.00 | 1.00 | 5.06 | − | 1764 | −11.9 | −0.4 | 0.14 | 7.1 | 3.97 | 26.5 | 2.3 | Quench | 39.87 | 0.96 | ||||
| DR08-130 | 925 | 200 | 0.88 | 0.82 | 4.52 | 287 | 1452 | −12.1 | −0.6 | −0.02 | 5.7 | 2.4 | 30.5 | 3.8 | 42.4 | 0.99 | |||||
| DR08-131 | 925 | 200 | 0.69 | 0.60 | 3.76 | 596 | 1060 | −12.3 | −0.8 | −0.30 | 15.9 | 1 | 3.8 | 4.3 | 31.29 | 8.2 | 0.4 | 64.89 | 0.67 | ||
| DR08-132 | 925 | 200 | 0.45 | 0.35 | 2.73 | 900 | 614 | −12.8 | −1.3 | −0.77 | x | x | x | x | x | x | x | x | |||
| DR08-133 | 925 | 200 | 0.34 | 0.25 | 2.27 | 1004 | 449 | −13.1 | −1.6 | −1.04 | x | x | x | x | x | x | x | ||||
| DR08-134 | 925 | 200 | 0.19 | 0.13 | 1.49 | 1138 | 226 | −13.7 | −2.2 | −1.66 | x | x | x | x | x | x | ? | ||||
| DR08-135 | 925 | 200 | 0.09 | 0.05 | 0.84 | 1215 | 93 | −14.4 | −2.9 | −2.45 | x | x | x | x | x | x | ? | ||||
| DR08-925 °C/150 MPa; t = 71, 5h | |||||||||||||||||||||
| DR08-158 | 925 | 150 | 1.00 | 1.00 | 3.85 | 0 | 1339 | −11.8 | −0.3 | 0.26 | 7.72 | 4.01 | 20.6 | 4.8 | 37.13 | 0.32 | |||||
| DR08-159 | 925 | 150 | 0.90 | 0.86 | 3.56 | 259 | 1154 | −11.9 | −0.4 | 0.13 | 7.74 | 3.72 | 3.74 | 22.7 | 3.1 | 41 | 0.5 | ||||
| DR08-160 | 925 | 150 | 0.65 | 0.52 | 2.72 | 998 | 698 | −12.3 | −0.8 | −0.31 | 7.44 | 7.76 | 1.73 | 3.59 | 2.3 | 12.8 | 18.4 | 0.9 | 54.92 | 0.33 | |
| DR08-161 | 925 | 150 | 0.54 | 0.45 | 2.52 | 1164 | 603 | −12.5 | −1.0 | −0.44 | X | X | X | X | X | X | X | X | |||
| DR08-162 | 925 | 150 | 0.30 | 0.21 | 1.66 | 1719 | 277 | −13.1 | −1.6 | −1.11 | X | X | X | X | X | X | X | ||||
| DR08-163 | 925 | 150 | 0.18 | 0.12 | 1.26 | 1897 | 165 | −13.6 | −2.1 | −1.58 | X | X | X | X | X | X | X | ||||
| DR08-925 °C/100 MPa; t = 64 h | |||||||||||||||||||||
| DR08-164 | 925 | 100 | 1.00 | 1.00 | 3.14 | 0 | 915 | −11.8 | −0.3 | 0.18 | 2.45 | 8.18 | 3.7 | 14.5 | 18.9 | 3.1 | 50.83 | 0.56 | |||
| DR08-165 | 925 | 100 | 0.88 | 0.84 | 2.87 | 168 | 772 | −12.0 | −0.5 | 0.03 | 5.33 | 7.6 | 1.09 | 4.03 | 6.39 | 21.3 | 2.9 | 5.5 | 54.14 | 0.4 | |
| DR08-166 | 925 | 100 | 0.71 | 0.62 | 2.44 | 424 | 571 | −12.3 | −0.7 | −0.23 | X | X | X | X | X | X | X | ||||
| DR08-167 | 925 | 100 | 0.47 | 0.37 | 1.86 | 741 | 341 | −12.7 | −1.2 | −0.68 | X | X | X | X | X | X | X | ||||
| DR08-168 | 925 | 100 | 0.33 | 0.24 | 1.46 | 921 | 217 | −13.1 | −1.6 | −1.07 | X | X | X | X | X | X | X | ||||
Ol: olvine, Mt: magnetite, ilm: ilmenite, Ap: apatite, Cpx: Clinopyroxene, Pl: plagioclase, Kr: Kaersutite/Amphibole, Bt: biotite.
XH2Oin =, initial H2O∕(H2O + CO2) in the charge.
XH2Ofin = final H2O∕(H2O + CO2) in the charge.
H2O wt%, water content in the melt in wt%.
CO2 content in the melt ppm.
log fO2(bar), logarithm of the oxygen fugacity calculated from the experimental fH2.
ΔNNO = log fO2 experiment-log fO2 of the NNO buffer calculated at experimental T–P after Pownceby and O’Neill [1994].
ΔQFM = log fO2 experiment-log fO2 of the QFM buffer calculated at experimental T after Chou [1978].
X, mineral phase identified by SEM but, its abundance could not be calculated.
fH2(bar), hydrogen fugacity of the experiment. See text for details.
Experimental olivine compositions (wt%)
| Charge | T (°C) | P (MPa) | n | SiO2 | TiO2 | Al2O3 | FeO* | MnO | MgO | CaO | Na2O | K2O | Sum | Fo% |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR08-142 | 950 | 400 | 3 | 33.5 | 0.2 | 0.2 | 43.8 | 1.1 | 19.9 | 0.7 | 0.1 | 0.0 | 99.5 | 44.8 |
| sd | 0.4 | 0.1 | 0.2 | 1.0 | 0.1 | 0.3 | 0.4 | 0.1 | 0.0 | 0.7 | 0.2 | |||
| DR08-143 | 950 | 400 | 2 | 32.9 | 0.3 | 0.1 | 47.3 | 1.2 | 17.6 | 0.9 | 0.1 | 0.0 | 100.5 | 39.9 |
| sd | 0.0 | 0.0 | 0.0 | 0.2 | 0.1 | 0.2 | 0.1 | 0.0 | 0.0 | 0.0 | 0.3 | |||
| DR08-144 | 950 | 400 | 1 | 32.8 | 0.5 | 0.7 | 48.9 | 1.3 | 16.4 | 0.8 | 0.1 | 0.1 | 101.7 | 37.5 |
| DR08-145 | 950 | 400 | 1 | 34.2 | 0.3 | 1.5 | 45.0 | 1.6 | 15.0 | 1.1 | 0.3 | 0.2 | 99.2 | 37.3 |
| DR08-155 | 925 | 400 | 2 | 32.7 | 0.2 | 0.1 | 50.3 | 1.8 | 13.9 | 1.3 | 0.1 | 0.1 | 100.5 | 33.0 |
| sd | 0.5 | 0.1 | 0.1 | 1.0 | 0.1 | 0.5 | 0.2 | 0.0 | 0.1 | 0.7 | 0.4 | |||
| DR08-157 | 925 | 400 | 1 | 32.7 | 0.5 | 0.5 | 49.3 | 1.4 | 13.5 | 1.6 | 0.1 | 0.1 | 99.6 | 32.8 |
| DR08-58 | 950 | 200 | 2 | 33.3 | 0.3 | 0.1 | 40.2 | 1.2 | 21.6 | 1.2 | 0.0 | 0.0 | 97.9 | 49.0 |
| sd | 0.4 | 0.3 | 0.1 | 0.3 | 0.1 | 0.6 | 0.5 | 0.0 | 0.0 | 0.2 | 0.9 | |||
| DR08-131 | 925 | 200 | 1 | 33.5 | 0.2 | 0.5 | 42.6 | 1.7 | 19.2 | 1.0 | 0.2 | 0.1 | 98.9 | 44.5 |
| DR08-132 | 925 | 200 | 2 | 32.1 | 0.8 | 0.6 | 42.4 | 1.5 | 18.6 | 1.3 | 0.2 | 0.1 | 97.5 | 43.9 |
| sd | 1.0 | 0.5 | 0.3 | 1.1 | 0.1 | 0.5 | 0.2 | 0.0 | 0.0 | 1.2 | 0.5 | |||
| DR08-160 | 925 | 150 | 1 | 34.8 | 0.1 | 0.0 | 40.6 | 1.4 | 22.4 | 0.5 | 0.0 | 0.0 | 99.8 | 49.6 |
| DR08-161 | 925 | 150 | 2 | 34.8 | 0.1 | 0.6 | 40.6 | 1.7 | 22.2 | 0.4 | 0.3 | 0.1 | 100.8 | 49.4 |
| sd | 0.1 | 0.1 | 0.1 | 0.5 | 0.3 | 0.3 | 0.1 | 0.0 | 0.0 | 0.8 | 0.6 | |||
| DR08-162 | 925 | 150 | 1 | 31.6 | 1.3 | 0.5 | 44.0 | 1.3 | 18.6 | 1.7 | 0.1 | 0.1 | 99.3 | 42.9 |
| DR08-164 | 925 | 100 | 2 | 34.3 | 0.1 | 0.4 | 38.8 | 1.4 | 22.8 | 1.8 | 0.1 | 0.0 | 99.8 | 51.1 |
| sd | 0.7 | 0.1 | 0.1 | 0.2 | 0.0 | 0.0 | 0.4 | 0.1 | 0.0 | 0.3 | 0.1 | |||
| DR08-165 | 925 | 100 | 1 | 32.1 | 1.0 | 0.2 | 40.4 | 1.3 | 22.0 | 1.6 | 0.0 | 0.1 | 98.7 | 49.2 |
| DR08-166 | 925 | 100 | 2 | 32.7 | 1.0 | 0.8 | 40.1 | 1.3 | 20.9 | 2.0 | 0.2 | 0.1 | 99.2 | 48.2 |
| sd | 0.6 | 0.4 | 0.2 | 0.3 | 0.1 | 0.3 | 0.2 | 0.1 | 0.0 | 0.5 | 0.1 |
n: number of analysis.
sd: standard deviation.
FeO*: Total iron reported as Fe2+.
Fo mole(%) = 100 Mg/(Mg + Fe*) in olivine.
Experimental amphibole compositions (wt%). Classification after Ridolfi [2021]
| Charge | T (°C) | P (MPa) | n | SiO2 | TiO2 | Al2O3 | FeO* | MnO | MgO | CaO | Na2O | K2O | Sum | Species | Si | AlIV | Ti | T sum | AlVI | Ti | Cr | Fe3+ | Mg | Fe2+ | Mn | C sum | Ca | Na | B sum | Na | K | A sum | OH | F | Cl | W sum | Charge | Mg/ (Mg + Fe2+) | Mg# | Al# | AlT | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR08-140 | 950 | 400 | 5 | 39.85 | 2.91 | 11.07 | 13.14 | 0.25 | 11.23 | 9.97 | 2.71 | 0.87 | 92.41 | Mg- hastingsite | 6.24 | 1.76 | 0.00 | 8.00 | 0.28 | 0.34 | 0.00 | 0.45 | 2.62 | 1.27 | 0.03 | 5.00 | 1.67 | 0.33 | 2.00 | 0.49 | 0.17 | 0.67 | 2.00 | 0.00 | 0.00 | 2.00 | 45.55 | 0.67 | 0.60 | 0.14 | 2.04 | |||||
| sd | 0.53 | 0.23 | 0.35 | 1.00 | 0.07 | 0.76 | 0.08 | 0.05 | 0.10 | 0.89 | ||||||||||||||||||||||||||||||||||||
| DR08-141 | 950 | 400 | 2 | 39.16 | 3.72 | 12.10 | 16.20 | 0.28 | 9.97 | 11.72 | 2.75 | 0.91 | 96.83 | Pargasite | 6.00 | 2.00 | 0.00 | 8.00 | 0.18 | 0.43 | 0.00 | 0.12 | 2.28 | 1.95 | 0.04 | 5.00 | 1.92 | 0.08 | 2.00 | 0.74 | 0.18 | 0.92 | 2.00 | 0.00 | 0.00 | 2.00 | 45.88 | 0.54 | 0.52 | 0.08 | 2.18 | |||||
| sd | 0.73 | 0.60 | 1.09 | 1.07 | 0.14 | 0.68 | 1.81 | 0.00 | 0.09 | 1.25 | ||||||||||||||||||||||||||||||||||||
| DR08-142 | 950 | 400 | 3 | 39.41 | 5.14 | 12.22 | 14.83 | 0.19 | 11.01 | 10.51 | 2.68 | 0.95 | 96.94 | kaersutite | 5.91 | 2.09 | 0.00 | 8.00 | 0.07 | 0.58 | 0.00 | 0.52 | 2.46 | 1.34 | 0.02 | 5.00 | 1.69 | 0.31 | 2.00 | 0.47 | 0.18 | 0.65 | 2.00 | 0.00 | 0.00 | 2.00 | 45.48 | 0.65 | 0.57 | 0.03 | 2.16 | |||||
| sd | 0.97 | 0.35 | 0.57 | 0.58 | 0.06 | 0.39 | 0.36 | 0.20 | 0.12 | 0.68 | ||||||||||||||||||||||||||||||||||||
| DR08-143 | 950 | 400 | 3 | 38.60 | 5.55 | 11.77 | 16.27 | 0.36 | 9.23 | 11.83 | 2.71 | 1.12 | 97.44 | kaersutite | 5.95 | 2.05 | 0.00 | 8.00 | 0.09 | 0.64 | 0.00 | 0.00 | 2.12 | 2.10 | 0.05 | 5.00 | 1.95 | 0.05 | 2.00 | 0.76 | 0.22 | 0.98 | 2.00 | 0.00 | 0.00 | 2.00 | 46.27 | 0.50 | 0.50 | 0.04 | 2.14 | |||||
| sd | 0.79 | 0.55 | 0.57 | 1.03 | 0.10 | 0.18 | 0.78 | 0.10 | 0.06 | 0.66 | ||||||||||||||||||||||||||||||||||||
| DR08-144 | 950 | 400 | 2 | 41.10 | 5.05 | 10.50 | 18.19 | 0.25 | 9.07 | 10.80 | 2.96 | 1.14 | 99.04 | low-Mg | 6.20 | 1.80 | 0.00 | 8.00 | 0.06 | 0.57 | 0.00 | 0.02 | 2.04 | 2.27 | 0.03 | 5.00 | 1.74 | 0.26 | 2.00 | 0.61 | 0.22 | 0.83 | 2.00 | 0.00 | 0.00 | 2.00 | 45.98 | 0.47 | 0.47 | 0.03 | 1.87 | |||||
| sd | 0.11 | 0.03 | 0.28 | 0.89 | 0.05 | 0.26 | 0.48 | 0.03 | 0.09 | 1.54 | ||||||||||||||||||||||||||||||||||||
| DR08-145 | 950 | 400 | ||||||||||||||||||||||||||||||||||||||||||||
| DR08-152 | 925 | 400 | 3 | 40.44 | 3.07 | 11.78 | 13.30 | 0.31 | 12.71 | 11.49 | 2.66 | 0.86 | 96.63 | Mg- hastingsite | 6.04 | 1.96 | 0.00 | 8.00 | 0.12 | 0.35 | 0.00 | 0.53 | 2.83 | 1.14 | 0.04 | 5.00 | 1.84 | 0.16 | 2.00 | 0.61 | 0.16 | 0.78 | 2.00 | 0.00 | 0.00 | 2.00 | 45.47 | 0.71 | 0.63 | 0.06 | 2.08 | |||||
| sd | 0.57 | 0.19 | 0.70 | 0.40 | 0.06 | 0.50 | 0.21 | 0.10 | 0.08 | 0.92 | ||||||||||||||||||||||||||||||||||||
| DR08-154 | 925 | 400 | 4 | 40.25 | 3.79 | 11.11 | 18.85 | 0.42 | 9.00 | 10.41 | 2.98 | 0.68 | 97.48 | Mg- hastingsite | 6.10 | 1.90 | 0.00 | 8.00 | 0.09 | 0.43 | 0.00 | 0.56 | 2.04 | 1.83 | 0.05 | 5.00 | 1.69 | 0.31 | 2.00 | 0.57 | 0.13 | 0.70 | 2.00 | 0.00 | 0.00 | 2.00 | 45.44 | 0.53 | 0.46 | 0.04 | 1.99 | |||||
| sd | 0.94 | 0.15 | 0.19 | 0.51 | 0.12 | 0.30 | 0.23 | 0.10 | 0.04 | 0.89 | ||||||||||||||||||||||||||||||||||||
| DR08-155 | 925 | 400 | 6 | 39.33 | 4.04 | 10.40 | 19.05 | 0.45 | 8.12 | 10.80 | 3.01 | 0.85 | 96.06 | low-Mg | 6.16 | 1.84 | 0.00 | 8.00 | 0.08 | 0.48 | 0.00 | 0.11 | 1.90 | 2.38 | 0.06 | 5.00 | 1.81 | 0.19 | 2.00 | 0.73 | 0.17 | 0.89 | 2.00 | 0.00 | 0.00 | 2.00 | 45.89 | 0.44 | 0.43 | 0.04 | 1.92 | |||||
| sd | 0.61 | 0.20 | 0.23 | 0.40 | 0.05 | 0.20 | 0.26 | 0.07 | 0.03 | 0.47 | ||||||||||||||||||||||||||||||||||||
| DR08-156 | 925 | 400 | 1 | 39.48 | 4.32 | 10.75 | 18.24 | 0.45 | 7.66 | 11.42 | 2.98 | 0.92 | 96.23 | low-Mg | 6.22 | 1.78 | 0.00 | 8.00 | 0.22 | 0.51 | 0.00 | 0.00 | 1.80 | 2.41 | 0.06 | 5.00 | 1.93 | 0.07 | 2.00 | 0.84 | 0.18 | 1.02 | 2.00 | 0.00 | 0.00 | 2.00 | 46.42 | 0.43 | 0.43 | 0.11 | 2.00 | |||||
| DR08-157 | 925 | 400 | 1 | 40.53 | 3.89 | 12.15 | 17.63 | 0.32 | 7.00 | 10.99 | 3.23 | 0.96 | 96.70 | low-Mg | 6.33 | 1.67 | 0.00 | 8.00 | 0.57 | 0.46 | 0.00 | 0.00 | 1.63 | 2.30 | 0.04 | 5.00 | 1.84 | 0.16 | 2.00 | 0.82 | 0.19 | 1.01 | 2.00 | 0.00 | 0.00 | 2.00 | 46.66 | 0.41 | 0.41 | 0.25 | 2.24 | |||||
| sd | ||||||||||||||||||||||||||||||||||||||||||||||
| DR08-56 | 950 | 200 | 5 | 39.495 | 4.4431 | 11.74 | 12.44 | 0.27 | 12.61 | 11.03 | 2.62 | 0.85 | 95.50 | kaersutite | 5.96 | 2.04 | 0.00 | 8.00 | 0.05 | 0.50 | 0.00 | 0.48 | 2.84 | 1.09 | 0.03 | 5.00 | 1.78 | 0.22 | 2.00 | 0.55 | 0.16 | 0.71 | 2.00 | 0.00 | 0.00 | 2.00 | 45.52 | 0.72 | 0.64 | 0.03 | 2.09 | |||||
| sd | 0.2655 | 0.2434 | 0.22 | 0.11 | 0.05 | 0.15 | 0.23 | 0.13 | 0.08 | 0.44 | ||||||||||||||||||||||||||||||||||||
| DR08-58 | 950 | 200 | 5 | 39.336 | 5.0109 | 11.10 | 14.51 | 0.28 | 10.84 | 11.56 | 2.68 | 0.98 | 96.30 | kaersutite | 6.04 | 1.96 | 0.00 | 8.00 | 0.04 | 0.58 | 0.00 | 0.00 | 2.48 | 1.86 | 0.04 | 5.00 | 1.90 | 0.10 | 2.00 | 0.70 | 0.19 | 0.89 | 2.00 | 0.00 | 0.00 | 2.00 | 46.03 | 0.57 | 0.57 | 0.02 | 2.01 | |||||
| sd | 0.1415 | 0.3008 | 0.36 | 0.69 | 0.07 | 0.08 | 0.53 | 0.12 | 0.10 | 0.88 | ||||||||||||||||||||||||||||||||||||
| DR08-129 | 925 | 200 | 3 | 38.76 | 3.58 | 12.34 | 13.13 | 0.32 | 11.82 | 11.33 | 2.65 | 0.86 | 94.78 | Mg- hastingsite | 5.94 | 2.06 | 0.00 | 8.00 | 0.17 | 0.41 | 0.00 | 0.40 | 2.70 | 1.29 | 0.04 | 5.00 | 1.86 | 0.14 | 2.00 | 0.65 | 0.17 | 0.82 | 2.00 | 0.00 | 0.00 | 2.00 | 45.60 | 0.68 | 0.62 | 0.07 | 2.23 | |||||
| sd | 0.12 | 0.16 | 0.08 | 0.08 | 0.07 | 0.28 | 0.26 | 0.07 | 0.16 | 0.17 | ||||||||||||||||||||||||||||||||||||
| DR08-131 | 925 | 200 | 3 | 39.08 | 3.78 | 11.19 | 16.27 | 0.33 | 10.49 | 10.75 | 2.83 | 0.84 | 95.57 | Mg- hastingsite | 6.00 | 2.00 | 0.00 | 8.00 | 0.03 | 0.44 | 0.00 | 0.55 | 2.40 | 1.54 | 0.04 | 5.00 | 1.77 | 0.23 | 2.00 | 0.61 | 0.16 | 0.78 | 2.00 | 0.00 | 0.00 | 2.00 | 45.45 | 0.61 | 0.53 | 0.01 | 2.03 | |||||
| sd | 0.05 | 0.08 | 0.00 | 0.31 | 0.03 | 0.21 | 0.10 | 0.13 | 0.02 | 0.14 | ||||||||||||||||||||||||||||||||||||
| DR08-158 | 925 | 150 | 3 | 40.12 | 3.67 | 11.78 | 15.39 | 0.26 | 11.38 | 11.22 | 2.82 | 0.68 | 97.34 | Mg- hastingsite | 6.01 | 1.99 | 0.00 | 8.00 | 0.09 | 0.41 | 0.00 | 0.53 | 2.54 | 1.40 | 0.03 | 5.00 | 1.80 | 0.20 | 2.00 | 0.62 | 0.13 | 0.75 | 2.00 | 0.00 | 0.00 | 2.00 | 45.47 | 0.65 | 0.57 | 0.04 | 2.08 | |||||
| sd | 0.83 | 0.23 | 0.25 | 0.30 | 0.10 | 0.28 | 0.23 | 0.02 | 0.15 | 0.84 | ||||||||||||||||||||||||||||||||||||
| DR08-159 | 925 | 150 | 3 | 40.63 | 3.68 | 11.43 | 15.42 | 0.36 | 11.21 | 11.07 | 2.89 | 0.69 | 97.38 | Mg- hastingsite | 6.09 | 1.91 | 0.00 | 8.00 | 0.10 | 0.41 | 0.00 | 0.45 | 2.50 | 1.48 | 0.05 | 5.00 | 1.78 | 0.22 | 2.00 | 0.62 | 0.13 | 0.75 | 2.00 | 0.00 | 0.00 | 2.00 | 45.55 | 0.63 | 0.56 | 0.05 | 2.02 | |||||
| sd | 0.16 | 0.14 | 0.06 | 0.55 | 0.13 | 0.11 | 0.30 | 0.10 | 0.09 | 0.29 | ||||||||||||||||||||||||||||||||||||
| DR08-160 | 925 | 150 | 3 | 39.45 | 3.93 | 10.65 | 15.66 | 0.37 | 10.56 | 11.71 | 3.01 | 0.86 | 96.19 | Pargasite | 6.10 | 1.90 | 0.00 | 8.00 | 0.04 | 0.46 | 0.00 | 0.00 | 2.43 | 2.02 | 0.05 | 5.00 | 1.94 | 0.06 | 2.00 | 0.84 | 0.17 | 1.01 | 2.00 | 0.00 | 0.00 | 2.00 | 46.00 | 0.55 | 0.55 | 0.02 | 1.94 | |||||
| sd | 0.38 | 0.11 | 0.38 | 0.43 | 0.09 | 0.43 | 0.62 | 0.04 | 0.10 | 0.76 | ||||||||||||||||||||||||||||||||||||
| DR08-161 | 925 | 150 | 1 | 40.30 | 4.19 | 10.89 | 16.32 | 0.44 | 10.64 | 11.03 | 2.89 | 0.85 | 97.56 | Mg- hastingsite | 6.08 | 1.92 | 0.00 | 8.00 | 0.02 | 0.48 | 0.00 | 0.38 | 2.39 | 1.68 | 0.06 | 5.00 | 1.78 | 0.22 | 2.00 | 0.63 | 0.16 | 0.79 | 2.00 | 0.00 | 0.00 | 2.00 | 45.62 | 0.59 | 0.54 | 0.01 | 1.94 | |||||
| DR08-164 | 925 | 100 | 4 | 39.99 | 4.23 | 11.07 | 14.57 | 0.28 | 11.14 | 11.70 | 2.81 | 0.71 | 96.51 | Pargasite | 6.10 | 1.90 | 0.00 | 8.00 | 0.09 | 0.49 | 0.00 | 0.05 | 2.53 | 1.81 | 0.04 | 5.00 | 1.91 | 0.09 | 2.00 | 0.74 | 0.14 | 0.88 | 2.00 | 0.00 | 0.00 | 2.00 | 45.95 | 0.58 | 0.58 | 0.04 | 1.99 | |||||
| sd | 0.18 | 0.25 | 0.16 | 0.24 | 0.10 | 0.14 | 0.25 | 0.10 | 0.05 | 0.29 | ||||||||||||||||||||||||||||||||||||
| DR08-165 | 925 | 100 | 2 | 39.74 | 4.36 | 10.53 | 14.83 | 0.34 | 10.85 | 12.21 | 2.91 | 0.83 | 96.60 | kaersutite | 6.13 | 1.87 | 0.00 | 8.00 | 0.04 | 0.51 | 0.00 | 0.00 | 2.49 | 1.91 | 0.04 | 5.00 | 2.02 | -0.02 | 2.00 | 0.89 | 0.16 | 1.05 | 2.00 | 0.00 | 0.00 | 2.00 | 46.25 | 0.57 | 0.57 | 0.02 | 1.91 | |||||
| sd | 0.54 | 0.16 | 0.34 | 0.12 | 0.05 | 0.09 | 0.59 | 0.07 | 0.00 | 0.68 | ||||||||||||||||||||||||||||||||||||
| DR08-166 | 925 | 100 | 2 | 39.54 | 4.46 | 10.17 | 14.92 | 0.25 | 10.59 | 12.29 | 2.88 | 0.93 | 96.04 | kaersutite | 6.17 | 1.83 | 0.00 | 8.00 | 0.04 | 0.52 | 0.00 | 0.00 | 2.46 | 1.95 | 0.03 | 5.00 | 2.05 | -0.05 | 2.00 | 0.93 | 0.19 | 1.11 | 2.00 | 0.00 | 0.00 | 2.00 | 46.41 | 0.56 | 0.56 | 0.02 | 1.87 | |||||
| sd | 0.27 | 0.12 | 0.68 | 0.05 | 0.12 | 0.21 | 0.72 | 0.01 | 0.04 | 0.79 |
n: number of analysis.
sd: standard deviation.
FeO*: Total iron reported as Fe2+.
Experimental biotite composition
| Charge | T (°C) | P (MPa) | n | SiO2 | TiO2 | Al2O3 | FeO* | MnO | MgO | CaO | Na2O | K2O | Sum | Mg# |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR08-140 | 950 | 400 | 2 | 35.02 | 3.67 | 14.25 | 11.51 | 0.10 | 15.30 | 0.10 | 1.40 | 7.19 | 88.55 | 70.33 |
| sd | 0.83 | 0.02 | 0.38 | 0.06 | 0.04 | 0.68 | 0.08 | 0.13 | 0.08 | 0.24 | ||||
| DR08-141 | 950 | 400 | 1 | 34.77 | 5.54 | 15.35 | 16.50 | 0.09 | 12.35 | 0.11 | 1.27 | 7.17 | 93.15 | 57.16 |
| DR08-145 | 950 | 400 | 1 | 38.21 | 7.24 | 14.21 | 17.26 | 0.13 | 8.51 | 3.04 | 1.63 | 6.46 | 96.70 | 46.78 |
| DR08-152 | 925 | 400 | 1 | 37.27 | 3.96 | 15.79 | 16.29 | 0.18 | 12.79 | 0.37 | 1.50 | 7.14 | 95.28 | 58.32 |
| DR08-130 | 925 | 200 | 1 | 34.77 | 5.81 | 15.40 | 18.24 | 0.11 | 12.23 | 0.55 | 1.24 | 7.18 | 95.53 | 54.44 |
| DR08-158 | 925 | 150 | 1 | 37.86 | 4.87 | 16.12 | 15.23 | 0.15 | 11.85 | 0.51 | 1.43 | 6.50 | 94.51 | 58.11 |
| DR08-159 | 925 | 150 | 1 | 34.11 | 5.56 | 14.27 | 16.37 | 0.26 | 13.18 | 0.27 | 1.34 | 6.73 | 92.08 | 58.93 |
n: number of analysis.
sd: standard deviation.
FeO*: Total iron reported as Fe2+.
Mg# = 100*(MgO/MgO + FeO*).
Experimental Magnetite and ilmenite compositions (wt%)
| Charge | T (°C) | P (MPa) | n | SiO2 | TiO2 | Al2O3 | FeO* | MnO | MgO | CaO | Na2O | K2O | Cr2O3 | Sum | X-Usp | X-Ilm |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR08-140 | 950 | 400 | 1 | 0.17 | 20.16 | 3.45 | 72.00 | 0.70 | 2.70 | 0.19 | 0.06 | 0.10 | 0.46 | 100 | 0.60 | |
| DR08-141 | 950 | 400 | 1 | 1.80 | 49.68 | 1.82 | 41.63 | 0.78 | 2.26 | 1.41 | 0.33 | 0.21 | 0.09 | 100 | 0.99 | |
| DR08-141 | 950 | 400 | 1 | 0.11 | 21.47 | 4.31 | 71.25 | 0.64 | 2.00 | 0.08 | 0.05 | 0.07 | 0.03 | 100 | 0.67 | |
| DR08-142 | 950 | 400 | 1 | 0.10 | 50.89 | 0.38 | 44.61 | 1.07 | 2.50 | 0.29 | 0.04 | 0.09 | 0.03 | 100 | 0.95 | |
| DR08-142 | 950 | 400 | 1 | 0.12 | 21.43 | 4.59 | 71.06 | 0.66 | 1.92 | 0.15 | 0.03 | 0.01 | 0.04 | 100 | 0.67 | |
| DR08-143 | 950 | 400 | 1 | 0.10 | 21.79 | 4.22 | 71.05 | 0.66 | 1.93 | 0.10 | 0.04 | 0.05 | 0.05 | 100 | 0.68 | |
| DR08-152 | 925 | 400 | 1 | 0.11 | 49.52 | 0.20 | 46.08 | 0.96 | 2.82 | 0.20 | 0.04 | 0.05 | 0.02 | 100 | 0.92 | |
| DR08-152 | 925 | 400 | 1 | 0.03 | 19.91 | 3.07 | 73.05 | 0.95 | 1.94 | 0.11 | 0.06 | 0.07 | 0.81 | 100 | 0.59 | |
| DR08-154 | 925 | 400 | 1 | 0.72 | 21.63 | 3.02 | 70.50 | 1.11 | 1.53 | 0.29 | 0.03 | 0.12 | 1.04 | 100 | 0.67 | |
| DR08-56 | 950 | 200 | 3 | 0.37 | 21.93 | 3.66 | 69.75 | 0.78 | 2.94 | 0.28 | 0.03 | 0.06 | 0.14 | 100 | 0.66 | |
| sd | 0.18 | 0.33 | 0.05 | 0.98 | 0.05 | 0.03 | 0.05 | 0.02 | 0.04 | 0.03 | ||||||
| DR08-58 | 950 | 200 | 1 | 0.33 | 24.63 | 3.20 | 67.79 | 0.78 | 2.74 | 0.33 | 0.00 | 0.15 | 0.00 | 100 | 0.74 | |
| DR08-129 | 925 | 200 | 1 | 0.46 | 20.15 | 3.45 | 71.65 | 0.92 | 2.41 | 0.31 | 0.04 | 0.07 | 0.53 | 100 | 0.61 | |
| DR08-131 | 925 | 200 | 1 | 0.32 | 20.06 | 3.88 | 70.74 | 0.96 | 2.14 | 0.26 | 0.00 | 0.07 | 1.58 | 100 | 0.62 | |
| DR08-158 | 925 | 150 | 1 | 0.71 | 18.75 | 3.57 | 72.26 | 0.79 | 2.37 | 0.92 | 0.07 | 0.03 | 0.53 | 100 | 0.57 | |
| DR08-159 | 925 | 150 | 1 | 0.44 | 18.16 | 3.61 | 73.69 | 0.87 | 2.34 | 0.76 | 0.00 | 0.08 | 0.04 | 100 | 0.54 | |
| DR08-160 | 925 | 150 | 1 | 0.28 | 20.01 | 2.82 | 72.36 | 0.94 | 2.27 | 0.23 | 0.00 | 0.02 | 1.06 | 100 | 0.59 | |
| DR08-161 | 925 | 150 | 1 | 0.11 | 21.76 | 2.27 | 70.29 | 1.13 | 2.21 | 0.20 | 0.04 | 0.07 | 1.91 | 100 | 0.63 | |
| DR08-165 | 925 | 100 | 1 | 1.89 | 18.78 | 3.22 | 69.89 | 0.98 | 2.41 | 0.34 | 0.20 | 0.15 | 2.14 | 100 | 0.61 | |
| DR08-165 | 925 | 100 | 1 | 0.06 | 51.45 | 0.18 | 41.95 | 1.57 | 4.58 | 0.18 | 0.00 | 0.02 | 0.02 | 100 | 0.94 | |
| DR08-166 | 925 | 100 | 1 | 1.67 | 19.18 | 2.92 | 70.17 | 0.94 | 2.14 | 0.38 | 0.17 | 0.14 | 2.29 | 100 | 0.61 | |
| DR08-166 | 925 | 100 | 1 | 0.17 | 50.35 | 0.18 | 43.95 | 1.47 | 3.50 | 0.26 | 0.05 | 0.07 | 0.00 | 100 | 0.93 |
Experimental clinopyroxenes composition (wt%)
| Charge | T (°C) | P (MPa) | n | SiO2 | TiO2 | Al2O3 | FeO* | MnO | MgO | CaO | Na2O | K2O | Total | En | Fs | Wo | Mg# |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR08-145 | 950 | 400 | 1 | 47.78 | 1.45 | 4.03 | 10.89 | 0.29 | 10.73 | 20.54 | 0.95 | 0.37 | 97.03 | 33.77 | 19.23 | 46.47 | 63.72 |
| DR08-160 | 925 | 150 | 3 | 49.09 | 0.95 | 2.96 | 11.78 | 0.62 | 11.71 | 20.76 | 0.72 | 0.08 | 98.65 | 34.86 | 19.67 | 44.43 | 63.94 |
| 0.60 | 0.19 | 0.16 | 0.57 | 0.09 | 0.13 | 0.19 | 0.09 | 0.04 | 0.66 | 0.15 | 0.70 | 0.82 | 0.85 |
n: number of analysis.
sd: standard deviation.
FeO*: Total iron reported as Fe2+.
Composition of experimental glasses normalized to 100% anhydrous (wt%)
| Charge | T (°C) | P (MPa) | n | SiO2 | TiO2 | Al2O3 | FeO* | MnO | MgO | CaO | Na2O | K2O | P2O5 | Total | Original sum | P.I. = (Na + K/Al) | Normative Nepheline (wt%) |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DR08-140 | 950 | 400 | 5 | 58.57 | 0.99 | 18.37 | 6.83 | 0.17 | 1.61 | 4.33 | 5.98 | 2.75 | 0.39 | 100 | 85.72 | 0.70 | |
| sd | 0.42 | 0.08 | 0.25 | 0.22 | 0.07 | 0.08 | 0.07 | 0.56 | 0.15 | 0.07 | 0.53 | ||||||
| DR08-141 | 950 | 400 | 7 | 59.95 | 0.68 | 19.60 | 5.71 | 0.15 | 1.00 | 2.90 | 6.35 | 3.24 | 0.43 | 100 | 93.86 | 0.72 | |
| sd | 0.58 | 0.08 | 0.10 | 0.25 | 0.08 | 0.03 | 0.03 | 0.34 | 0.10 | 0.08 | 0.78 | ||||||
| DR08-142 | 950 | 400 | 3 | 60.50 | 0.68 | 19.97 | 6.13 | 0.15 | 1.00 | 2.90 | 6.53 | 3.24 | 0.19 | 100 | 95.13 | 0.72 | |
| sd | 0.61 | 0.18 | 0.18 | 0.21 | 0.06 | 0.20 | 0.04 | 0.29 | 0.07 | 0.01 | 0.83 | ||||||
| DR08-143 | 950 | 400 | 1 | 60.68 | 0.51 | 18.36 | 4.79 | 0.15 | 0.58 | 1.77 | 7.60 | 5.24 | 0.32 | 100 | 88.50 | 1.00 | 7.2 |
| sd | |||||||||||||||||
| DR08-152 | 925 | 400 | 3 | 60.41 | 0.78 | 18.98 | 5.66 | 0.15 | 0.97 | 3.71 | 6.34 | 2.59 | 0.42 | 100 | 88.08 | 0.70 | |
| sd | 0.34 | 0.13 | 0.23 | 0.46 | 0.11 | 0.16 | 0.06 | 0.18 | 0.05 | 0.20 | 0.36 | ||||||
| DR08-154 | 925 | 400 | 1 | 62.53 | 0.42 | 18.20 | 5.15 | 0.03 | 0.51 | 1.83 | 7.40 | 3.54 | 0.39 | 100 | 95.23 | 0.88 | |
| DR08-56 | 950 | 200 | 2 | 58.01 | 0.95 | 18.59 | 7.08 | 0.29 | 1.50 | 3.87 | 6.25 | 3.19 | 0.30 | 100 | 92.90 | 0.74 | |
| sd | 0.44 | 0.09 | 0.09 | 0.21 | 0.01 | 0.07 | 0.01 | 0.16 | 0.03 | 0.02 | 0.93 | ||||||
| DR08-58 | 950 | 200 | 2 | 58.58 | 0.77 | 18.50 | 7.41 | 0.22 | 0.86 | 2.57 | 6.45 | 4.22 | 0.41 | 100 | 94.58 | 0.83 | 0.1 |
| sd | 0.53 | 0.05 | 0.07 | 0.37 | 0.11 | 0.00 | 0.07 | 0.08 | 0.09 | 0.15 | 0.28 | ||||||
| DR08-129 | 925 | 200 | 2 | 58.84 | 0.62 | 18.71 | 6.96 | 0.18 | 0.73 | 3.47 | 6.40 | 3.16 | 0.91 | 100 | 94.39 | 0.75 | |
| sd | 0.09 | 0.04 | 0.01 | 0.59 | 0.01 | 0.07 | 0.15 | 0.06 | 0.23 | 0.09 | 0.34 | ||||||
| DR08-130 | 925 | 200 | 2 | 59.00 | 1.09 | 18.96 | 6.55 | 0.17 | 0.67 | 3.16 | 6.32 | 3.24 | 0.83 | 100 | 93.87 | 0.74 | |
| sd | 0.38 | 0.71 | 0.50 | 0.02 | 0.11 | 0.01 | 0.01 | 0.16 | 0.03 | 0.22 | 1.34 | ||||||
| DR08-131 | 925 | 200 | 1 | 59.23 | 0.49 | 18.39 | 7.16 | 0.05 | 0.49 | 1.76 | 7.30 | 4.96 | 0.16 | 100 | 97.57 | 0.95 | 6.4 |
| DR08-158 | 925 | 150 | 9 | 60.78 | 0.68 | 19.29 | 5.02 | 0.16 | 1.01 | 3.04 | 6.51 | 3.21 | 0.31 | 100 | 92.67 | 0.74 | |
| sd | 0.34 | 0.09 | 0.34 | 0.21 | 0.08 | 0.03 | 0.07 | 0.28 | 0.14 | 0.05 | 0.68 | ||||||
| DR08-159 | 925 | 150 | 9 | 60.81 | 0.74 | 19.18 | 4.82 | 0.18 | 0.92 | 2.78 | 6.77 | 3.47 | 0.33 | 100 | 93.44 | 0.78 | |
| sd | 0.56 | 0.07 | 0.25 | 0.56 | 0.05 | 0.10 | 0.14 | 0.20 | 0.10 | 0.08 | 0.47 | ||||||
| DR08-160 | 925 | 150 | 2 | 60.52 | 0.63 | 19.25 | 4.92 | 0.16 | 0.80 | 2.73 | 6.75 | 3.80 | 0.44 | 100 | 95.98 | 0.79 | |
| sd | 0.82 | 0.04 | 0.13 | 0.10 | 0.15 | 0.06 | 0.68 | 0.02 | 0.28 | 0.45 | 1.23 | ||||||
| DR08-164 | 925 | 100 | 3 | 61.71 | 0.53 | 18.63 | 5.00 | 0.11 | 0.73 | 2.28 | 6.71 | 3.99 | 0.31 | 100 | 95.89 | 0.83 | |
| sd | 0.03 | 0.03 | 0.23 | 0.15 | 0.04 | 0.05 | 0.11 | 0.17 | 0.18 | 0.07 | 0.94 | ||||||
| DR08-165 | 925 | 100 | 2 | 61.81 | 0.59 | 19.39 | 5.02 | 0.00 | 0.74 | 2.10 | 6.44 | 3.78 | 0.14 | 100 | 94.98 | 0.76 | |
| sd | 0.07 | 0.04 | 0.45 | 0.18 | 0.00 | 0.02 | 0.02 | 0.13 | 0.25 | 0.15 | 0.35 |
n: number of analysis.
sd: standard deviation.
FeO*: Total iron reported as Fe2+.
P.I: peralkaline index calculated as the (Na + K)/Al in moles.
A typical experiment contained five or six capsules, each loaded with a different H2O∕CO2 ratio. Run duration varied between 64 and 74 h, depending on temperature. Experiments were terminated by using the drop quench device and then switching off the power supply. After the experiments, capsules were checked for leaks, opened, and half of the run product was embedded in a probe mount with an epoxy resin and polished for optical observation, and carbon coated for subsequent EMPA and scanning electron microscopy (SEM) characterisation.
3.4. Water content, fH2, fO2 in the capsules
The variation of the XH2Oin in the capsules allowed us to explore different water fugacities, and thus different H2Omelt (Table 2). The amount of dissolved water (XH2Oin) in the glass of the different H2O-saturated charges was calculated using the H2O-solubility model of Jiménez-Mejías et al. [2021] which is suited for basanitic–phonotephritic compositions. The water contents of charges with XH2Oin < 1 ran at the same temperature and pressure than the H2O-saturated charge, were calculated by mass balance using the constraint of equilibrium fugacities of H2O and CO2 species between melt and fluid, along with the H2O–CO2 solubility model of Jiménez-Mejías et al. [2021].
The fH2 of the experiments was determined using an empirical calibration curve established in previous experimental works, relating the H2 pressure loaded initially to the autoclave at room temperature to the final fH2 as determined using NiPd sensors run at similar P–T–fO2 [Andújar and Scaillet 2012, Andújar et al. 2015, Romano et al. 2018]. Once the prevailing fH2 is known, the fO2 can be determined by using the dissociation constant of water [Robie et al. 1979], knowing the fH2O at the experimental temperature and pressure. fH2O was calculated as fH2O = XH2Oin ∗ fH2Oo where fH2Oo is the fugacity of pure water [Burnham et al. 1969]. Afterwards, the fO2 for each capsule was calculated (Table 2) (see Andújar and Scaillet [2012] for further details). The experiments were conducted at oxygen fugacities around NNO ± 0.3 at H2O-saturation. As fO2 decreases with XH2Oin [e.g., Andújar and Scaillet 2012, Freise et al. 2009, Scaillet et al. 1995, Webster et al. 1987], at fixed T, P, fH2, for water-undersaturated charges the prevailing oxygen fugacity is necessarily <NNO ± 0.3. As a result, in a single experimental series, the decrease in XH2Oin from 1 to 0.08 decreased the fO2 up to about 2.5 log units below the NNO buffer (or ∼1.8 log units below the Fayalite-Magnetite-Quarz buffer, FMQ; Table 2).
3.5. Analytical techniques
Experimental runs were first characterized using a Zeiss Merlin Compact electron microscope equipped with an EDS micro-analysis system (Bruker-Quantax-XFlash6) at ISTO. EDS spectra allowed a first order identification of the minerals present in each charge. Experimental phases were analyzed using a Cameca SX-Five EPMA with an acceleration voltage of 15 kV, a sample current of 6 nA, and a counting time of 10 s. For glasses, a defocused beam of 20 μm was used whereas for minerals a focused beam was employed instead. Sodium and potassium were analyzed first and a Phi-Rho-Z correction procedure was applied. Major elements calibration was done using the following standards: albite (Na, Si), orthoclase (K), andradite (Ca), apatite (P), chromite (Cr), corundum (Al), magnesium oxide (Mg), hematite (Fe) and pyrophanite (Mn, Ti). The relative analytical errors are 1% (SiO2, Al2O3, CaO), 3% (FeO, MgO, TiO2) and 5% (MnO, Na2O, K2O, P2O5).
In the case of experimental plagioclase (Pl), the small size of this phase in the charges (usually ⩽3 μm) along with the presence of small (⩽0.5 μm) Fe–Ti oxides-apatite inclusions inside the crystals, made difficult the proper analysis of this phase with EPMA. To circumvent this problem, Pl composition was obtained via using a SEM-EDS calibrated analysis: albite, orthose and andradite EMPA standards were analyzed first with a focused beam (1 μm), a voltage of 15 kV and counting times of 15 s for the different elements (Na, K, Si, Al, Ca, Na, K). SEM-EDS compositions of the standards were found to be within ⩽5% to those obtained by EMPA (see Supplementary Table). Consequently, experimental plagioclases were determined by SEM-EDS in each charge, using the same conditions and the average SEM-EDS calibrated compositions fulfilling the stoichiometric requirements for this phase (see Supplementary Table).
It should be noted that, compared to EMPA analyses, which are characterized by lower standard deviations (see Tables 3–8), SEM-EDS compositions are slightly more variable (see Supplementary Table), with a variation of ∼2–3 mol% on end-member molecules. However, despite this, we can still use these compositions for comparison purposes.
3.6. Attainment of equilibrium
The crystallization experiments performed in this work share similar phase stabilities, textural and compositional characteristics to those found in equivalent studies performed on alkali-rich basalts [e.g., Andújar et al. 2015, Freise et al. 2009, Iacovino et al. 2016] using a similar procedure. As in these works, the following observations show that near-equilibrium conditions were attained in our experiments: the euhedral shape of crystals, the homogeneous distribution of phases within the charges, the smooth variation of phase proportions and compositions with changes in experimental conditions, and the small sum of residuals of mass-balance (generally ⩽1; Table 2). Since each experimental charge is a closed-system, the low-residuals indicate that there was no mass loss or gain during the experiments, and that no major mineral phase has been omitted in our calculations.
4. Results
Phase proportions were obtained by mass-balance calculations of the charges using the bulk rock composition and the composition of the different mineral and glass phases. Results are provided in Table 2.
4.1. Phase relationships
The main mineral phases crystallizing in our runs are Bt (biotite), Amp (Kaersutite-type), Ol, Mt, Ilm, Ap, Pl and Cpx, with different glass proportions. Variations in temperature, pressure and H2Omelt directly affect the stability and relationships of the different minerals. These changes are displayed in two isothermal-polybaric sections described below (Figure 3).
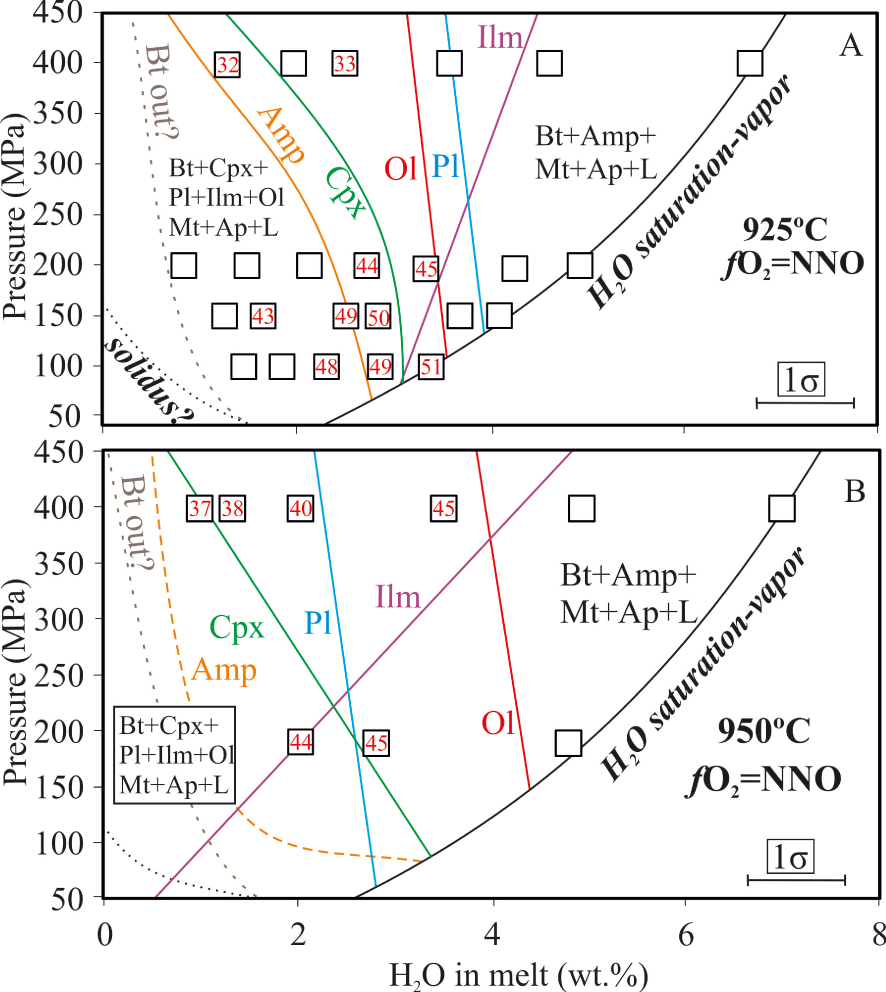
Isothermal phase relationships of the DR08 sample at (A) 925 °C and (B) 950 °C for different pressures and water contents in the melt. fO2 ∼ NNO: oxygen fugacity at water-saturation conditions. The decrease in XH2O (hence H2Omelt) results in a decrease in fO2 in the capsules (see text for details). Mt: magnetite, Bt: biotite, Cpx: clinopyroxene, Ilm: ilmenite, Amp: amphibole, Pl: plagioclase, Ap: apatite; Ol: olivine, L: liquid. Dashed lines are estimated phase boundaries. Numbers inside squares indicate the forsterite content (Fo) in moles of the crystallizing olivines (see text for details). Masquer
Isothermal phase relationships of the DR08 sample at (A) 925 °C and (B) 950 °C for different pressures and water contents in the melt. fO2 ∼ NNO: oxygen fugacity at water-saturation conditions. The decrease in XH2O (hence H2Omelt) results in a ... Lire la suite
At 925 °C, the water-rich part of the diagram (H2Omelt > 4 wt%) is dominated by the assemblage Bt+Amp+Mt+Ap+melt, followed by Ilm, Pl and Cpx with decreasing H2Omelt (Figure 3A). Cpx coexists in a relatively narrow H2Omelt region with Amp, both being involved along with Pl and melt, in a reaction-relationship that results in the disappearance of Amp at lower H2Omelt (Figure 3A). Bt is stable everywhere in the P–H2Omelt space explored. However, the decrease of its modal proportion with H2Omelt (Table 2) and the consequent enrichment of the residual liquid in K2O point towards its instability in the driest part of the system (Figure 3A). Hence, at 925 °C and for H2Omelt < 1 wt%, the mineral assemblage that dominates the explored P-H2Omelt region is Ol+Cpx+Pl+Ap+Mt+Ilm.
At this temperature and for pressures between 100 and 200 MPa, the Amp-out and Cpx, Ol, Ilm and Pl-in curves display steep or even vertical slopes, all occurring in a narrow H2Omelt interval between 2.5–3 wt% (Figure 3A). Above 200 MPa, Cpx-in and Amp-out curves shrink towards lower H2Omelt (⩽2 wt%) whereas Ilm stability shifts towards higher H2Omelt. Pressure does not have any major effect on Ol and Pl saturation curves.
At 950 °C, the general topology is broadly similar to that at 925 °C (Figure 3B), with Pl and Cpx appearing at H2Omelt < 2–3 wt%, as anticipated, and Ol crystallizing first relative to Pl.
4.2. Phase proportions
Since experiments were performed at relatively low temperatures, liquidus conditions were not reached during the experiments. Charges always contained different amounts of crystals, which vary between 29 and 66 wt% (Figure 4A). In general, at fixed P–T conditions, the crystal proportions increase as H2Omelt decreases (Figure 4A). Amp is the dominant phase whenever present, with proportions of up to 50–70 wt% of the crystallizing assemblage (Figure 4B); as H2Omelt decreases, the Amp content decreases below 15 wt%. With decreasing H2Omelt, Mt, Bt and Ap proportions steadily decrease while Ol, Ilm, Cpx and Pl increase. However, compared to Amp, any of these phases never exceed 15–25 wt% (Table 2).
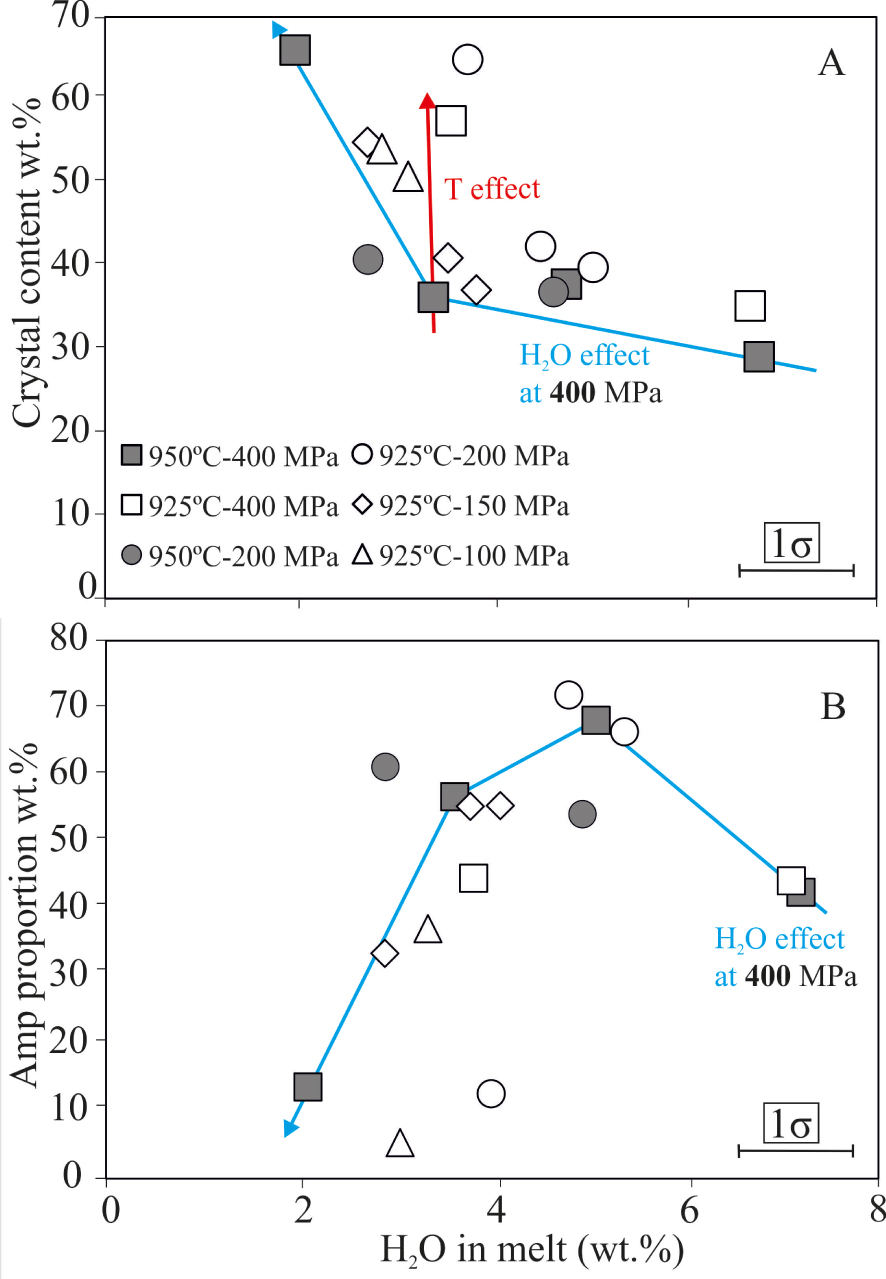
(A) Total crystal content of charges versus H2Omelt. (B) Amph proportion in the fractionating crystal assemblage versus H2Omelt. Vertical errors equal to symbol size.
4.3. Phase compositions
Experimental phase compositions (minerals and glass) are displayed in Tables 3–9, in supplementary and their variation with experimental variables (P–T and H2Omelt) is discussed in the following sections.
4.3.1. Olivine
Ol crystallizing in our charges varies between Fo51−33, with relatively constant contents of MnO (1.4 ± 0.2 wt%), depending on H2Omelt, temperature and pressure conditions (Table 3). At a given P and T, a decrease in H2Omelt decreases the Fo and increases the CaO content of Ol, similarly to the trends observed in other basaltic compositions [Andújar et al. 2015, 2017, Berndt et al. 2005, Di Carlo et al. 2006]. This is well exemplified at 400 MPa and 950 °C where a decrease in H2Omelt of 2.5 wt% produces a diminution of 8 mol% of Fo (Figure 5A) and an increase of ∼0.5 wt% in the CaO content of Ol (Figure 5B). A drop of 25 °C results in a relatively modest variation of Fo content, generally <5 mol% (Figure 5A). Series conducted at 925 °C allow to assess the effect of pressure on Ol composition. Considering a fixed H2Omelt (e.g. 2.5–3 wt%), runs conducted between 100–150 MPa crystallized Ol with a similar composition (e.g. ∼Fo50). Increasing P by 50 MPa (so up to 200 MPa) produces changes in Ol composition that are equivalent to those observed with a T diminution (i.e., decrease of 8 mol% in Fo; Figure 5A). At higher pressures, the effect of this parameter in olivine composition is more pronounced, since at 400 MPa, crystallizing Ol are highly more fayalitic (Fo33) compared to their shallower counterparts (e.g., Fo45−50 P⩽200 MPa). The average Ol–liquid exchange (KdFe*–Mg; using FeO* as total iron) is 0.29 ± 0.1 (varying between 0.25 and 0.44).
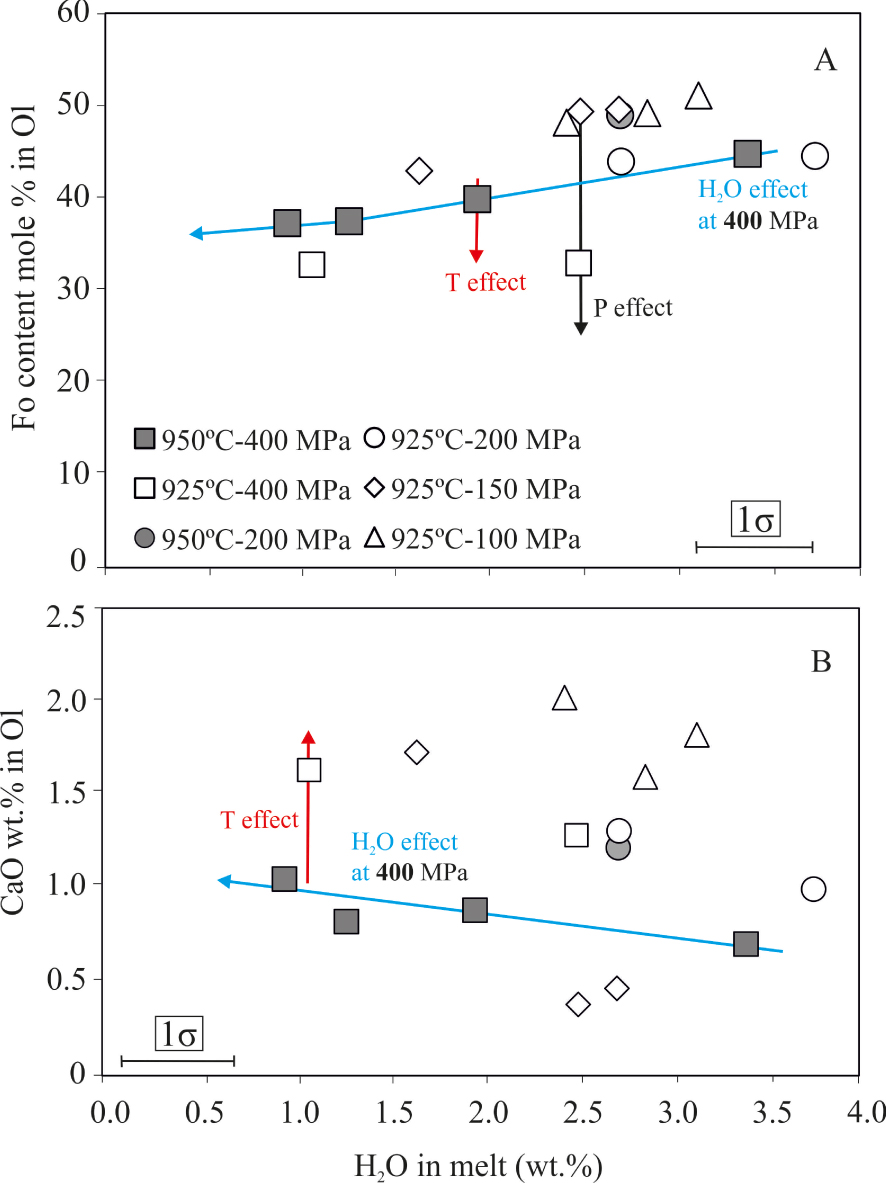
Composition of experimental olivine. (A) Fo content, (B) CaO (wt%) in Ol versus H2Omelt. Vertical errors are equal to size symbols.
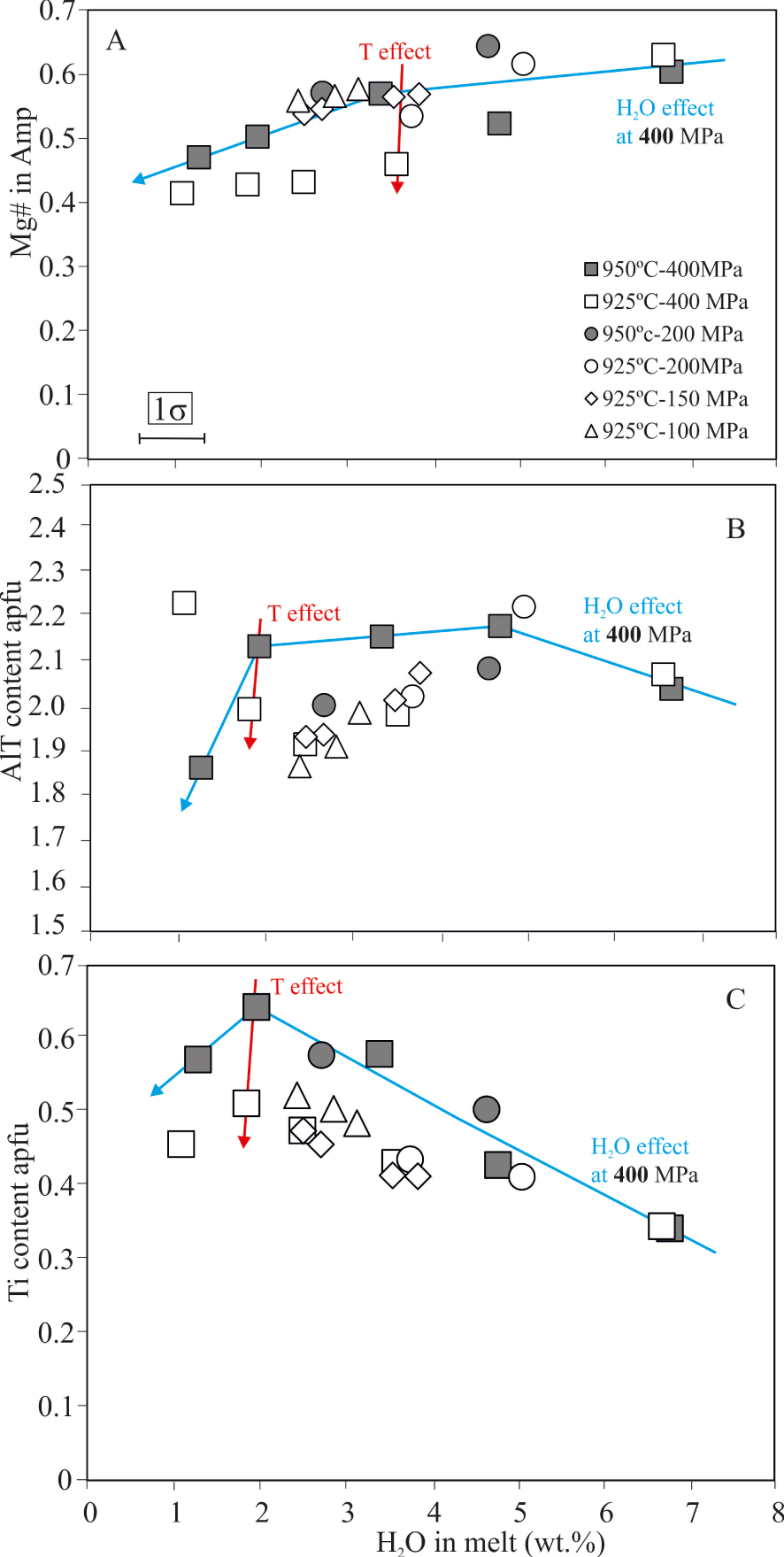
Composition of experimental amphibole. (A) Al total cations in atoms per formula unit (apfu), (B) Ti content and (C) Mg# = (Mg∕Mg + Fetotal) versus H2Omelt. Vertical errors equal to symbol size.
4.3.2. Amphibole
Based on Ridolfi [2021], Amp that were synthetized in our runs vary between pargasite, Mg-hastingsite and kaersutite (Table 4). At a fixed P, amphibole Mg#(calculated with Fe* in atoms per formula unit, apfu), Altot and AlIV decrease by ∼20% relative (Figure 6A,B), while Ti increases, as H2Omelt decreases (Figure 6C; Table 4). In series run at P⩽200 MPa, a decrease of 25 °C does not significantly affect the composition of Amp. The variations of Altot, AlIV and AlVI with temperature and pressure have been widely used as a basis of empirical thermometers and barometers to infer crystallization conditions of Amp-bearing magmas. In this work, however, neither AlIV nor AlVI show obvious correlations with pressure/temperature changes. Only Amp crystallized at 950 °C, 2–5 wt% H2Omelt and 400 MPa have higher Altot contents (0.1–0.2 apfu) than their 200 MPa counterparts (Figure 6B).
4.3.3. Biotite
Representative analyses are presented in Table 5. The Mg#(calculated with FeO*) varies between 70 and 47 when H2Omelt decreases from 7 to 1 wt%. TiO2 content is inversely correlated with water content, the maximum (7 wt%) being achieved at ∼1 wt% H2Omelt and conditions of 950 °C–400 MPa. Water-saturated charges (DR08-140 and DR08-152) at 400 MPa show a difference of 10% in Mg#when T is decreased by 25 °C. Otherwise, under water-rich conditions and at constant T (e.g. 925 °C), the decompression of the system from 400 to 200 MPa produces Ti-rich biotites. The limited set of data does not allow us to provide further constraints on the effect of pressure or temperature on Bt composition.
4.3.4. Fe–Ti oxides
Mt has a relatively constant TiO2 content at ∼22 ± 2.5 wt%, with more variable FeO* (68–74 wt%). When present, Ilm was often too small to obtain reliable compositions by EPMA. Nevertheless, analyzed Ilm in 5 charges have almost constant TiO2, at 50 wt%, and FeO* contents in the range 42–46 wt% (Tables 6–7). As for other phases, changes in experimental parameters affect Mt and Ilm compositions. The effect of H2Omelt is significant: for example, in charges annealed at 950 °C–400 MPa, a decrease of 4 wt% H2Omelt decreases Mg#by ∼4% (Figure 7A), FeO* by 1.5 wt% and increases %Ulvöspinel from 60 to 70% (Figure 7B). The other experimental series display quite similar trends (Figure 7). A decrease in temperature of 25 °C produces similar changes. As for olivine, the effect of pressure on Mt composition is enhanced within the interval 200–400 MPa, low pressure Mt being enriched in Mg#and depleted in FeO* and %Ulvöspinel (compare series run at 950 °C–400 and 200 MPa in Figure 7).
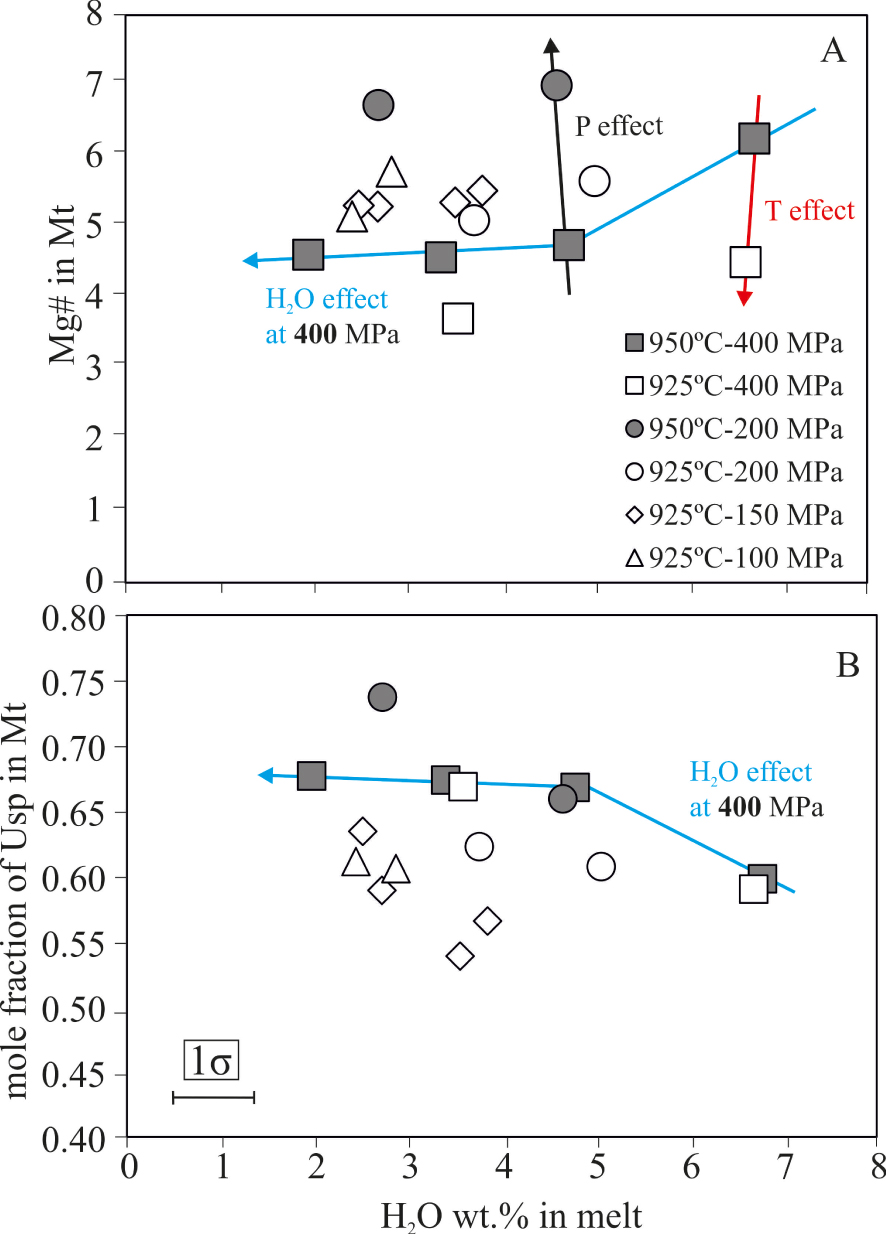
Composition of experimental magnetite. (A) Mg# = (MgO/MgO + FeO*), (B) Ulvöspinel content [calculated as in Andersen and Lindsley 1998] versus H2Omelt. Vertical errors equal to symbol size.
4.3.5. Plagioclase
Pl are mostly andesines with compositions between An27–An42 [Deer et al. 1972]. The most evolved Pl (oligoclase) crystallized instead in charges DR08-132, 155 and 161. As already observed in previous experimental studies performed on hydrous basaltic or basaltic andesite melts, Pl composition is highly sensitive to changes in H2Omelt [e.g. Andújar et al. 2015, 2017]. The most important compositional variations are observed in charges run at 925 °C and P⩽150 MPa, where the decrease in 2 wt% H2Omelt results in a reduction of 10% in the mole fraction of An (see Supplementary Table). Similar variations in the CaO content of the experimental plagioclases can be produced when pressure decreases from 200 to 150 or 100 MPa at a given H2Omelt, an effect that is again in agreement with the results of previous works [e.g. Andújar et al. 2015, 2017].
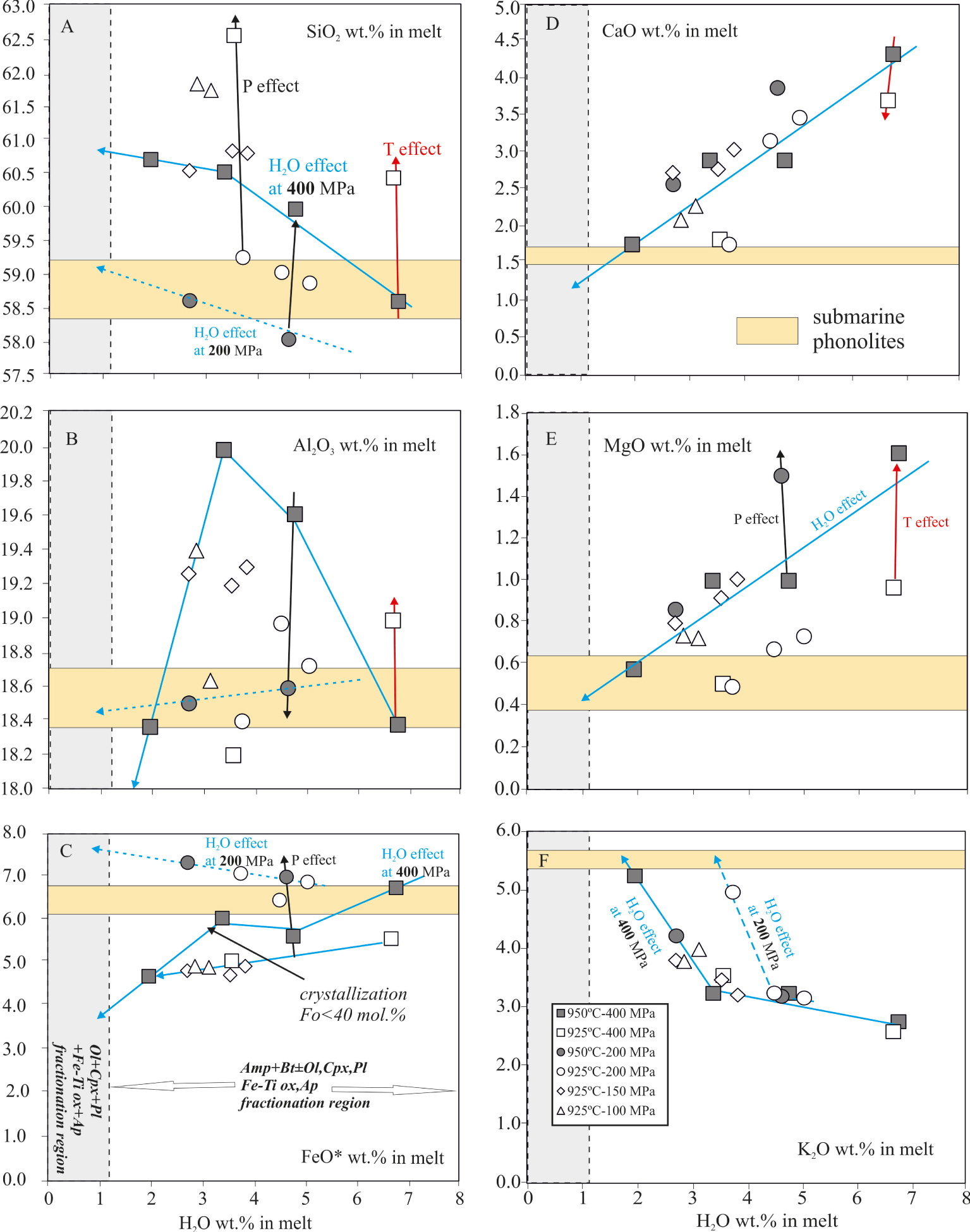
Experimental glass compositional variations of major and minor oxides versus H2Omelt. For each graph, the yellow horizontal bar shows the natural compositions of phonolites dredged on the Horseshoe site (Table 1). Grey vertical bar shows the fractionation region for Ol+Cpx+Mt+Ilm+Ap (see text for details). Vertical errors equal the symbol sizes.
4.3.6. Experimental glasses
Residual glass compositions (calculated on a 100% anhydrous basis) have benmoreitic–trachytic affinities (according to the classification of Le Bas and Streckeisen [1991] with P.I. (peralkaline indice, =Na+K/Al in moles) at 0.7–1 (Figures 8–9, Table 9). Only charges DR08-143 and DR08-131 produced liquids that straddle the phonolite–trachyte boundary line (Figure 9), having a P.I. = 0.95–1, normative nepheline (6.5–7%) and containing 2–4 wt% H2Omelt (Table 8). The production of benmoreitic–trachytic–phonolitic liquids reflects the significant degree of crystallization of the charges. In the range of temperatures explored in this work, crystallization is dominated by Amp+Bt along with different proportions of Fe–Ti oxides+Ap for H2Omelt in the range 2–7 wt% (Figures 3–4, Table 2). The onset of Pl+Ol+Cpx crystallization results in a drastic decrease of Mt, Amp and Bt proportions until the breakdown of the two last phases which occurs at ⩽2 wt% H2Omelt (Figure 3). The combination of the above factors increases both the SiO2 and the alkalinity of the residual melt, which reaches the phonolitic field (Figure 9A). In parallel, TiO2 (not shown), CaO and MgO decrease (Figure 8B,D). In contrast, FeO* may even slightly increase when H2Omelt decreases (Figure 8C), though by a modest amount (usually <0.5 wt%). This Fe-enrichment is due to the changing proportions of hydrous Fe–Mg phases and Mt (Table 2). In comparison, a decrease of 200 MPa lowers FeO* by ∼1.5–2 wt% for a given H2Omelt (Figure 8C). Below 3 wt% H2Omelt, the liquid produced at 400 MPa are even more depleted in FeO* (Figure 8C). This is due to the fact that, at 200 MPa, the precipitation of a relatively Fe-poor Ol (Fo45−40) enhances the iron-enrichment process. In contrast, at 400 MPa, the crystallization of significant amounts of relatively fayalitic-rich Ol (e.g., Fo⩽38) counteracts the depleting role of Mt at higher H2Omelt (Figures 3, 8C). Such a difference explains why charges at 200 MPa yield melts with the highest analyzed FeO* contents (7.4 wt%) compared to their high-pressure counterparts, which achieved only a maximum of 6.7 wt% FeO*, and under very high H2Omelt.
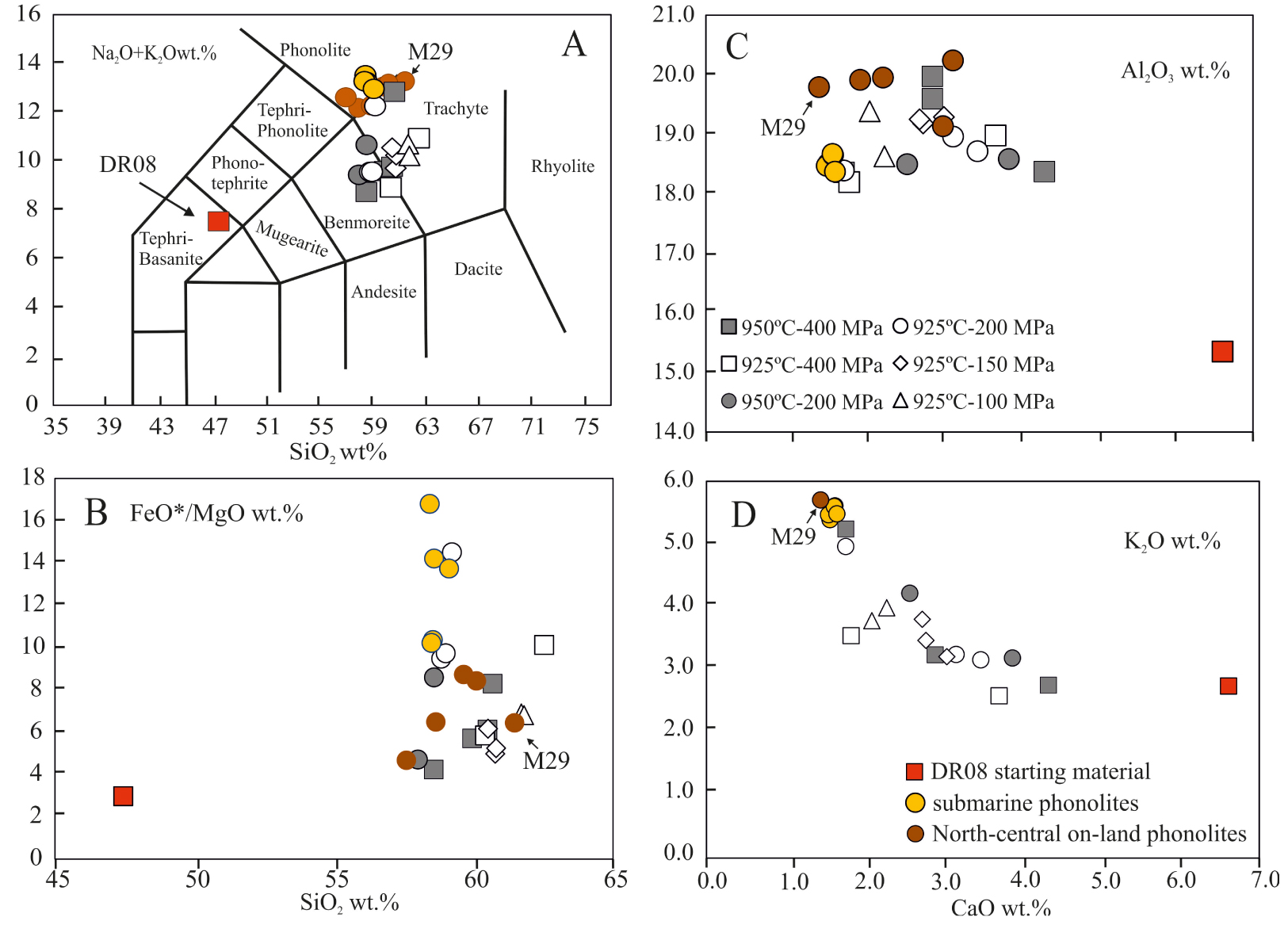
(A) Total alkalis (Na2O + K2O) versus SiO2 diagram [after Le Bas and Streckeisen 1991] showing the composition of the experimental glasses, the DR08 starting material, and onland/offshore phonolites at Mayotte (Table 1). (B) FeO*/MgO versus. SiO2 wt%, (C) Al2O3 versus CaO wt%, (D) K2O versus CaO wt%. Legend as in previous figures. Vertical errors are equal to symbol size.
Another striking point concerns the charges annealed at 100–150 MPa since these yield residual melt compositions overlaping those produced at higher P (⩾200 MPa), not only in terms of iron but also in the other major elements (Figure 8). In fact, despite the presence of moderate proportions of olivine with compositions Fo51−43 (Figure 5A, Table 3), which according to 200 MPa data promotes the Fe-enrichment of the melt, the co-crystallization of ∼8 wt% of Fe-rich magnetite and different amounts of Cpx+Pl±Bt±Amp inhibit the Fe-enrichment process and drive the residual melt towards more Si- and Al-rich and low P.I. compositions [Figures 8–9; Table 8; Botcharnikov et al. 2008, Giehl et al. 2013, Scaillet and MacDonald 2006, Toplis and Carroll 1995]. The effect of P variation is not restricted to FeO*; as pressure increases, the SiO2, CaO increase with decreasing Al2O3 content, MgO and alkalis being somewhat less affected (Figure 8).
It should be also noted that between 7 and 3 wt% H2Omelt, the precipitation of Amp along with Bt in amounts, representing 60–80% of the total crystal cargo, increases the SiO2 and Al2O3 content but buffers K2O at ∼3 wt% (Figure 8F). Below this H2Omelt, the crystallization of Ol+Cpx+Pl, along with a drastic drop of Amp+Bt+Mt proportions, increases the potassium content of the melt by ∼50% relative (Figure 8F). Na2O follows a similar trend but the observed enrichment is less significant compared to K2O. The evolution of Al2O3 is well illustrated in series run at 200–400 MPa where Al2O3 increases when H2Omelt decreases until the onset of Pl crystallization (Figure 8B). Pl crystallization in relative high proportions (up to ∼30 wt%; Table 2) decreases the Al2O3 and accelerates the evolution of the residual melts towards alkali-rich compositions.
Aside from water and pressure, temperature is also an important parameter controlling melt composition. At lower temperatures (i.e. 925 °C) and for a fixed H2Omelt, residual liquids tend to be enriched in SiO2 and Na2O but depleted in TiO2, MgO and CaO when compared to melts produced at 950 °C. FeO*, K2O and Al2O3 do not show any clear relationship with T in charges run at P⩽200 MPa. Only those at 400 MPa and 950 °C have higher Al2O3 and FeO compared to their colder counterparts (Figure 8).
In summary, the fractionation of a relatively hydrous basanite under low oxygen fugacity conditions (fO2⩽ FMQ; Table 2), at temperatures between 925–950 °C and 200–400 MPa (corresponding to depths of 6–12 km) produces Fe-rich phonolitic liquids containing 2–4 wt% dissolved H2O. In the next section, we use the composition of the natural phonolites dredged along the Mayotte submarine ridge or collected on-land to shed light on the conditions needed to generate this type of evolved melts at Mayotte.
5. Discussion
As stated in the previous sections, most of the seismo-volcanic activity recorded during the 2018–2021 eruptive episode (and up to date) occurred along the 60 km long submarine volcanic ridge located eastward of Mayotte (Figure 1B). Dredged samples along this ridge define a bimodal distribution of basanites and phonolites [Berthod et al. 2021a, b], phonolites mostly outcropping in the Horseshoe region (Figure 1). Submarine mafic magmas are chemically and petrologically similar to the on-land counterparts from the moderately silica-undersaturated Comoros chemical trend [Bachèleryet al. 2016, Berthod et al. 2021b, Pelleter et al. 2014]. Dredged phonolites have major and trace elements that are also compatible with this magmatic lineage. However, when compared to their on-shore equivalents, they are slightly enriched in Fe–Mg–Ca and have lower Al-total alkalis content, both akin to the somewhat less evolved character of these magmas [Pelleter et al. 2014]. This is also reflected by differences between submarine and on-shore phonolites in their mineralogy. Whereas offshore phonolites are moderate porphyritic magmas with small crystals (⩽200 μm) of fayalitic Ol (Fo ⩽ 32 mol%), Pl (An ⩽ 24 mol%), Ap and Fe–Ti oxides microcrystals [Ox; Berthod et al. 2021b], those on land are characterized by the presence of an amphibole-bearing assemblage consisting of Cpx+Amp+Afs+Ap+Ox±nepheline (Nph) [Debeuf 2004, Pelleter et al. 2014]. It is worth noting that Ol-bearing phonolites are not restricted to the submarine domain since they were also identified onshore at Mayotte [Berthod et al. 2021b, Debeuf 2004]. Based on our experimental results, the differences in phase assemblage and composition observed between these two groups can potentially be related to changes in the evolution conditions of the parental basanitic magma (P–T–H2O), as we discuss below.
5.1. Production of phonolitic liquids at the WNW–ESE submarine volcanic ridge of Mayotte
The low porphyritic character of the submarine phonolites dredged in the vicinity of the active Horseshoe structure of Mayotte [Berthod et al. 2021a, b] allows us to consider the bulk-rock composition as representative of liquids. Accordingly, the experimental results detailed above are useful for understanding the conditions for the production of phonolitic melts at this site (Figures 8–9, Tables 1 and 8). Although glasses produced at conditions of 925 °C–200 MPa and 950 °C–400 MPa with 2–7 wt% H2Omelt broadly reproduce the SiO2, Al2O3, MgO and CaO contents of natural phonolites (Figure 8), only the residual glasses of charges DR08-143 (950 °C–400 MPa; ∼2 wt% H2Omelt) and DR08-131 (925 °C–200 MPa; ∼4 wt% H2Omelt) closely match the phonolitic affinities (P.I. ∼ 0.9–1) of the natural products (Figure 9A; Table 1). Both charges have the same mineral assemblage and phase proportions (∼65 wt% of Ol+Pl+Amp+Mt+Ilm+Ap±Bt; Table 2), but differ in the composition of the phases (see above). In detail, only the 200 MPa experiment closely reproduces the FeO* and SiO2 contents (and other oxides) as well as the FeO*/MgO ratios (Figure 9B). The glass produced at 400 MPa has a FeO* content ∼2 wt% lower and a higher SiO2 with respect to the dredged phonolites (Figure 9B). Our data therefore show that the crystallization of DR08 basanite at 925 °C–200 MPa (corresponding to depths of 6–8 km) and under relatively reduced conditions (fO2 ∼ FMQ; Table 2) generates phonolitic melts (with up to 3.8 wt% H2Omelt) with FeO* contents and FeO*/MgO ratios similar to phonolites sampled offshore. Whereas such T–P–H2Omelt estimates are in agreement with the conditions set for most of the phonolites and evolved peralkaline magmas worldwide [Andújar et al. 2008, 2010, 2013, Andújar and Scaillet 2012, Di Carlo et al. 2010, Giehl et al. 2013, Moussallam et al. 2013, Romano et al. 2018, Scaillet and MacDonald 2006, Scaillet et al. 2008], they strongly differ from those inferred by Berthod et al. [2021b], in particular with respect to pressure conditions: as stated previously, these authors conclude that the fractionation of a 80 wt% dry mineral assemblage from a hydrous basanite (2.3 wt% H2Omelt) at mantle depths (18–20 km, 600 MPa) produces the Fe-rich phonolites of the ridge. However, in this model, no role is given to the hydrous phases like amphibole or biotite in the evolutionary process. Assuming that H2Omelt (2.3 wt%) determined in olivine melt inclusions in DR08 sample by Berthod et al. [2021b] is representative, a fractionating assemblage lacking Amp with such an H2Omelt requires a maximum pressure of ∼200 MPa (Figure 3). Our results show, however, that basanite crystallization under such wet conditions (i.e., ending with residual liquids with H2Omelt > 6 wt%, as required by the largely incompatible behaviour of H2O, for 80 wt% crystallization of a basanite with 2.3 wt% H2Omelt) produces mostly trachytic melts (Figure 9A), true phonolites requiring drier conditions ( <3–4 wt%). This mismatch is seemingly due to the fact that MELTs does not incorporate amphibole, which is an important fractionating phase in hydrous basanite as our experiments demonstrate.
It should be also stressed here that the differences between MELTs calculations and our results not only concern the role of hydrous phases in the fractionating process, but also the composition of feldspar that is in equilibrium with these Fe-rich phonolitic melts. Whereas the calculations of Berthod et al. [2021b] predict the precipitation of at least 30% of alkali feldspar (anorthoclase) to produce the phonolitic melts, the experimental charges yielding to such type of liquids are instead characterized by the presence of relatively primitive Plagioclase (oligoclase) with composition ∼27 mol% in the fractionating mineral assemblage. The submarine phonolites have a dominant alkali feldspar population at ⩽An15 mol%, but also contain a few more calcic cores (An24 mol%) [Berthod et al. 2021b]. The latter is close to that of experimental Pl (An27±3 mol%) produced in charge DR08-131 (925 °C–200 MPa), which is as shown above, in equilibrium with a Mayotte-type phonolitic liquid. Hence, the more calcic Pl cores in the natural phonolite can be considered as crystals inherited from the fractionation process involving the crystallization of the mineral assemblage Ol+Pl+Amp+Mt+Ilm+Ap±Bt down to the phonolitic production stage. The less calcic and dominant variety of feldspar microphenocrysts in the phonolite is likely the result of a small increment of crystallization that occurred after the phonolitic liquid was extracted from its parental basanitic magma, perhaps due to an adiabatic cooling effect upon decompression that took place when the phonolitic melt had left the basanitic reservoir.
Hence, we conclude that conditions of 950/925 °C–200 MPa with H2Omelt⩽3–4 wt% are best suited for the production of Fe-rich phonolites at Mayotte. Deeper conditions produce significantly different compositions, as stated previously (Figure 8).
5.2. The production of the north-central Mayotte Karthala type phonolites
As stated in previous sections, the compositions of the magmas at Mayotte clearly define two distinct magmatic series. Since our starting basanitic composition belongs to the moderate-silica undersaturated series (i.e. Karthala type), our results primarily apply to this specific compositional lineage (Figure 2).
Detailed mineralogical information concerning the phonolitic products of Mayotte is scarce, however: only the works of Pelleter et al. [2014] and Späth et al. [1996] provide bulk-rock information and few mineral data from these evolved compositions (Table 1). For comparison purposes, we have only considered the bulk-rock compositions having MgO contents up to 0.4 wt%, since these are the maximum amounts of our experimental melts (Table 8). The phonolites-trachytes with MgO contents lower than ∼0.25 wt% represent the most evolved products of the Karthala LLD [Pelleter et al. 2014, Späth et al. 1996]. In this case, the liquids produced in our experiments possibly represent the parental compositions from which the Mayotte on-land phonolites derive.
Compared to phonolites from the submarine ridge, those from Mayotte island are characterized by a contrasted mineralogy [Amp+Cpx+Mt+Ilm+ Afs+Ap±Nph) and have slightly lower Fe-contents (between 3.7 to 5.1 wt%; Pelleter et al. 2014]. Rare Ol+Afs bearing phonolites [i.e., equivalent to the dredged products; Berthod et al. 2021b], and aphyric glassy samples [sample M29 in Pelleter et al. 2014; Table 1] can be also found in the northern-central parts of Mayotte.
Apart from sample M29, the Mayotte phonolites are reproduced by the 200–400 MPa series products on TAS, FeO*/MgO, CaO, Al2O3 versus SiO2 Harker diagrams (Figure 9), suggesting that these melts could have been potentially generated at ∼300 MPa (9 km depth). This pressure is about 100 MPa (3 km) higher (deeper) than that determined for the conditions of production of their 2018–2021 submarine equivalents. However, both the lower iron content of the Mayotte island phonolites compared to the submarine ridge products and the presence of Amp in these compositions (Amp stability is enhanced with pressure; Figure 3), support deeper conditions for generation of the on-land phonolites.
The M29 aphyric sample has the highest SiO2 (61.5 wt%) and the lowest FeO* (3.2 wt%) contents of all phonolites considered in this work (Table 1). According to our results, the production of this phonolitic composition will require ponding conditions of a DR08 type basanite at pressures ⩾400 MPa. In fact, the melt from the charge DR08-143 run at 400 MPa closely matches the total alkalis and SiO2 contents of this phonolite (Figure 9A). However, when it comes to FeO*/MgO ratio and TiO2 and CaO contents, the best fits are obtained by charges produced at 925 °C–100 MPa (Figure 9, Table 8). The absence of phenocrysts in the natural sample does not allow us to use the mineralogical information to discriminate between these conditions. However, we cannot exclude that this aphyric phonolite was produced by the fractionation of a basanitic magma at low-pressure (⩽100 MPa) and under relatively dry (⩽3 wt%) and hot (?) conditions.
As stated above, the existence of phonolites, with MgO contents <0.4 wt% containing a mineral assemblage Amp+Cpx+Pl+Fe–Ti oxides [Debeuf 2004, Pelleter et al. 2014], suggests that, even if these magmas evolved from liquids similar to those produced in this work, their composition and mineralogy [low-Mg number of amphiboles, Debeuf 2004] do not necessary reflect the production level: they could reflect shallower final ponding conditions. This observation highlights the need for detailed petrographic data on Mayotte phonolites, especially those on-land, if we are to define more precisely the pre-eruptive conditions of Khartala-type phonolites.
5.3. Various levels of phonolite production/storage at Mayotte
The crystallization experiments, performed on a representative evolved basanitic sample from the 2018–2021 eruption, allowed us to define the best conditions (T–P–fO2–fH2O) for producing the characteristical compositional variability of phonolites from the north-central parts of the Grand Terre island, Petite Terre and NE active submarine ridge of Mayotte. In particular, these experiments show the ease of producing phonolitic melts in the shallow crust, as observed in many other settings. Phonolitic melts containing up to ∼3–4 wt% of H2Omelt are produced by the extensive crystallization of at least 65 wt% of a mineral assemblage consisting of Ol+Pl+Amp+Cpx+Bt+Mt+Ilm+Ap, at crustal levels ⩽12–15 km. Pressure (hence depth) appears to be an important factor controlling the final FeO* content of residual phonolitic liquids. This oxide could be potentially used as a geobarometer (for this type of magma series) to infer the generation/storage depths of corresponding magmas.
A conceptual model summarizing our view on Mayotte phonolites is shown in Figure 10. Considering the above water-contents, the differentiation of basanitic magmas at depths ⩽4–5 km produces residual liquids with trachyte–benmoreite affinities. The Fe-enriched phonolite compositions found in the Mayotte system are instead produced at around 6–8 km, whereas at greater depths, the fractionation process results in the generation of phonolitic melts which are progressively enriched in SiO2–Al2O3 and depleted in FeO*.
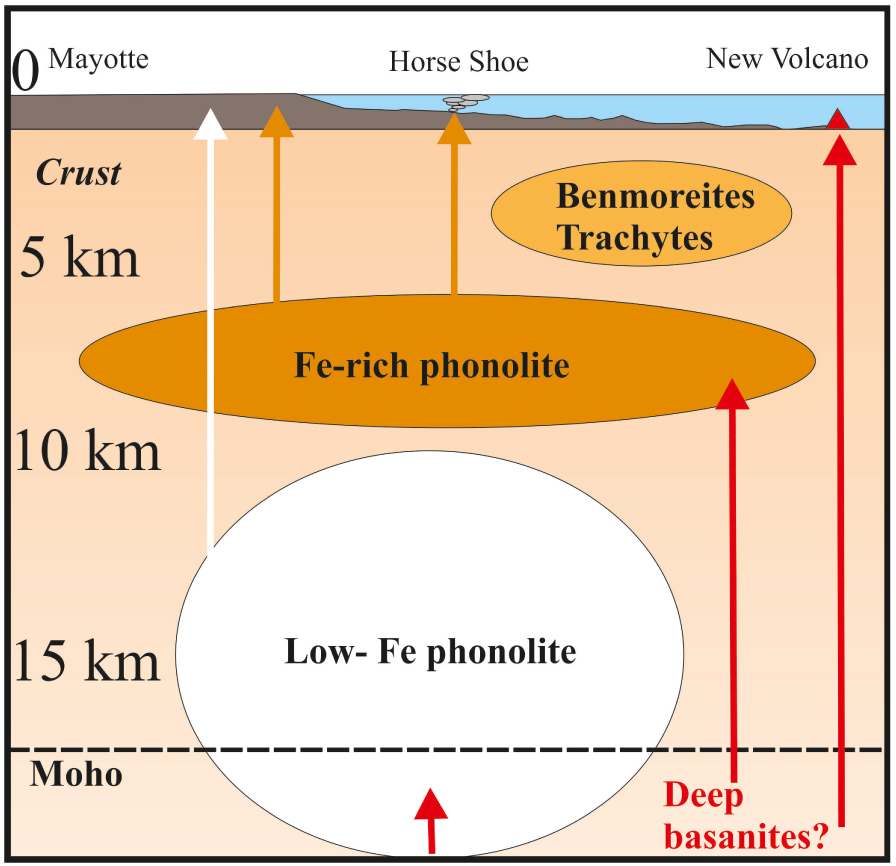
Conceptual model of the plumbing system operating at Mayotte for producing Fe-rich, Low-Fe phonolites and benmoreite–trachyte compositions (see text for details). Moho location from Dofal et al. [2021].
Hence, the production of phonolitic melts with the Karthala affinities of Mayotte island occurs at multiple depths above the local Moho [16–17 km, Dofal et al. 2021]. However, the presence of highly evolved phonolitic magmas (i.e. MgO poor) suggests that these magmas experienced a later episode of crystallization and fractionation at shallower levels, prior to their eruption. The occurrence of relatively shallow phonolitic reservoirs below the Horseshoe region (Figure 1C) would have permitted their subsequent rapid emptying and collapse during past eruptions, which would readily explain the presence of a caldera-looking depression at this point, since relatively shallow depths of magma reservoirs favors the formation of caldera structures [e.g. Martíet al. 2008].
Our results contrast with the conditions inferred from mineral geobarometers and MELTS simulations, which for producing the Fe-rich phonolites from Mayotte, predict the crystallization of a dry mineral assemblage at significantly higher pressures than those determined in this work (600 versus 200 MPa). This difference highlights the still poor resolution that these two methods have for predicting the depths of magma evolution in alkaline systems, and calls for additional calibration of these tools.
Conflicts of interest
Authors have no conflict of interest to declare.
Acknowledgments
The authors thank Patricia Benoist for the technical support with SEM and EMPA. We thank the scientific party of the MAYOBS 2 (DR08 sampling) campaign that was conducted by several French research institutions and laboratories (IPGP/CNRS/BRGM/IFREMER/IPGS). All marine operations are performed as part of the MAYOBS set of campaigns (https://doi.org/10.18142/291), under the supervision of REVOSIMA (Réseau de Surveillance Volcanologique et Sismologique de Mayotte; REVOSIMA), a partnership between the Institut de Physique du Globe de Paris (IPGP), the Institut Français de Recherche pour l’Exploitation de la Mer (IFREMER), the Bureau de Recherches Géologiques et Minières (BRGM), the Centre National de la Recherche Scientifique (CNRS) and the REVOSIMA consortium. We thank the captain and crew of the R/V Marion Dufresne (TAAF/IFREMER/LDA). R. Cioni and an anonymous reviewer are thanked for the detailed reviews of this work. We want also to thank the editorial handling of editor Jérôme Van der Woer.
Funding
This work was supported by the Institut des Sciences de la Terre d’Orléans (ISTO-CNRS), the LABEX VOLTAIRE (ANR-10-LABX-100-01) and the EQUIPEX PLANEX (11-EQPX-0036). Since June 2019, all activities on Mayotte, including MAYOBS campaigns, are funded by le Ministère de l’Enseignement Supérieur, de la Recherche et de l’Innovation (MESRI), le Ministère de la Transition Ecologique (MTE), le Ministère des Outremers (MOM), le Ministère de l’Intérieur (MI), and le Ministère des Armées with the support of the DIRMOM (Direction Interministérielle aux Risques Majeurs en Outremer) and the MAPPPROM (Mission d’appui aux politiques publiques pour la prévention des risques majeurs en Outremer).
Vous devez vous connecter pour continuer.
S'authentifier