

1 Background
1.1 Health issues
Arsenic is a trace element, whose fate and bioavailability for plants and humans strongly depend on its speciation. Arsenic speciation and coordination are themselves strongly linked to redox conditions of the aqueous media, whether present in an aquifer or in a cell (Charlet and Polya, 2006; Rosen, 2002). Ever since Nero used arsenic to poison Claudius and Britannicus, arsenic has been well known for its acute toxicity to human beings, although it has also been used (and is still used) in the treatment of acute promyelocytic leukemia (Tamas and Wysocki, 2001). More recently, epidemiological studies in Chile and South-East Asia have demonstrated its chronic toxicity in countries where daily chronic ingestion occurs via contaminated water and rice (Meharg and Rahman, 2003; Smith et al., 2006). Consumption of drinking water containing five or 50 times the European Union (EU) and World Health Organization (WHO) Maximum Contaminant Level (MCL = 10 μg/L for As) induces a lung cancer risk equivalent to that of a passive or active smoker, respectively (Smith et al., 2006). Other arsenicosis symptoms include hyperkeratosis, various forms of cancer (skin, bladder, and kidney), cardiovascular troubles, stillbirth and spontaneous abortion. Worldwide, 150 million people are at risk due to arsenic, among whom 110 million are living in South-East Asia deltas (Ganga-Bramapoutra, Mekong and Red River deltas), but other people at risk are living in desert areas and often depend on hydrothermal springs for their drinking water supply (e.g. inhabitants of Los Angeles in the USA and Antofagasta in Chile), or are located downstream from mining activities (Brammer and Ravenscroft, 2009; Polya and Charlet, 2009; Ravenscroft et al., 2009; Smith et al., 2006; Winkel et al., 2008). In Bangladesh alone, 21.4% of all deaths and 23.5% of deaths linked to chronic diseases have recently been shown to be linked to the consumption of water with an arsenic concentration larger than the WHO MCL (Argos et al., 2010).
Arsenic bioavailability depends on its oxidation state, and each oxidation state (V, III or–III) corresponds to a specific coordination, which will, in turn, dictate the fit of a given species within a given mineral structure, or the ability of arsenic to cross a given biological barrier. The most oxidized form, As(V), corresponds to oxoanions with a tetrahedral structure (arsenate ions). In suboxic to mild reductive environments, the dominant form is As(III), which corresponds to aqueous arsenite species characterized by a pyramid geometry (trigonal coordination). In strongly reductive biological environments (in human liver or red cells) arsenic may be biotransformed to arsenide (As(-III)) species (Gailer et al., 2002; Manley et al., 2006; Tamas and Wysocki, 2001) For instance, E. coli cells have been recently demonstrated to be able to produce the highly toxic gaseous form arsine, AsH3(g), as well as other methylated As(-III) species (Yuan et al., 2008). Moreover, a variety of methylated As(III) and As(V) species (O’Day, 2006), as well as (seleno)glutathione-As(III) complexes are commonly produced in living organisms, these latter molecules playing a key role in As detoxification processes (Gailer et al., 2002; Manley et al., 2006; Miot et al., 2008, 2009; Rosen, 2002). Similar reduction mechanisms occur in anoxic surface environments (peat bogs, paddy fields, stratified lakes)
1.2 Mineralogical versus biological control of As scavenging and release
Within the complexity of deltaic hydrology and geochemistry (Metral et al., 2008; Polizzotto et al., 2008), the predominant nano mineral phases change over distances of 100 m (A. Foster, Pers. Comm.). Wherever overlying sandy soils allow a direct vertical recharge of the groundwater, down to 60 meters in less than 60 years (Lawson, 2010), Fe(III) oxyhydroxide and oxyhydrosulfate nanoparticles may induce the trapping of arsenic in these oxic/suboxic grey sediments (Metral et al., 2008). On the other hand, wherever an overlying impermeable soil leads to anoxia in the aquifer located below, mobilization of arsenic occurs in the aquifer. The Fe(II/III)-rich nanophases formed in these conditions (e.g., mackinawite and magnetite) are comparatively poor arsenic sorbents. The concentration of arsenic in groundwater is therefore strongly related to the mineralogy, pH, and Eh as well as to bacterial activity (Burnol et al., 2007; Fendorf et al., 2010; Van Geen et al., 2004) (Fig. 1).
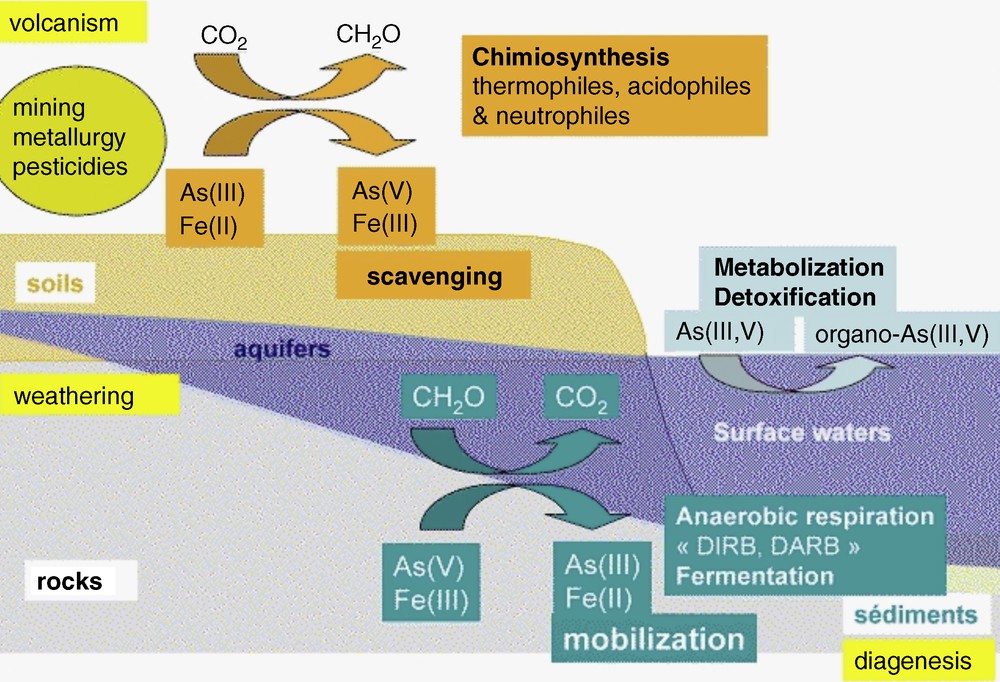
Simplified sketch of arsenic cycling in the environment, showing the influence of microbial metabolisms on iron and arsenic oxidation states. Oxidation reactions are generally associated to scavenging of As(V) by insoluble Fe(III)-oxyhydroxides and oxyhydroxysulfates, while the reductive dissolution of the latter phases, for instance by Dissimilatory Iron and/or Arsenic Reducing bacteria (DIRB, DARB) is generally associated to the release of the highly toxic As(III) form in groundwater.
Cycle simplifié de l’arsenic dans l’environnement, qui montre l’influence des métabolismes microbiens sur les degrés d’oxydation – et donc sur la mobilité – de l’arsenic et du fer. Les réactions d’oxydation sont généralement associées au piégeage de As(V) par les oxyhydroxydes insolubles de Fe(III) et par les oxyhydroxysulfates, tandis que la dissolution de ces solides par réduction du fer et/ou de l’arsenic par les bactéries réductrices, est en général associée à la libération de As(III), forme hautement toxique de l’arsenic, dans les eaux souterraines.
Even if a full understanding of the mobilization of arsenic in groundwater is far from being achieved, a large body of recent literature delineates the mechanisms controlling the fate of arsenic in soils and aquifers. Field and laboratory investigations performed over the last twenty years on oxic soils and mining environments have shown arsenic to be scavenged in oxic conditions by Fe(III) oxyhydroxides (e.g., Charlet et al., 2007; Morin and Calas, 2006 and references theirein), while in anoxic conditions Fe(II) and As(III) tend on the long term to be released in the aqueous phase (Tufano and Fendorf, 2008), even though the process can be delayed by arsenic sorption on Fe(II)-rich phases (green-rusts, magnetite, pyrite, troilite and mackinawite) (Bostick and Fendorf, 2003; Burnol and Charlet, 2010; Ona-Nguema et al., 2009; Root et al., 2007).
Consumption of dissolved oxygen and nitrate precedes the reduction and dissolution of Mn- and Fe-oxides (see e.g., Burnol and Charlet, 2010) and in West Bengal groundwater, the consumption of nitrates precedes the reduction of sulfate and Fe oxides (Burnol and Charlet, 2010). However, on a short term, Fe and As releases may not occur simultaneously (Burnol et al., 2007; Islam et al., 2004; Van Geen et al., 2004). Van Geen et al. (2004) have shown that truly anoxic conditions may indeed not be required for the release of As from reducing grey sediment in Bangladesh. In two months long experiments run with unamended gray sediments from Bangladesh, they observed a gradual release of the equivalent of 0.5 to 1.0 μg/g As to the dissolved phase even in the presence of some dissolved oxygen (∼1 mg/L). They showed consequently that a release of significant amounts of arsenic may occur without the need for extensive Fe dissolution, suggesting that the release of As and Fe is decoupled. Burnol et al. (2007) demonstrated by microcosm studies and dynamic equilibrium modeling that this discrepancy is controlled by chemistry rather than by microbiology. When arsenic-rich, 1–5 nm wide ferrihydrite particles (Michel et al., 2007) are dissolved by the iron-reducing bacteria, aqueous Fe2+ concentration increases and Eh first decreases while no arsenic appears in solution (Fig. 2). This process is interpreted as an immediate re-adsorption of the As(V), released by the reductive dissolution of the As-rich ferrihydrite coprecipitate, on other ferrihydrite particles. When Eh reaches the As(V)/As(III) boundary limit, As(V) is reduced to As(III). Since, under these experimental conditions, this form is more weakly adsorbed than As(V) in presence of carbonates, As appears in solution, in an apparent “decoupled” manner, but in fact thermodynamically perfectly coupled to the release of Fe(II) in solution (Fig. 2). In an alternative scenario, a Fe-bearing phase may dissolve and release As, while another As-poor, Fe-bearing phase (e.g., a phosphate-like vivianite) could precipitate kinetically (Burnol et al., 2007). This alternative scenario could account for the lack of evidence for As being released into solution during the early Fe release (G.E. Brown, Pers. Comm.). Microorganisms are clearly playing a central role in As release, as demonstrated by experiments run with the same sediment treated with a cocktail of penicillin G, chloramphenical and streptomycin antibiotics (Guillard reagent), in which no arsenic release was observed in these mesocosms (Van Geen et al., 2004).
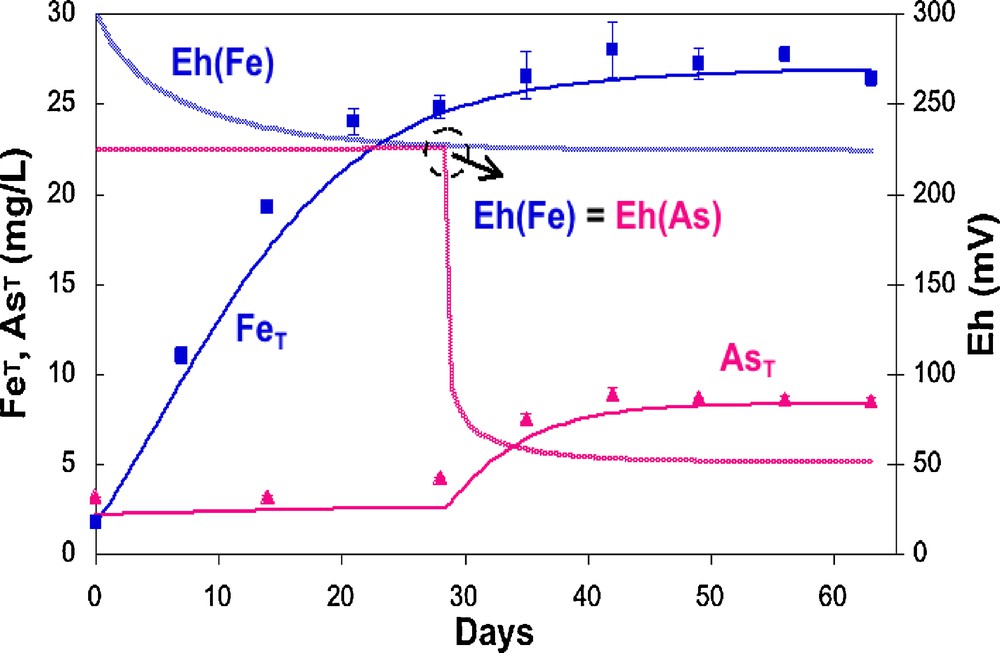
Decoupled release of Fe(II) and As(III) in a sediment incubation microcosm experiment (symbols). The model curve is computed according to a dynamic equilibrium model including equilibrium chemistry, for As-rich ferrihydrite nanoparticle dissolution, and kinetics, for microbial induced reduction (from Burnol et al., 2007).
Libération découplée du Fe(II) et de As(III) dans des expériences d’incubation de sédiment en microcosme (symboles). La courbe est calculée selon un modèle d’équilibre dynamique intégrant chimie d’équilibre, pour la dissolution des nanoparticules de ferrihydrite riche en arsenic, et cinétique de réduction microbienne.
(d’après Burnol et al., 2007).
Microbially driven reductive dissolution of ferric iron hydroxide (goethite, ferrihydrite or lepidocrocite) in the presence of organic matter and CO2 can be written simplistically as:
CH2O + 7CO2 +4 FeOOH + H2O = 4 Fe2+ + 8 HCO3−.
The age of DIC (HCO3− ions) and DOC (“CH2O”) may be differentiated by a combination of 13C/12C and 14C/12C measurements. In Cambodia and in Indian delta groundwaters, the water DIC is younger than the DOC (Lawson, 2010), therefore not being formed only by the above chemical reaction. DOC is also not primarily derived from modern surface organic matter. Since it is negatively charged, DOC may not only act as electron donor, but also as a direct competitor with arsenic anionic species for sorption on iron oxyhydroxide particles.
The bicarbonate ions produced in the above reaction may further lead to the formation of siderite (FeCO3). Cambodian and Indian arsenic affected groundwaters are often oversaturated by an order of magnitude with respect to siderite precipitation (Kocar and Fendorf, 2009; Nath et al., 2009). In presence of excess bisulfide ions, mackinawite (FeS) and other FeSx (with 1 < x < 2) may form. Flow-through reactor studies performed on soil synthetic aggregates (made of ferrihydrite, quartz sand, Shewanella sp. iron-reducing bacteria and agarose) lead to contrasting results depending on whether As is present or not in water. When the system was amended with a 0.3 mM lactate solution without arsenic, the development of an anoxic environment in the heart of the aggregate was observed after three months of bacterial activity (Pallud et al., 2010a, 2010b) with micron-sized siderite particles at the heart of the aggregate. However, when arsenic was added to the lactate solution, no siderite was formed (Masue-Slowey et al., 2011). Some magnetite was also produced at the surface of the aggregate but not in the core region. These dynamic experiments demonstrate differential secondary product formation: siderite, together with α/γ FeOOH, forms in the core region of the aggregate where more reducing conditions prevail, and magnetite forms at the surface of the aggregate. The nanosize of most of these products also explains why XRD rarely provides evidence for the formation of magnetite and siderite in SE Asia deltaic aquifers, while diffuse reflectance measurements point to the presence of Fe(II)-rich solid phases (Van Geen et al., 2004).
On the other hand, in soils where oxic conditions prevail, in situ speciation of As investigated by combining X-ray Absorption Spectroscopy (XAFS) with selective chemical extractions demonstrated As to be mostly present as surface complexes on iron oxides in soils (e.g., Morin and Calas, 2006 and references therein). Although, the relative importance of phyllosilicates as arsenic sorbents is generally difficult to evaluate in soils, the lower affinity of As(III) for Al-bearing phyllosilicate minerals compared to iron oxyhydroxides (Goldberg, 2002) is responsible for the increased As(III) mobility in iron-depleted anaerobic media.
In extreme environments as acid mine drainage (Benzerara et al., 2008; Morin and Calas, 2006) and geothermal springs (Inskeep et al., 2004) XAFS studies have revealed a similar coupling between arsenic and iron chemistry. In both contexts, microbial oxidation of Fe(II) and As(III) leads to the formation of amorphous As(V)-Fe(III) hydroxysulfate compounds with similar local structure (Morin et al., 2007). The solubility of these compounds directly decreases with increasing Fe/As ratio (Fig. 3). As(V)-Fe(III) hydroxysulfate minerals are frequently associated with biological substances, and may be considered as potential markers of microbial activity in extreme acidic environments. The role of microbial oxidation is especially important for the kinetics of Fe(II) oxidation, which is low in acidic environment and may directly influence the nature of the biogenic minerals formed (Egal et al., 2009). The ability of arsenic resistant anaerobic iron oxidizing microorganisms to immobilize As by sorption on biogenic Fe(III) oxyhydroxides in anoxic conditions, has been further recently demonstrated in laboratory experiments (Hohmann et al., 2010).
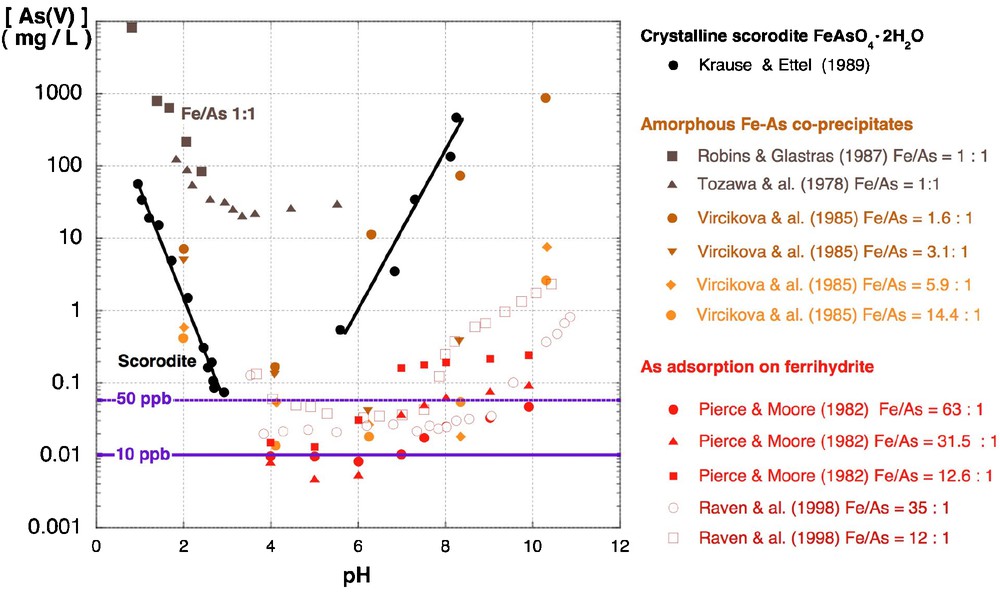
Solubility of crystalline and amorphous hydrated ferric arsenate mineral phases compared to the As(V) concentration at equilibrium with As(V) sorbed onto ferrihydrite. At low Fe/As ratio the crystalline phase is less soluble than the amorphous or nanocrystalline phases. The As solubility significantly decreases with increasing the Fe/As ratio, and the lowest “solubility” values are obtained for aqueous As(V) at equilibrium with As(V) sorbed on ferrihydrite surface, which yield dissolved As concentrations lower than the WHO Recommended Drinking Limit (10 ppb). (See Krause and Ettel (1989); Pierce and Moore (1982); Raven et al. (1998); Robins and Glastras (1987); Tozawa et al. (1978); Vircikova et al. (1985)).
Solubilité des phases cristallines et hydratées-amorphes d’arséniates ferriques, comparée à la concentration en As(V) à l’équilibre avec l’As(V) adsorbé sur ferrihydrite. À faible rapport Fe/As. la phase cristalline est moins soluble que les phases amorphes ou nanocristallines. La solubilité de As décroît significativement pour des rapports Fe/As plus élevés. La plus faible « solubilité » est observée pour l’équilibre avec As(V) adsorbé à la surface de la ferrihydrite, pour qui la concentration en As soluble est inférieure aux limites de potabilité de l’OMS (10 ppb). (Voir Krause et Ettel (1989) ; Pierce et Moore (1982) ; Raven et al. (1998) ; Robins et Glastras (1987) ; Tozawa et al. (1978) ; Vircikova et al. (1985)).
2 Sorption: Kd and species-specific mechanisms of As sorption
Sorption of As onto a mineral particles can be characterized macroscopically by the observed Kd (g/L) value, defined as the ratio measured at a given pH and ionic strength between the total aqueous As concentration (mol/L) and the solid As concentration (mol/g). Table 1 reports Kd values measured at pH 7 or pH 7.5 (i.e., in the pH range of most SE Asia As-contaminated groundwaters) at various solid/solution ratios. Clearly, the As(V) and As(III) Kd values differ. For example, Kd values are sometimes higher for As(III) (for sorption on ferrihydrite, goethite) than for As(V), and sometimes lower (for sorption on mackinawite, siderite, magnetite or biotite). Magnetite and mackinawite have similar Kd values. Those Kd values for Fe(II) rich minerals are particularly important since pure Fe(III) oxyhydroxides (e.g. ferrihydrite and goethite) are seldom observed in anoxic deltaic aquifers throughout SE Asia, but instead diffuse reflectance spectra consistently show the presence of fine-grained Fe(II)-bearing minerals in these porous media (Metral et al., 2008).
Kd d’As(III) et d’As(V) sur les phases qui contiennent du Fe(II)-Fe(III) évalué à partir des expériences des fronts d’adsorption (pH 7 et 7,5).
| Fe(II)-Fe(III)-bearing phases | Solid (g/L) | Kd L/g As(III) at pH 7 (pH 7.5) |
Kd L/g As(V) at pH 7 (pH 7.5) |
Ref. |
| HFO | 0.03 | 85.72 (87.79) | 49.3 (37.59) | Inskeep et al., 2004 |
| Goethite | 0.5 | 14.46 (16.1) | 8.05 (5.39) | Inskeep et al., 2004 |
| mackinawite | 0.044 | 2 | 9 | Islam et al., 2004 |
| siderite | 2.5 | 0.28 (0.36) | 3.36 (1.86) | Jain et al., 1999 |
| magnetite | 3.1 0.5 |
0.08 (0.20) 1.85 (2.02) |
(8.89) – |
Jain et al., 1999
Inskeep et al., 2004 |
| fougerite | 4.5 | 0.12 (0.42) | – | Jain et al., 1999 |
| vivianite | 2.5 | – | 0.18 (0.18) | Jegadeesan et al., 2010 |
| Muscovitea | 4.1 | 0.36 (0.13) | 0.36 (0.13) | Jonsson and Sherman, 2008 |
| Biotitea | 4.25 | 0.97 (0.31) | 3.4 (0.9) | Jonsson and Sherman, 2008 |
a Derived from constant capacity (CC) modeling of adsorption edge experiments.
Recent studies using XAFS spectroscopy, neutron diffraction, HRTEM, and DFT molecular modeling, have revealed, as will be discussed in the following sections, the formation of a large variety of arsenic surface complexes upon sorption onto ferric oxyhydroxides (Ona-Nguema et al., 2005; Wolthers et al., 2005a), nanomaghemite (Auffan et al., 2008; Morin et al., 2008), nano-magnetite (Morin et al., 2009; Wang et al., 2008; Yean et al., 2005), iron hydroxycarbonates (Auffan et al., 2007), mackinawite (Wolthers et al., 2005a), calcite (Roman-Ross, 2006) and gypsum (Fernandez-Martinez et al., 2008). Arsenite forms a specific tridentate, triple corner-sharing surface complex both with magnetite (Morin et al., 2009; Wang et al., 2008) and maghemite (Auffan et al., 2008a), which explains, in part, the high adsorption affinity of arsenite for these substrates. In addition, a “nano” effect is observed for magnetite, which may sorb 0.021, 0.388 and 1.532 mmol g−1 of arsenite, i.e. 5–6 μmol m−2 to 18 μmol m−2, for particle sizes decreasing from 200 nm to 20 and 12 nm, respectively (Auffan et al., 2008b; Morin et al., 2009; Yean et al., 2005). Although the origin of this increased reactivity is still a matter of debate, it is attributed either to an increased surface tension (Auffan et al., 2007, 2009) or to surface precipitation of arsenite (Morin et al., 2009). Polymeric arsenite surface complexes may also form on green-rusts and may play an important role in delaying the release of arsenic in suboxic soils (Auffan et al., 2007; Wang et al., 2010). Even though electron transfer between structural Fe(II) and arsenate species is not observed on green-rusts (Wang et al., 2008), arsenate may be reduced by Fe2+ sorbed on micas and clays (Charlet et al., 2005). In the case of carbonate and sulfate minerals, isomorphic substitution of the constitutive anion by the appropriate arsenic oxoanion may enhance sorption as well (Fernandez-Martinez et al., 2008; Roman-Ross et al., 2006).
2.1 Ferrihydrite, goethite, and As(V) adsorption: alternative spectroscopy and modes of sorption
Thanks to more than two decades of laboratory studies, sorption of arsenate at the surfaces of common ferric oxyhydroxide minerals, especially goethite and ferrihydrite, has been demonstrated to be one of the most efficient trapping processes for dissolved arsenate (Manning et al., 1998; Waychunas et al., 1993), and many water purification processes utilize this process (after a first step of chemical or biotic oxidation of arsenite to arsenate). Several studies addressing arsenic speciation in contaminated environments have also shown that this process is active in the field (e.g., Foster et al., 1998; Morin and Calas, 2006) and references therein). Comparison of natural As-bearing soils with polluted ones have shown that arsenate binding to poorly ordered ferric oxyhydroxides, such as ferrihydrite, retards As transfers toward surface- and ground-waters in the oxic zone whenever crystalline arsenate minerals are not less stable and therefore do not retain arsenate ions in long-term weathering processes (Cances et al., 2008; Morin et al., 2002) (Fig. 3). Less data are available on the behavior of the toxic As(III) form, but extensive research has been conducted on the interactions of this species with the ferric oxyhydroxide surfaces (e.g. Manning et al., 1998). An XAS-based study conducted by Ona-Nguema et al. (2005) compared the modes of As(III) sorption onto 2-line ferrihydrite, hematite, goethite, and lepidocrocite. Sorption experiments and spectroscopic data acquisition were performed under anoxic conditions in order to minimize As(III) oxidation due to reactive oxygen species (ROS). These EXAFS data indicate that As(III) surface complexes on hematite and ferrihydrite are similar, but they differ significantly from those on goethite and lepidocrocite. The main difference is the absence of bidentate edge-sharing complexes (2E) at the surface of the two latter minerals. This 2E complex, which is characterized by a short As-Fe distance of 2.9 Å, appears to be specific to the As(III)O3 pyramid geometry, since it has been demonstrated by several recent studies that this complex actually does not form in the case of the tetrahedral As(V) species (e.g., Cances et al., 2005, 2008; Morin et al., 2008; Sherman and Randall, 2003; Wang et al., 2010). It was indeed shown that, in the case of As(V), As-O-O and As-O-O-O multiple scattering path contributions to the EXAFS had been long misinterpreted as being due to a 2E complex (e.g. Cances et al., 2005; Sherman and Randall, 2003). These findings are consistent with the structure of ferric arsenate and arsenite minerals in which the 2E linkage is only observed for arsenite (e.g., Morin et al., 2007).
Despite these significant advances in understanding the mode of arsenic binding at the surface of ferric oxyhydroxides, several questions are still open and may have important implications for an accurate modeling As sorption reactions. The main recent important finding provides evidence for both outer-sphere and inner-sphere As(V) complexes at the hematite/aqueous solution interface demonstrated using Resonant Anomalous X-ray Reflectivity (RAXR) by Catalano et al. (2008). These RAXR results show that about 35% of the sorbed arsenic occurred in the outer-sphere form in the hematite samples studied. The exact nature of these outer-sphere complexes is still poorly constrained, but several lines of evidence suggest that they could correspond to hydrogen-bonded species, which would explain the fact that they are not displaced with increasing ionic strength. Such outer-sphere complexes are difficult to detect using EXAFS spectroscopy in the presence of inner-sphere As(V) complexes because their presence is manifested by average As(V)-OH2 distances that do not differ significantly from the average As(V)-O distances in inner-sphere As(V) complexes to allow their unambiguous distinction. In addition, although the As(V)-Fe(III) distances of outer-sphere As(V) complexes at hematite/water interfaces are significantly longer than those of inner-sphere As(V) complexes, the integrated intensity of this feature in the radial distribution function should be significantly weaker than that of the As(V)-Fe(III) pair correlation due to inner-sphere complexes. As a result, the presence of outer-sphere As(V) complexes at Fe(III)-oxide/aqueous solution interfaces could have been underestimated in past laboratory and field studies or missed altogether. Grazing-incidence EXAFS (GI-EXAFS) spectroscopy is also able to detect outer-sphere complexes, as demonstrated by a study (Bargar et al., 1996) on Pb(II) sorption onto α-Al2O3 (0001) single crystal surface in contact with water. A major limitation of both RAXR and GI-EXAFS studies, however, is that they can only be conducted on species sorbed on single crystal substrates, which are not representative of the fine-grained, high surface area minerals that are typical of most natural environments. More experimental and theoretical work is needed to better understand the nature and importance of these outer-sphere complexes for both As(V) and As(III).
2.2 Magnetite and maghemite: nanoparticle size and site specific effects on As sorption
As discussed above, iron oxides and iron oxy-hydroxides are strong sorbents for many metals and metalloids such as arsenic. At given physicochemical conditions (pH, ionic strength metal ion concentration, reactive surface area of sorbents), the sorbed quantity of the metal or metalloid will depend on the nature of the iron mineral. Although the thermodynamic constants of sorption will vary as a function of iron oxide or oxy-hydroxide composition (Table 1), the adsorbed quantity can also be size dependent for a given mineral. Indeed, for the specific case of magnetite, a potentially important sorbent for arsenic in reducing environments as well as in putative remediation processes, especially for the reduced and highly toxic As(III) form, the quantity of arsenic per gram strongly increases from 0.02 mmol/g of arsenic for 300 nm particles up to 0.38 mmol/g for 20 nm magnetite and 1.8 mmol/g for even smaller 11 nm wide magnetite particles (Yean et al., 2005). Such a significant increase in amount of arsenic adsorbed on nanometric particles may be related to strong modifications of other properties like surface structure and surface reactivity, as size decreases (Auffan et al., 2008b; Ha et al., 2009). All of these potential modifications of particle properties as size reaches the nano domain have been generalized as ‘nano-effects’ and are at the origin of the exiting emerging scientific field of nanotechnology.
Concerning the arsenic adsorption example, identification of a nano-effect cannot result from a comparison of the amount of As adsorbed per mass of particles. Indeed such strong increases are not surprising since the specific surface area (SSA) of particles is inversely proportional to the size of particles, assuming that nanoparticle aggregation is minimal. For instance for the latter example, the SSA increases from 3.7 to 60 to 98.8 m2/g for 300, 20, and 11 nm magnetite particle diameters, respectively. Therefore, to further investigate the possibility of a ‘true’ nano-effect, one must compare the adsorbed quantity not per mass but per surface area (Auffan et al., 2007). When sorption is expressed per unit of surface area, magnetite particles of 300 and 20 nm diameter adsorbed similar amounts of As (i.e., 3.5 As/nm2) suggesting similar adsorption mechanisms. Therefore, the difference in As uptake between the 20 and 300 nm magnetite particles is only related to the SSA. In stark contrast, the adsorption capacity increases particles get smaller than 20 nm, and 11 nm-diameter magnetite adsorbs three times more As per square nanometer (10.9 As/nm2) than does 20 and 300 nm-diameter magnetite.
We have recently observed a similar effect for arsenite adsorption at the surface of maghemite (Auffan et al., 2008a, 2008b). In that study, 6 nm-diameter maghemite particles adsorbed up to 8.1 ± 0.8 As per nm2. Such a ‘nano’ effect raises questions about the mechanisms of metal ion adsorption on nano-sized mineral particles, particularly the possibility that the surface atomic structure of nanoparticles in the smallest size range is potentially different from that of larger sized particles of the same material, which could lead to significant differences in reactivity. A combination of sorption experiments and characterization studies of the evolution in nanoparticle structure and As local atomic environment has lead to the identification of two main phenomena that may help explain the origin of these observed nano-effects. The first factor is related to a size-dependent structural modification of the surface of particles. Brice-Profeta et al. (2005) have shown that the occupation rate of the maghemite tetrahedral site by Fe ([FeTd]) decreases as particle size decreases. This study has also demonstrated the existence of a preferential iron octahedral layer at the nano-maghemite surface, i.e. a deficit of FeTd at the surface of very small maghemite nanoparticles. X-ray diffraction revealed that 10% of FeTd sites are vacant before As(III) adsorption. The adsorption of As(III) led to an increase in the occupancy of the surface FeTd sites, as revealed by X-ray diffraction, suggesting possible As adsorption at this very specific maghemite crystallographic surface sites. EXAFS at the As K edge further indicated that As was chemically sorbed but with a surprising high Fe coordination number (3.1 ± 0.6). A careful examination of the coordination of the [FeTd] site on the {111}, {011}, and {100} surface planes strongly suggested that As filled the [FeTd] surface sites through the formation of tridentate, hexanuclear, corner-sharing (3C) surface complexes. Morin et al. (2009) found the same As(III) complex at the surface of magnetite in sorption as well as co-precipitation experiments (Wang et al., 2008) (Fig. 4). At higher surface coverage, arsenite adsorbs on Fe octahedral [FeOh] surface sites through monodentate trinuclear complexes supposedly in a lattice position.
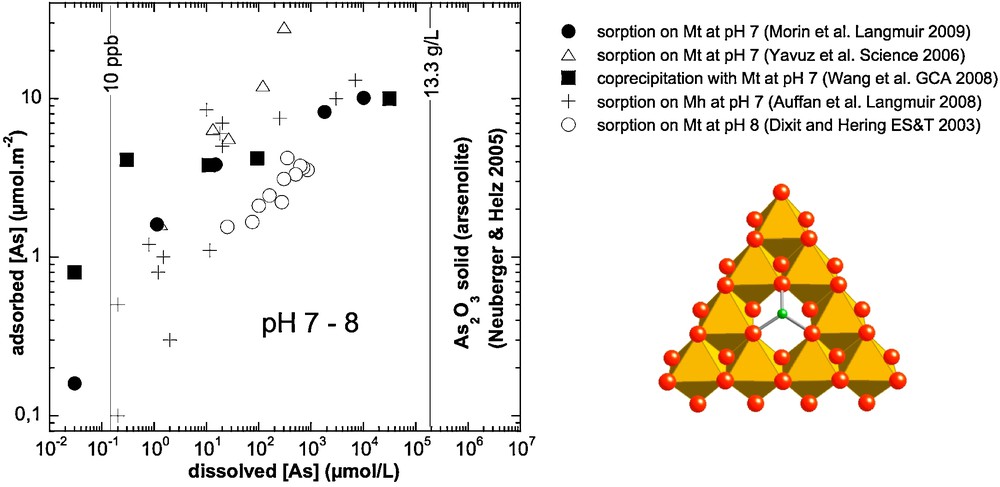
Isotherm data for As(III) sorption on magnetite (Dixit and Hering, 2003), nanomagnetite (Neuberger and Helz, 2005; Yavuz et al., 2006) and nanomaghemite (Auffan et al., 2008a), compared with data for As(III) coprecipitated with magnetite (Wang et al., 2008). The structure of a tridentate As(III) surface complex on the (111) face of magnetite/maghemite responsible for the high affinity of As(III) for these mineral surfaces (Auffan et al., 2008a; Neuberger and Helz, 2005; Wang et al., 2008) is deduced from EXAFS data and displayed on the bottom right. Arsenic and oxygen atoms, and iron octahedra are displayed in green, red and orange color, respectively.
Isothermes d’adsorption de l’As(III) sur magnétite (Dixit et Hering, 2003), nanomagnétite (Neuberger et Helz, 2005; Yavuz et al., 2006) et nanomaghémite (Auffan et al., 2008a), comparés à la solubilité de la magnétite coprécipitée avec l’As(III) (Wang et al., 2008). La structure du complexe de surface tridentate déduite des données EXAFS, complexe formé par As(III) à la face (111) de la magnétite et/ou maghémite et responsable de la très forte affinité de As(III) pour ces surfaces minérales (Auffan et al., 2008a; Neuberger et Helz, 2005; Wang et al., 2008) est donné en bas à droite. Les atomes d’arsenic et d’oxygène, ainsi que les octaèdres de fer sont représentés respectivement en vert, rouge et orange.
Even if adsorption of As(III) at the highly reactive vacant surface [FeTd] sites on magnetite and maghemite can explain the uptake of ∼ 2 As/nm2, it cannot explain the maximum amount observed for As adsorption (8 As/nm2). Other factors need to be taken into account to help understand this unusually high level of As(III) uptake. Indeed nanoparticles are thermodynamically unstable compared with their microscopic counterparts. Adsorption of ions at the surface of particles decreases the energy (ΔG) of a system by ΔG = 3 VmΔγ/r, where Vm is the molar volume, Δγ is the difference in interfacial energy before and after adsorption, and r is the radius of the particles. Therefore the adsorption of a dense arsenite layer decreases – via radius increase – the energy of the system more than when adsorption occurs on larger particles of 20 or 300 nm. Whereas in macroscopic systems adsorption is mainly governed by chemical affinity and electrostatic bond strengths, for nanoparticles the decrease of free energy must be taken into account. This driving force is predominant in crystal growth. In our past studies, the adsorption of AsIII in the vacant [FeTd] lattice positions at the nano-maghemite surface can be compared to a crystal growth mechanism in which AsIII mimics the [FeTd] atoms. This may explain the high density of As adsorbed at the surface of nano-maghemite.
2.3 Magnetite: surface precipitation of As
The large amount of As adsorbed at the surface of iron oxide nanoparticles may also have another origin. Morin et al. (2009) have shown that in the case of 11 and 34 nm nano-magnetite particles, the high surface coverage is due to the formation of an amorphous As(III)-rich surface precipitate. This explains why nanocrystalline magnetite (< 20 nm) exhibits higher efficiency for arsenite sorption than larger magnetite particles, sorbing as much as ∼10 μmol/m2 of arsenite (Yavuz et al., 2006).
The sorption mechanism may then be related to a modification of the nanoparticle surface involving the dissolution of a fraction of the surface iron atoms and coprecipitation of an amorphous solid with arsenite. Such a mechanism has been observed for Co(II) sorption on γ-Al2O3 (Towle et al., 1997), for Ni(II) sorption on Al2O3 (Scheidegger et al., 1997), and in the case of selenite adsorption on magnetite (Missana et al., 2009). It may be less important for As(III) adsorbed on nano-maghemite particles because Fe(III) solubility is several orders of magnitude lower than that of magnetite Fe(II). Indeed, HRTEM-EDXS analyses of sorbed and co-precipitated magnetite samples revealed the formation of an amorphous As(III)-rich surface precipitate which dominates As(III) speciation at surface coverages exceeding the maximum site density of vacant tetrahedral sites on the magnetite {111} surface (5.3 μmol/m2). The origin of this surface precipitate is still poorly understood in the case of sorption experiments. It may be due to the partial dissolution of surface Fe(II) precipitated with As(III). The comparison between sorption and coprecipitation experiments suggests that the surface precipitate might be similar to the sorption complex since the dissolved As(III) concentration converges toward similar values in both cases at high As surface coverage (Fig. 4). Although such a surface precipitate helps explain the exceptional As(III) sorption capacity of nanomagnetite, it causes a dramatic increase of dissolved As concentration at high surface coverage (up to ∼10 μmol/m2).
For lower As surface coverage, recent XAFS studies (Morin et al., 2009; Wang et al., 2008) have demonstrated that, when sorbed on, or coprecipitated with, magnetite at neutral pH, As(III) forms dominantly inner-sphere, tridentate, hexanuclear, corner-sharing surface complexes (3C) in which AsO3 pyramids occupy vacant tetrahedral sites on octahedrally terminated {111} surfaces of magnetite. A similar geometry was observed by Kirsch et al. (2008) for Sb(III) sorption complexes on magnetite, as well as by Auffan et al. (2008) for As(III) sorption complexes on nanomaghemite (see previous section). Upon As(III) sorption on magnetite below surface coverages of 0.2 μmol/m2, the observed dissolved As(III) concentration is below the Maximum Concentration Level recommended by the World Health Organization (10 μg/L) (Neuberger and Helz, 2005).
Another remarkable property of magnetite toward arsenite sorption found by Ona-Nguema et al. (2010) is its ability to rapidly oxidize As(III) to the less toxic As(V) form upon sorption onto nanomagnetite under oxic conditions at neutral pH. Comparison of As(III) sorption in the presence or absence of Fe(II) and under oxic or anoxic conditions indicates that As(III) is likely oxidized by ROS forming upon oxidation of Fe(II) by dissolved oxygen. Such oxidation reactions involving ROS help to explain the rapid As(III) oxidation in aerated water in presence of zero-valent iron or dissolved Fe(II) (Hug and Leupin, 2003 and references theirein). The study by Ona-Nguema et al. (2010) showed that As(III) oxidation also occurs in the presence of green-rust. Oxidation reactions involving ROS should be considered as potentially important at redox boundaries in the environment and may influence the redox cycling of pollutants.
2.4 As sorption on green rust: trimer surface species
Green rusts (GRs), [FeII(1−x)FeIIIx (OH)2]x+ (CO3, Cl, SO4)x- are particularly relevant to reducing environments because they occur in hydromorphic soils and in anoxic iron-rich sediments, and as intermediate Fe(II)-Fe(III) mineral in the corrosion pathway of zero-valent iron. This mineral may further influence arsenic mobility in groundwaters since it is a common product of the microbial reduction of ferric oxyhydroxides (e.g., Ona-Nguema et al., 2010 and references therein). After the pioneering work of Randall et al. (2001) which showed the adsorption of As(V) on the edges of GR particles using XAS, only a few studies have addressed the mechanisms of arsenic sorption on – or coprecipitation with – GRs and related layered iron hydroxides. Thoral et al. (2005) have shown that nanosized Fe(OH)2 are able to bind As(III) via surface complexes forming on the edges of the octahedral layer, after co-precipitation of As(III) with Fe(II) at high As loading. Jonsson and Sherman (2008) proposed a similar surface sorption mechanism for As(III) on GR, corresponding to edge and double corner As(III) surface complexes.
More recently, we proposed a new mode of As(III) sorption on such layered iron-hydroxides, which relies on the formation of oligomeric As(III) surface species bound to Fe octahedra via corner sharing linkage (Ona-Nguema et al., 2005). These species were first proposed in the case of As(III) co-precipitation with GR and nanosized Fe(OH)2 phases obtained via the bioreduction of As(V)-sorbed lepidocrocite by Shewanella putrefaciens (Ona-Nguema et al., 2005) (Fig. 5). Smaller polymeric species as pairs have been also proposed by Wang et al. (2010) for As(III) sorption on GR. According to the theoretical studies of concentrated As solutions by Tossell (1997), the most stable configuration for such oligomers would be a (H3AsO3)3 trimer ring. On the basis of the small amount of EXAFS data, no definitive clue can be given yet to distinguish between polymeric As(III) surface species and classical edge- and double corner-sharing surface complexes. However, the formation of sorbed oligomeric As(III) species on iron oxides is supported by the almost systematic occurrence of such polymers in the crystal structures of iron arsenite minerals (Fig. 5), and could be regarded as an alternative mode of As(III) sorption in modeling studies.
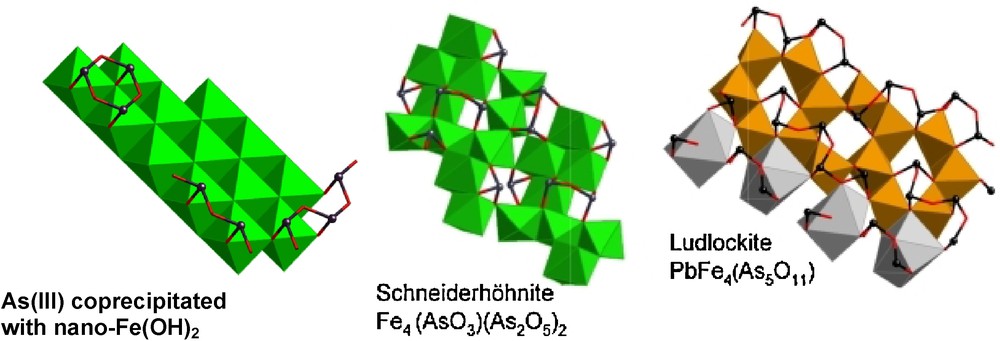
Possible model for oligomeric As(III) species coprecipitated with nanocrystalline Fe(OH)2 particles (after (Ona-Nguema et al., 2005)) compared to As(III) dimers and small chains in the structure of schneiderhönite (Hawthorne et al., 1987) and ludlockite (Cooper and Hawthorne, 1996), respectively. Iron(II) and (III) coordination octahedra are displayed in green and orange color, respectively. Arsenic(III) ions are displayed as black spheres. Lead(II) coordination polyhedron is displayed in gray color.
Modèle d’oligomères formés par As(III) et coprecipités avec des particules nanocristallines de Fe(OH)2 (d’après (Ona-Nguema et al., 2005)), comparé aux dimères et courtes chaînes d’As(III) présents respectivement dans les structures de schneiderhönite (Hawthorne et al., 1987) et de ludlockite (Cooper and Hawthorne, 1996). Les octaèdres de coordination du fer (II) et (III) sont représentés respectivement en vert et en orange. Les ions As(III) sont représentés par des sphères noires. Les polyèdres de coordination du plomb(II) sont indiqués en gris.
Eventually, although GR and related mineral phases are able to adsorb As(III) at their surfaces, the intrinsic affinity of As(III) for these phases is lower than that observed for magnetite (Jonsson and Sherman, 2008), especially at slightly acidic pH. Consequently, although these phases should be able to retard As(III) mobilization in anoxic groundwater, they are expected to be much less efficient than magnetite. However, magnetite hardly forms in natural low temperature aqueous media, due to the presence of numerous crystal growth inhibitors such as organic acids, silica, and phosphate. In addition, prolonged reducing conditions in well drained soils lead to Fe(II) leaching and subsequent mobilization of arsenic after complete dissolution of iron minerals (Fakih et al., 2009; Tufano and Fendorf, 2008).
2.5 As sorption on mackinawite
In ambient anoxic sufidic environments such as aquifers, sediments, or closed marine basins (e.g., the Black Sea) and in presence of ferrous iron, mackinawite, FeS, is the first iron sulfide to precipitate and it constitutes a major component of the empirically defined “acid volatile sulfides” characteristic of such media (Rickard and Morse, 2005). Structural characterization of FeS nanoparticulate minerals by analysis of Bragg XRD peaks is limited since diffraction patterns are dominated by broad diffuse scattering (Wolthers et al., 2005a,b). This diffuse component results from small domain sizes as well as from surface relaxation, strain, and complex disorder (Gilbert et al., 2004). In the frame of studies related to the structural characterization of mackinawite and its reactivity towards As, we used pair distribution function (PDF) analysis of both the structure and particle size of the nanoparticles (Scheinost et al., 2008). The domain size parameter of mackinawite was fit, resulting in 5.2 nm large domains. The specific surface area calculated from this particle size is 270 m2/g (Scheinost et al., 2008). PDF analysis of a freshly precipitated mackinawite showed an average particle size of 2 nm, which increased with aging to 4–5 nm (Michel et al., 2005). Using high-resolution transmission electron microscopy, laminar rectilinear prisms of 2 to 5.7 nm in thickness and 3 to 10.8 nm in length were observed for a similar sample (Ohfuji and Rickard, 2006).
The surface chemistry of mackinawite and its reactivity to As were also investigated. Acid-base titrations show that the point of zero charge (PZC) of disordered mackinawite lies at pH ∼7.5 (Wolthers et al., 2005a), and mackinawite is not stable below pH 6, the reason why it is one of the main components of the “Acid Volatile Sulfides” (Rickard and Morse, 2005). The hydrated disordered mackinawite surface can be best described by strongly acidic monocoordinated and weakly acidic tricoordinated sulfurs. The mono-coordinated sulfur site > Fe-SH determines the acid-base properties at pH ∼ PZC. At higher pH, the tricoordinated sulfur determines surface charge changes. Total site density is four sites nm−2. The surface chemistry of FeS and its acid-base titration data were adequately described using a surface complexation model by Wolthers et al. (2005a). Arsenate, AsO43−, sorption onto mackinawite is fast. As(V) sorption decreases above the point of zero surface charge of FeS and follows the pH-dependent concentration of positively charged surface species (Wolthers et al., 2005b). No redox reaction was observed between the As(V) ions and the mineral surface over the time span of the experiments. These observations suggest that As(V) predominantly forms an outer-sphere complex at the surface of mackinawite. As(III) sorption is not strongly pH-dependent and can be expressed by a Freundlich isotherm. Sorption is fast, although slower than that of As(V). As(III) also forms an outer-sphere complex at the surface of mackinawite. In agreement with previous spectroscopic studies, complexation at low As(V) and As(III) concentration occurs preferentially at the mono-coordinated sulfide edge sites. Stronger sorption of As(V) than As(III), and thus a higher As(III) mobility, may be reflected in natural anoxic sulfidic waters when disordered mackinawite controls arsenic mobility in ambient sulfidic environments.
2.6 As sorption and coprecipitation with carbonates (calcite and siderite)
Calcite is a ubiquitous mineral and most river waters are at equilibrium with calcite (Stumm and Morgan, 1981). Furthermore, many As-contaminated groundwaters are at equilibrium with siderite (FeCO3) and calcite (CaCO3) (Charlet et al., 2005; Nath et al., 2009). The sorptive capacity of siderite towards As is low (Table 1, (Jonsson and Sherman, 2008)). Although the specific surface area of these two minerals is low compared to the above-discussed minerals, the abundance of carbonate minerals, justifies a close look at their surface chemistry (Wolthers et al., 2008) and specifically at their reactions with arsenic.
Sorption of As(III) by calcite was investigated as a function of metalloid concentration, time, and pH (Roman-Ross et al., 2006). The adsorption mechanism was investigated at a macroscopic level and the coprecipitation at a nanoscopic level to determine which As species can be incorporated in bulk calcite by substitution at CO32− sites (Roman-Ross et al., 2006). The arsenic sorption isotherm, i.e. the log ΓAs(III) vs. log ([As(OH)3̊]/Assat) plot is S-shaped and has been modeled using an extended version of the surface precipitation model (Farley et al., 1985; Wolthers et al., 2008). At low concentration, As(OH)3̊ is adsorbed by complexation to surface Ca surface sites, as previously shown by the X-ray standing wave technique (Wersin et al., 1989). The inflexion point of the isotherm, where As(OH)3̊ sorption is limited by the amount of surface sites, yields six sites nm−2, in good agreement with crystallographic data. Beyond this value, the amount of sorbed arsenic increases linearly with solution concentration, up to the saturation of arsenic with respect to the precipitation of CaHAsO3(s), and is interpreted in terms of formation of an ideal solid solution.
The solid solutions formed by calcite and As(III) were examined by high-resolution, synchrotron-based X-ray diffraction, and neutron diffraction, and more recently by electron spin resonance (ESR) spectroscopy. The experimental results were compared with results from molecular modeling. The use of Density Function Theory (DFT) allows modeling the effect of the HAsO32− substitution at CO32− sites on the expansion of the unit cell volume (Fig. 6b). This effect of As substitution on calcite unit cell parameters follows Vegard's law. This allows inferring a value for the amount of As incorporated in the bulk of the mineral; the average value obtained was of [As] = 30 ± 6 mmol/kg. These results extended those published by Cheng et al. (1999) on the incorporation of AsO33− on the calcite surface to the bulk. Arsenate, where As has a tetrahedral coordination, also substitutes for CO32− groups in calcite (Alexandratos et al., 2007).
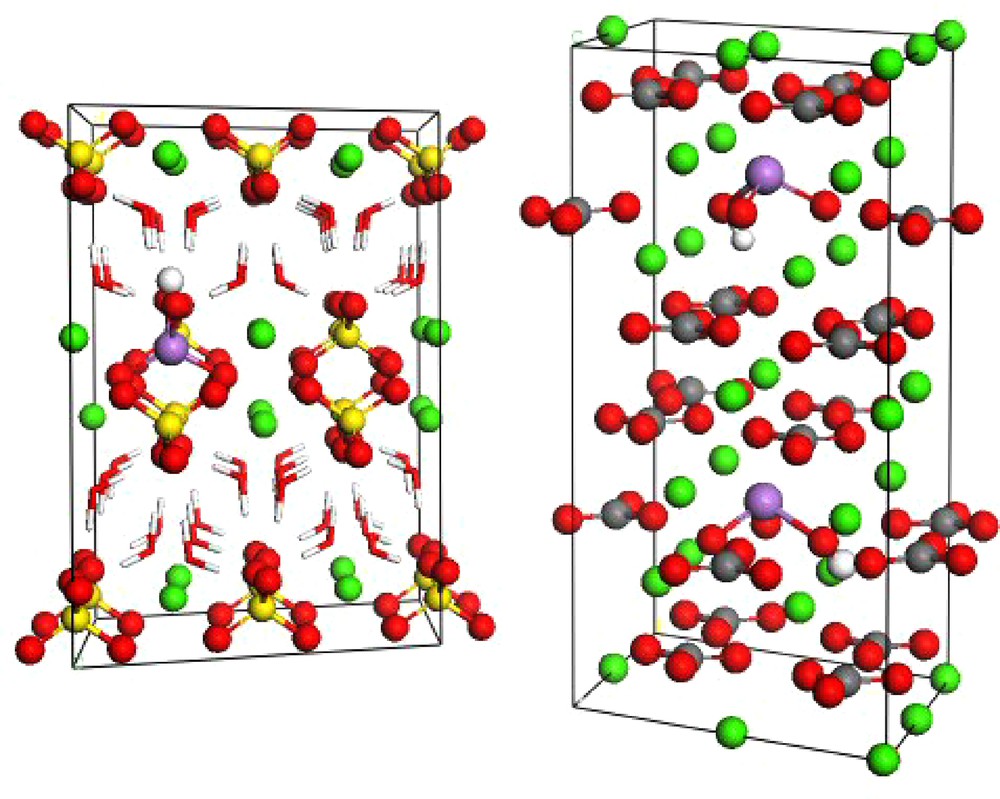
Supercells of gypsum (a, left) and calcite (b, right) showing substitutions of sulfate by arsenate (As(V); gypsum) and carbonate by arsenite oxyanions (As(III); calcite). The green spheres correspond to calcium atoms; red are oxygen; grey are carbon; yellow are sulfur and the red and white sticks in the gypsum structure are structural water molecules.
Super-maille de gypse (a, à gauche) et de calcite (b, à droite) montrant les substitutions du sulfate par les ions arséniate (As(V) dans le gypse) et du carbonate par les ions arsénite (As(III) dans la calcite). Les sphères vertes correspondent aux atomes de calcium, les rouges aux atomes d’oxygène, les grises aux atomes de carbone; les jaunes aux atomes de soufre et les bâtons rouges et blancs dans la structure du gypse représentent les molécules d’eau structurales.
2.7 As coprecipitation in gypsum
Gypsum is a common sulfate mineral that precipitates in oxic acidic environments. The ability of gypsum (CaSO4·2H2O) to host arsenic atoms in its crystalline structure has been demonstrated through experimental structural studies on the solid solutions formed upon co-precipitation with arsenic (Fernandez-Martinez et al., 2008). Neutron and X-ray diffraction methods showed an enlargement of the gypsum unit cell proportional to the concentration of arsenic in the solids and to the pH solution value. The substitution of sulfate ions (SO42−) by arsenate ions is more likely under alkaline conditions, where HAsO42− species predominates. A theoretical DFT model of the arsenic-doped gypsum structure reproduces the experimental volume expansion. EXAFS measurements of the local structure around the arsenic atom in the co-precipitated solids confirm solid-state substitution and allow some refinement of the local structure, corroborating the simulated structure. These results suggest a predominant substitution of CO32− ions by HAsO42− species (Fig. 6a). The possibility that other species such as AsO43− may be incorporated in the bulk structure of gypsum was also evaluated. The presence of layers of water in the gypsum structure opens the possibility that some hydronium could be present, compensating the extra charge brought by the unprotonated arsenate species. EXAFS results combined with molecular modeling of the As local structure disregarded this possibility. The charge redistribution within the structure upon substitutions of either the protonated or the unprotonated arsenate species is studied by means of Mulliken Population Analyses, demonstrating an increase in the covalency of the interaction between Ca2+ and AsO43−, whereas the interaction between Ca2+ and HAsO42− remains predominantly ionic.
3 Artifacts in sampling, preparation and spectroscopic analysis
3.1 Potential oxidation of As(III) and Fe(II) from sampling to analysis
The quality of both field and lab work related to Fe(II)-rich minerals and their redox reaction with arsenic species depends on the control of oxidation artefacts which may occur at any step of the work. Lab work may only be performed in a glove box filled with nitrogen. Although some studies use a mixed H2/N2 atmosphere, the “inert” role of H2 remains an open question, as hydrogen may act as a reductant, e.g. for selenium(IV) ions sorbed on clay mineral particle (Charlet et al., 2005). The oxygen partial pressure (pO2) must be monitored continuously by an O2 sensor and never allowed to exceed 1 ppm. This oxygen partial pressure is still high compared to anoxic aquifer conditions and every precaution must be taken to minimize the presence of oxygen which may be introduced, e.g. by the standard solutions. The latter should be introduced inside the glovebox using dry salts and water previously boiled and degassed outside the glovebox. Use of additional O2 traps (such as Fe(OH)2 suspensions or reduced Cr solutions, open to the glovebox atmosphere, are advised to further reduce the O2 content in the glovebox atmosphere. When these anoxic conditions are not met, artifacts are observed. For instance As(III) rapidly oxidizes in the presence of green-rust and magnetite upon drying in air, which may be explained by the formation of ROS upon reaction of surface Fe(II) with dissolved oxygen (Ona-Nguema et al., 2010).
Extreme care must also be taken to prevent sample oxidation during the transport from the glove box to the spectroscopy facilities. Small aliquots (a few mL) of suspensions are typically filtered (Millipore filter 0.022 lm) and the wet pastes then transferred to Mössbauer or XAFS sample holders. The sample holders are sealed with Kapton tape (XAFS spectroscopy) or with epoxy resin (Mössbauer spectroscopy) and placed in small plastic boxes. All these steps are performed in the glove box. The samples are then immediately shock-frozen with liquid N2 and must be transported to the spectroscopy facilities in a Dewar flask filled with liquid N2. At the synchrotron facility, they are transferred within 2 min from the Dewar to, for example, a closed-cycle He cryostat with He atmosphere and 15 K temperature, used for XAFS measurements. At the Mössbauer facility, the samples are transferred within 1 min from the Dewar to the Mössbauer bath cryostat with a He gas atmosphere.
Working in the field with anoxic conditions at the middle of a delta with 40 ̊C temperature is even more a challenge. Core samples are particularly delicate to retrieve and to handle. Lake sediments are best obtained like in Sweden in winter time, with the so-called sword technique. The hollow sword is introduced gravimetrically in the sediment. It is then filled with liquid CO2. The sediment freezes around the sword and is brought back frozen to the surface. Since sampling occurs usually in winter from a hole drilled in surface ice the frozen sediment can be directly transferred to the cold room. The same concept is soon to be used to retrieve samples from delta sediments. A specially designed coring tube once it reaches the desired depth may be sealed on both ends, before retrieval, by freezing with liquid CO2 or N2. It is brought back, with its two frozen ends, to the surface where it is immediately introduced in a field campaign glovebox. Pore water, bacteria and particles can then be obtained at each depth without artifact.
The collection of water and particles without oxygen or bacterial contaminations can be done in a cheaper way, using the needle sampler technique (Van Geen et al., 2004). Unconsolidated deposits are drilled in India or Bangladesh using the manual “handflapper” method with 3-m sections of PVC or galvanized iron (GI) pipe (Van Geen et al., 2004). A group of five local drillers can drill that way down to 30 m in a given day. At depth intervals of 2–4 m, the drilling is interrupted. The drilling pipes are removed and an empty cylinder is screwed on the bottom pipe. Vacuum is made in this cylinder and a hollow needle is squeezed into the rubber cork which closes the sampling cylinder. The pipes are reintroduced into the borehole, until they touch the bottom of the drill hole. The needle then penetrates a depth ∼ 0.3 m below the bottom of the drill hole. It is mechanically pushed inside the cylinder. The slurry sample (∼ 100 mL) made of groundwater, bacteria and fine particles are then sucked into the cylinder. Once brought back at the surface, and immediately after collection, the headspace of the needle-sampler is purged with N2. About 5–10 mL of groundwater contained in the needle-sampler is then filtered under a gentle N2 pressure through a 0.45 μm syringe filter and into acid-leached polyethylene scintillation vials (PolySeal Cap.) for further element total concentration analysis. In order to preserve arsenic speciation in water, additions of phosphoric acid are further often recommended and this conservation method is discussed in (Daus et al., 2006). Another aliquot of groundwater is filtered into scintillation vials that have been rinsed with MQ water only for anionic species measurement. Sediment contained in the N2 purged needle-sampler is stored in the dark until further processing on the evening of collection.
3.2 Beam-induced speciation changes: As oxidation on Fe minerals and reduction with organic matter
A major limitation of XAFS for studying redox- sensitive elements such as arsenic in dilute samples comes from beam damage. Indeed, although 3rd generation synchrotron X-ray sources allow one to reach very low detection limits in concentration (XANES ≈ 0.1 ppm and EXAFS ∼ 10 ppm) on very powerful undulator (e.g., ID26 at ESRF) and wiggler beamlines (e.g., BL 11-2 at SSRL), and about ten times higher on focused BM beamlines (e.g., FAME at ESRF), the high photon flux on the sample may easily change the redox state of the analyzed element during the course of a measurement. In the case of biological or organic samples, the beam can also alter the integrity and molecular structure of the sample. This issue is even more crucial in the case of a focused beam on the micrometer- or nanometer-scale. Beam damage artifacts are especially important in the case of arsenic. Photo-oxidation and photo-reduction can both be observed and depend on the presence of electron acceptors and donors in the sample matrix, both reactions being activated by the beam. This activation is explained by the intense ionization of the sample under the X-ray beam, which leads to the photo-emission and migration of electrons that can facilitate electron recombination between electron donors and acceptors. In general, thermodynamically favorable but kinetically-limited oxidation and reduction reactions are activated under the beam, as for instance As(III) oxidation by Fe(III) and As(V) reduction by organic molecules. In a given matrix, the proportion of oxidized or reduced As directly depends on the total As concentration and reaches a plateau that might be related to a limiting distance between the electron donor and acceptor atoms. Fortunately, the rates of such beam-activated electron recombinations can be dramatically lowered by decreasing the temperature during beam exposure. For instance, the oxidation of As(III) sorbed Fe(III)-oxides is very fast under the beam at ambient temperature but can be significantly slowed at cryogenic temperatures (preferably below 20 K) (Fig. 7). XAFS data that requires the averaging of several scans can then be recorded by moving the position of the beam spot between each scan (Morin et al., 2008 and references therein). In addition, the quick scan mode (< 1 mn) is required at very low As concentration (< 100 ppm) since significant oxidation of As can occur in a few tens of seconds.
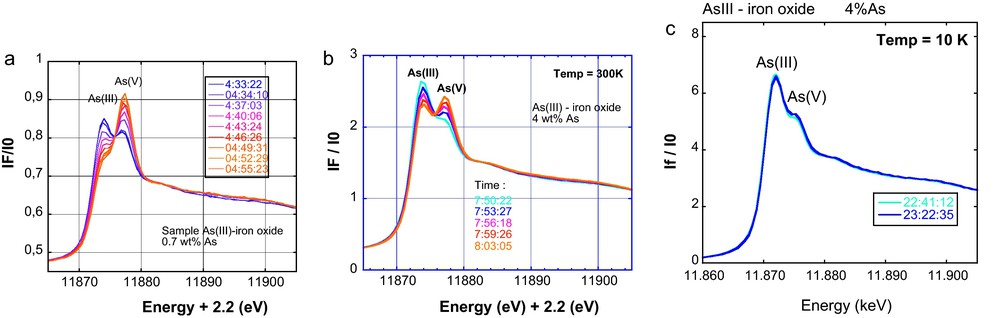
XANES spectra at the As K-edge, for As sorbed onto ferrihydrite recorded in 2002 on ID26 and FAME-BM30B ESRF beamlines. At room temperature, successive XANES scans in quickscan mode (20 sec per scan) shows that As(III) oxidizes rapidly under the beam. Such rapid photo-induced oxidation hinders the recording of EXAFS data within a large energy range. The oxidation rate decreases when increasing the As/Fe ratio in the sample, from (a) to (b) and when the recording temperature decreases, i.e. (b) to (c). At cryogenic temperature (c), the oxidation rate is low enough to record a 40 min long EXAFS scan without significantly modifying the As oxidation state (<10% oxidation).
Spectres XANES enregistrés à l’ESRF en 2002 sur les lignes ID26 et FAME-BM30B au seuil K de As, pour des échantillons d’As adsorbé sur la ferrihydrite, A température ambiante, les scans successifs de XANES en mode scans rapides (20 secondes par scan) montrent que As(III) est rapidement oxydé sous faisceau. Une telle oxydation photo-induite rapide interdit l’enregistrement de spectres EXAFS sur une grande gamme d’énergie. La vitesse d’oxydation décroît quand le rapport As/Fe de l’échantillon augmente, de (a) à (b), et quand la température d’enregistrement des spectres décroît, de (b) à (c). A température cryogénique, (c), la vitesse d’oxydation est suffisamment faible pour permettre l’enregistrement d’un spectre EXAFS de 40 minutes sans modification notable de l’état d’oxydation de l’arsenic (< 10 % de As est oxydé).
4 Water treatment
4.1 Drinking water requirements
Because of a constant increase of public awareness concerning the importance of water quality, water regulations have continued to lower the MCL for pollutants. In the case of arsenic, the WHO reduced in 2002 the arsenic standard for drinking water from 50 μg/L to 10 μg/L. Reinforcement of regulations generates a strong need to improve processes to remove pollutants from water and to control water-treatment by-products. A wide range of physical-chemical and biological methods are already used to extract organic and/or inorganic contaminants from polluted waters. Among the most common methods such as coagulation-flocculation, membrane processes and adsorption, the use of inorganic salts as coagulation-flocculation agents such as AlCl3-Al13 (Bottero et al., 1980, 1982; Masion et al., 2000) and FeCl3-Fe24 (Bottero et al., 1993, 1994; Vilge-Ritter et al., 1999) polycation species is the most efficient and less expensive process for the removal of colloids and organics in water treatment. But such an approach has the disadvantage of generating a high volume of sewage sludge and results in difficulties to reuse the metals they content.
For water treatment, use a decontamination process that does not generate residuals is an advantage. In the case of arsenic removal, the use of magnetically assisted chemical separation (MACS) may be of great help (Ngomsik et al., 2005). Indeed MACS involve superparamagnetic particles (iron oxide microspheres of 0.1 to 25 μm of diameter) that can be recovered easily, leading to no residual production since iron oxide can be regenerated after adsorption. However, even if micron-sized adsorbents have an internal porosity that increases their specific surface area (SSA), the diffusion within the particles limits their adsorption efficiency. Then the 10 μg/l As MCL is often difficult to achieve by classical techniques
4.2 Nanoparticle–based adsorption treatment
As previously discussed, magnetic iron oxide nanoparticles (magnetite and maghemite) represent a new generation of environmental remediation technologies that could provide cost-effective solutions for water and industrial liquid waste treatments. The fact that nano-magnetic iron oxide may adsorb a larger amount of arsenic per unit surface area than other larger particle size adsorbents may be advantageous in reaching the arsenic MCL, even if size-dependent reactivity may be a limiting factor. Two main advantages exist: (i) a large specific surface area (SSA); and (ii) the separation of metal-loaded magnetic nano-adsorbents can be achieved via an external magnetic field. For instance, the SSA of an oxide nanoparticle of 10 nm in diameter is ≈ 100 times larger than the SSA of an oxide particle of 1 μm. Surface hydroxyl groups are the chemically reactive entities at the surface of solids in an aqueous environment. A higher SSA increases the number of available functional groups on the nanoparticle surface. Consequently, for a given mass, the maximum adsorption capacity of ions in solution is higher for nanoparticles than for micron-sized particles.
Most of the published work concerning arsenic removal has focused on the use of nano-metal oxides or oxyhydroxides (mostly iron and aluminium). Nano-akaganeite (Deliyanni et al., 2003), ferrihydrite (e.g., Dixit and Hering, 2003; Jain et al., 1999; Ona-Nguema et al., 2005; Raven et al., 1998; Waychunas et al., 1993; Wilkie and Hering, 1996), and other iron nano oxy-hydroxides (e.g., goethite, lepidocrocite (Farquhar et al., 2002; Manning et al., 2002; Sun and Doner, 1998; Wilkie and Hering, 1996), nano zerovalent iron (Kanel et al., 2005), nano-TiO2 (Jegadeesan et al., 2010) etc., have been tested as adsorbents for arsenic removal. Nevertheless, the most promising nanoparticles need to combine strong adsorption efficiency and magnetic properties for easy removal and regeneration. Accordingly, magnetite and maghemite remain on top of the list.
4.3 Limitations in recycling nanoparticles
Even if a strong affinity is required between the adsorbent and arsenic to reach the arsenic MCL in water, this may be a drawback in reuse of adsorbents. Indeed, the improvement of water treatment processes requires the attainment of a high level of adsorbent material reuse and recycling. However, a strong linkage between arsenic and the adsorbent, similar to that between As and nano-maghemite or nano-magnetite, may limit the regeneration step. Desorption necessitates breaking the chemical linkage between arsenic and the particle surface. For such reactions, acidic or basic baths are needed, which can efficiently desorb arsenic, but such treatments also may alter the adsorbent. Use of corrosive solutions may dramatically increase maintenance costs.
Moreover, for high arsenic concentration waters, the high solubility (10 mM H3AsO3 at pH 7) of the amorphous surface precipitate present at the surface of nano-magnetite is responsible for a dramatic increase of dissolved As concentrations at high As(III) surface coverage. This mechanism might lower the process efficiency, limits its use to not too highly contaminated waters and affect the adsorbent renewal capacity.
5 Conclusion and outlook
The sorption mechanisms on a variety of minerals of importance for delta aquifer environment have been reviewed. New mechanisms have been identified, which include the formation of (i) an amorphous As(III)-rich surface precipitate on the surface of magnetite, (ii) a strong tridentate inner-sphere complexes in vacant tetrahedral sites of magnetite and maghemite nanoparticles and (iii) an outer-sphere complexes formed on the surface of hematite. The relevance of each of these mechanisms in natural oxic and anoxic waters and in water treatment or decontamination systems is still to be understood. However, these fundamental investigations have shed light onto fundamental aspects of the mineral/water interfaces, bringing new ideas and thinking about the growth properties of iron oxide nanoparticles to minimize surface tension, the properties of the electric double layer on mineral/water interfaces and the co-existence of inner and outer-sphere complexes, or the existence of polymeric species of As(III), that had been classically underestimated.
This review suggests that, although the mechanisms by which toxic arsenic aqueous species are trapped in natural and engineered environments on mineral particles are now fairly understood, the integration of these mechanisms in an aquifer broad scale geochemical perspective still needs additional work. Of particular interest are studies that include the redox properties of As(III)/(V) and Fe(II)/(III) phases. The fact that “amorphous” Fe(II)-Fe(III)-bearing phases are the predominant As scavengers makes understanding the redox coupling between these two elements as being an essential know-how for the development of effective remediation strategies. In this sense, the ongoing efforts to understand the geochemical behavior of As in Southeast Asian groundwaters are good examples of combined fundamental and applied investigations. Studies made during the last 5 years have lead to critical achievements in our understanding the coupling between Fe(III) oxyhydroxides reductive dissolution and the mobilization of As(III) toxic species in groundwaters. More research in this field is needed to understand the relative influence of Fe(II)-bearing solid phases on the redox properties and on the availability of arsenic in these environments.
Acknowledgements
We gratefully acknowledge Georges Calas and Gordon E. Brown Jr., for having invited us to participate to this special issue, and for their precious help in the editing of this manuscript. LC acknowledges partial financial support by IUF, ANDRA, AQUATRAIN and CALIBRE EU networks and by EC2CO CNRS/INSU program. The work done by GM was funded by EC2CO-CYTRIX CNRS/INSU program, by ACI/FNS grant #3033, and by SESAME IdF grant #1775. Support was also provided by NSF grant CHE-0431425 (Stanford Molecular Environmental Science Institute). The XAS work was performed at the European Synchrotron Radiation Facilities, Grenoble, France and Stanford Synchrotron Radiation Lightsource, Stanford, USA. We are grateful to the machine and beamline groups whose outstanding efforts have made these experiments possible.