

1 Introduction
Mercury (Hg) is a natural element, present in every major compartment of our planet at low concentrations. Fig. 1 illustrates the relative proportions of Hg in key Earth surface reservoirs, and fluxes between reservoirs. Primary natural Hg emissions to the atmosphere, estimated at 500 Mg/year, result from aerial and sub-aerial volcanism and gradual degassing of soil systems (Fitzgerald and Lamborg, 2004; Selin, 2009). Man-made activities have modified the natural cycling of Hg by intentional and unintentional use of this metal. Intentional use includes Hg mining and the use of Hg in industrial processes, notably chlor-alkali plants and other chemical industries. The intentional use of liquid Hg by artisanal gold miners is also estimated to induce large losses to the environment, −727 Mg in 2010, (UNEP, 2013). Unintentional emissions of Hg are generated predominantly by coal-fired power plants, cement production, and pyrometallurgy. In these three industrial sectors, Hg is initially present at trace levels in feed coal, limestone, clay, and metal ores and is liberated to flue gas under high-temperature process conditions. The current best estimates for total primary anthropogenic Hg emissions are in the 1960–2800 Mg/year range (Pirrone et al., 2010; Selin et al., 2008; UNEP, 2013).
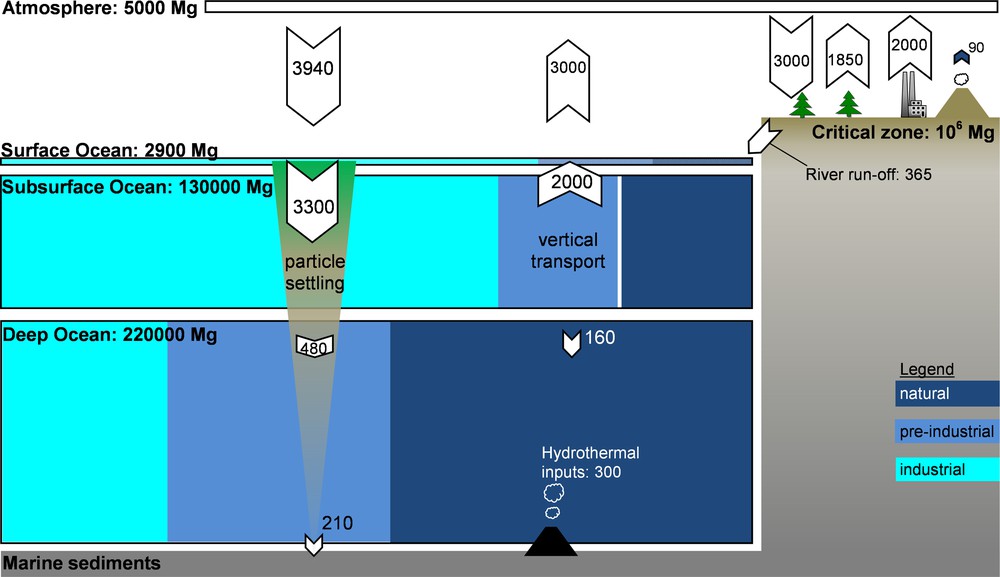
Box model of the modern-day global mercury cycle, based on Amos et al. (2013). Reservoir sizes (Mg) and fluxes (Mg/year) are approximately drawn to scale, except for marine sediments and the critical zone. Ocean reservoir box colors represent the estimated proportions of industrial emissions from 1850 to 2008 (), pre-industrial anthropogenic emissions from 2000 BC to 1850 (), and background natural emissions () (Amos et al., 2013). The diagram highlights the importance of legacy mercury at the Earth's surface environment. The sea-floor hydrothermal flux is from Fitzgerald and Lamborg (2004). The modern mercury cycle is not at steady state, and anthropogenic mercury emissions are accumulating primarily in the oceans and in continental critical zones.
Geographically, anthropogenic emissions are dominated by Asia (68% in 2005; Fig. 2), while the combined European and North American emissions have fallen to a 22% share in 2005 (UNEP, 2013). Past anthropogenic Hg emissions, deposited to soils and surface oceans have a tendency to be re-emitted back to the atmosphere. A recent study suggests that the contribution of these historical or so-called legacy Hg emissions to modern Hg exposure is much larger than thus far appreciated (Amos et al., 2013). By modeling how 4000 years of historic Hg emissions propagate through Earth surface reservoirs, these authors estimate that legacy Hg constitutes 60% of modern Hg deposition. Similarly, accumulated legacy Hg in the surface ocean causes North American and European (31%) marine Hg burdens to outweigh Asian contributions (18%).
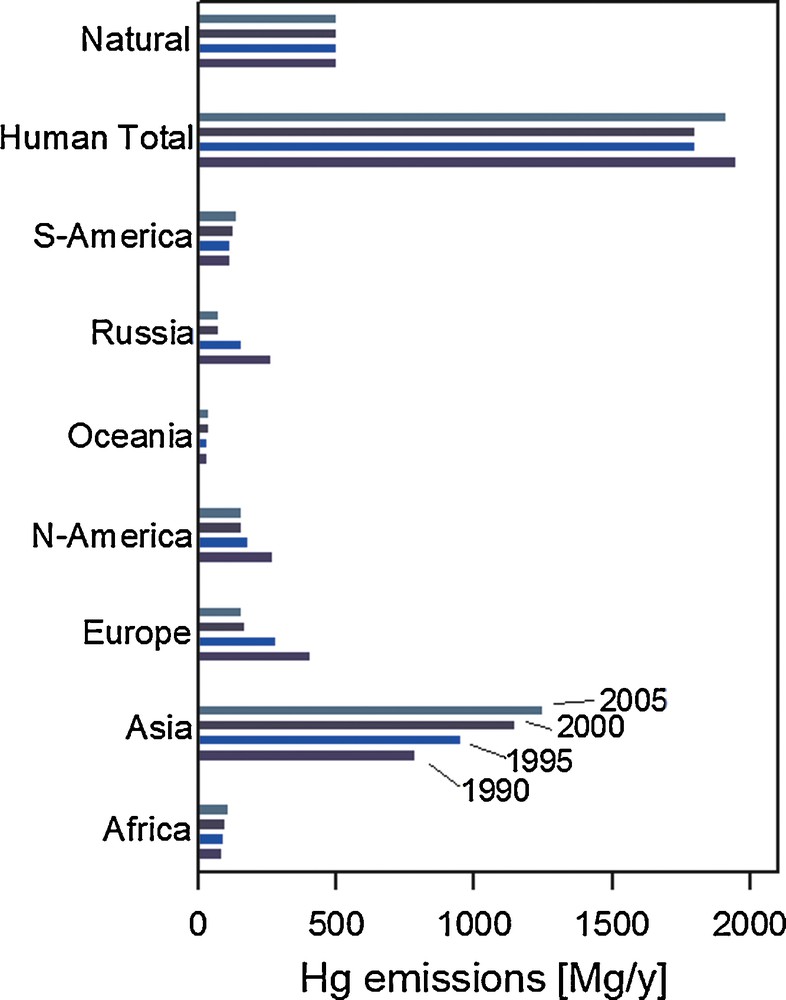
Revised estimates of anthropogenic mercury emissions to air (Mg/year) in 1990, 1995, 2000 and 2005 in different continents/regions. Based on (AMAP/Wilson et al., 2010) and (Selin et al., 2008).
Industrial accidents related to intentional use of Hg, such as the release of mono-methylmercury (MMHg) to the Minamata Bay in Japan in the 1950s have raised awareness about the toxicity of Hg (McAlpine and Araki, 1958). Subsequent toxicological and epidemiological research has found that low-level exposure to MMHg, mostly from seafood, may impair the neurological development of the unborn and of infants (NRC/NAS, 2000; Suzuki et al., 1991). Populations at particularly high risk are those that rely on aquatic biota for their protein intake, such as indigenous people living in the Arctic region (AMAP, 2011).
Parallel research has identified MMHg as a widely dispersed natural form of Hg that is produced by microbes in oxygen-poor aquatic environments, such as sediments (Compeau and Bartha, 1985; Jensen and Jernelov, 1969). MMHg has the unfortunate property of bioaccumulating and biomagnifying in aquatic food chains. Consequently, MMHg concentrations increase stepwise from plankton to copepods to small fish and large top predators, including seabirds, marine mammals, and humans. In developed countries, MMHg exposure to humans results predominantly from marine fish consumption (Sunderland, 2007).
Atmospheric circulation allows a rapid and efficient spreading of emitted Hg across the Earth's surface. Anthropogenic Hg emissions are predominantly (53%) in the form of gaseous elemental Hg (GEM), followed by gaseous oxidized Hg (GOM, 37%), and particulate Hg (PHg, 10%, < 2.5 μm fraction) forms (Pacyna et al., 2006). GEM has a long atmospheric lifetime, as it is poorly reactive under typical atmospheric conditions. Eventually, GEM is oxidized in the atmosphere and the divalent forms of Hg (PHg, and GOM) are deposited by wet and dry pathways to the Earth's surface. With anthropogenic GEM emissions outweighing pre-industrial GEM emission by a factor 4 since over 200 years, inorganic Hg deposition to remote ecosystems has tripled since pre-industrial times (Lindberg et al., 2007; Streets et al., 2011). Increased Hg deposition is thought to stimulate Hg methylation by microbes and in turn amplify food web MMHg dynamics and top predator MMHg exposure (EPA US, 1997; Harris et al., 2007). The relationship between inorganic Hg emissions and biota MMHg levels and MMHg exposure is, however, all but linear (AMAP, 2011). Major uncertainties continue to exist on the exact sites and mechanisms of MMHg production in the natural environment. Recent research has also questioned the role that climate and climate change may play in the biogeochemical cycle of Hg (AMAP, 2011; Stern et al., 2012). In the following, we will draw attention to those areas of research where paradigms are shifting or where major outstanding questions remain.
2 Mono-methylmercury toxicology and epidemiology
2.1 Mono-methylmercury reference dose
All aspects of environmental Hg research and policy are intricately linked to our best knowledge of MMHg toxicity. Thus far, developmental neurotoxicity of the fetus has been considered to be the most critical endpoint. Although increasing evidence is showing positive association between MMHg exposure and cardiac functions, there is presently “insufficient to possible evidence” that MMHg exposure increases the risk of cardiovascular disease in adults (FAO/WHO, 2010; Mergler et al., 2007). A reference dose for fetal MMHg neurotoxicity has been established by the National Research Council of the US National Academy of Sciences (NRC/NAS, 2000). Reference dose can be defined as the “estimate of daily MMHg exposure to a human population that is likely without appreciable risk of deleterious effects during a lifetime”. The NRC/NAS analysis was based on three well-known epidemiological studies in the Seychelles Islands, Faroe Islands and New Zealand, that all investigated mother–infant pairs in fish-eating communities. In the Faroe Islands and New Zealand studies, MMHg exposure was associated with poor neurodevelopmental results, while no relation with such results was observed in the Seychelles study (Davidson et al., 1998; Grandjean et al., 1997; Kjellstrom et al., 1989). The NRC/NAS concluded that the strengths of the Faroe study made it the most appropriate to determine the reference dose (NRC/NAS, 2000).
The NRC/NAS recommendations led the United States Environmental Protection Agency (US EPA) to adopt a 58 μg/L Hg in cord blood benchmark dose level, i.e. the concentration at which a doubling of the percent of children performing at the lowest 5% on a neurological test took place. An uncertainty factor of 10 was then applied to take into account physiological and pharmacokinetic variability, resulting in a 5.8 μg/L Hg level in cord blood. This value corresponds to a reference dose of 0.1 μg Hg per kg bodyweight per day (μg/kg bw/day). For monitoring purposes, a 5.8 μg/L Hg level in cord blood corresponds to 1.4 μg/g total Hg in human hair, an approximation that is based on a maternal hair:blood Hg ratio of 250 (μg/g per mg/L; NRC/NAS, 2000). It has been found that 16% of American women of childbearing age surpass these limits (Mahaffey et al., 2004). Similarly, a limited survey of hair Hg levels in the city of Toulouse (France) suggests that about 20% of women of childbearing age have Hg levels exceeding the NRC/NAS hair reference dose level of 1.4 μg/g (Fig. 3).
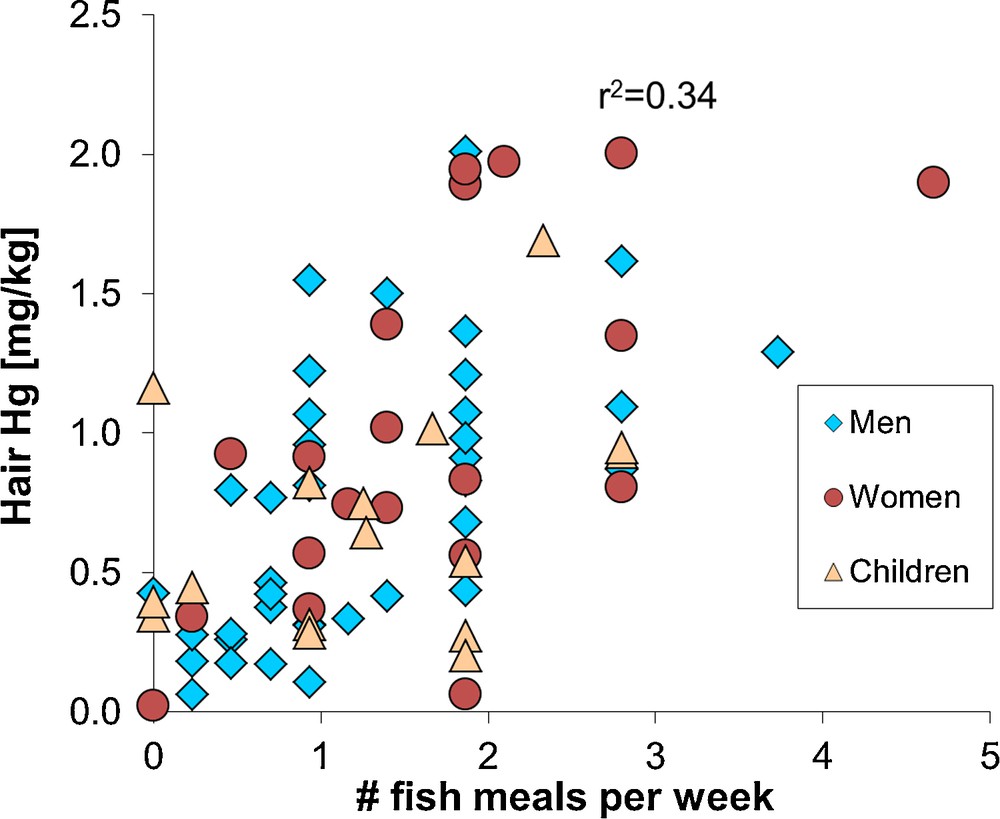
Hair total mercury levels in a human population (Toulouse, France), as a function of the number of fish meals consumed each month. The correlation is significant (r2 = 0.34, P < 0.05). Similar results have been observed for larger French cohorts (Pichery et al., 2012). Four out of 20 women (20%) of childbearing age have hair mercury levels in excess of 1.4 μg/g, which is the level corresponding to the United States Environmental Protection Agency reference dose for mono-methylmercury exposure. None of the female hair mercury levels exceed the French ANSES reference dose level of 3.2 μg/g. All analyses made by combustion – atomic absorption analysis (Milestone DMA-80) at the OMP-GET laboratory; analysis uncertainty is 10% 2RSD.
Public health institutions provide fish consumption advisories to women of childbearing age and young children based on the NRC/NAS reference dose of 0.1 μg/kg bw/day or on the World Health Organization (WHO) provisional tolerably weekly MMHg intake of 1.6 μg/kg bw/week (i.e. 0.23 μg/kg bw/day). The difference between the NRC/NAS and WHO reference dose resides in the different uncertainty factors of 10 and 6.4, respectively. The WHO, the US EPA or the French ANSES (previously AFSSA) recommend women and young children to, first, avoid consumption of large predator fish such as tuna, shark, swordfish, and privilege consumption of small fish or farmed fish such as herring, salmon, mackerel, and second to limit the number of fish meals to 1–2 per week. Fig. 3 shows that these recommendations are coherent: women and children that consume less than two fish meals per week generally have less than 1.4 μg/g Hg in their hair. Not all health institutions give fish consumption advisories for adults (other than women of childbearing age), because the MMHg reference dose is difficult to determine due to confounding of childhood and adult exposure effects (Mergler et al., 2007). The WHO recommends a provisional tolerably weekly MMHg intake of 3.3 μg/kg bw/week, which corresponds to approximately 7 μg/g Hg in human hair.
2.2 Risks and benefits of fish consumption
Continued study of the Seychelles main cohort has not revealed consistent neurotoxic evidence from neither prenatal, nor postnatal MMHg exposure. On the contrary, at ages of 5, 10 and 17 years, improved test performance has been associated with increased prenatal MMHg exposure, and was suggested to reflect the nutritional benefits of fish consumption (Davidson et al., 1995, 2011; Myers et al., 2003). Seafood contains n–3 polyunsaturated fatty acids (PUFAs) which are essential for child neurodevelopment (Mozaffarian and Rimm, 2006). In order to test the influence of maternal PUFA status during pregnancy on neurodevelopment, a subsequent Seychelles cohort has been followed since 2001. Indeed at age 9 and 30 months, maternal hair PUFAs had a positive, and hair MMHg a negative effect on infant psychomotor development (Stokes-Riner et al., 2011). Language test scores improved with maternal hair PUFA levels and showed no relation with MMHg for the same cohort at 5 years of age (Strain et al., 2012). The authors suggest that higher fish consumption, resulting in higher maternal prenatal PUFA status, is associated with beneficial developmental effects rather than detrimental effects resulting from the higher fetal exposure to MMHg. Over the last decade, researchers have slowly started to recognize the need to balance the risks and benefits of fish consumption (Hellberg et al., 2012; Mahaffey et al., 2011; Mozaffarian and Rimm, 2006). In 2010, the FAO/WHO conducted an expert analysis of the risks and benefits of MMHg and PUFA from fish consumption by directly comparing the respective loss and gain of intelligence quotient points (IQ) (FAO/WHO, 2010). Based on the FWO/WHO analysis, we illustrate on Fig. 4 how the positive IQ effects of PUFA outweigh the negative IQ effects of MMHg for typical fish consumption. The right panel shows the same calculation, but for a scenario using the upper uncertainty limit for MMHg neurotoxicity and consumption of fish with a total Hg content of 1 μg/g, i.e. the upper legal level for commercialization of top predator fish species in the European Union and North America. Our latter worst-case scenario, the consumption of three top predator fishmeals per week by pregnant women, still generates a net beneficial outcome on fetal neurodevelopment.

Risk-benefit analysis based on (FAO/WHO, 2010). Relationship between maternal fish consumption and fetal neurodevelopment, approximated as the gain and loss in intelligence quotient (IQ) points due to n–3 polyunsaturated fatty acids (PUFAs), benefits and mono-methylmercury (MMHg) exposure. Positive IQ effects of PUFA outweigh the negative IQ effects of MMHg for typical fish consumption. Corresponding maternal hair mercury (Hg) levels (mg/kg) are also illustrated assuming a ratio of blood to hair MMHg of 1:250. Left panel: calculation for a typical fish diet: 0.16 mg Hg per kg of seafood (Mahaffey et al., 2011), a median 5.5 mg PUFA per kg seafood, 60 kg body weight, 100 g servings (FAO/WHO, 2010), and a central IQ decrement of −0.465 points per μg/g Hg in maternal hair (Pichery et al., 2012). Right panel: calculation for 1.0 mg Hg per kg of seafood (tuna, shark, swordfish), and a maximum IQ decrement of −0.62 points per μg/g Hg in maternal hair (Pichery et al., 2012); all other parameters identical. Note that we updated the original FAO/WHO central and upper IQ regression coefficients of −0.18 and −0.70 by the values of Pichery et al. (2012) (−0.465 and −0.62) following a discussion in the literature (Grandjean et al., 2012).
Behavioral studies on the effectiveness on MMHg related fish consumption advisories show varying results. For example, the well-publicized 2001 US federal advisory on fish consumption by pregnant women resulted in a significant decrease in fish consumption (from 1.8 to 1.5 fish meals per week) in one study (Oken et al., 2003). A second US study did not show a decrease in fish consumption, but did note a decrease in maximum exposure levels (Mahaffey et al., 2009). A French study showed a decrease from 3.2 to 2.8 fish meals per week by women following a fish consumption advisory (Verger et al., 2007). Our quantitative risk-benefit analysis on Fig. 4 suggests that French fish consumption is optimal while the US advisory is aggravating an already suboptimal fish consumption by women. Therefore, taking into account both the risks and benefits of seafood consumption, we suggest that current nation-wide fish advisories may be too conservative and require modification. Nevertheless, we also emphasize that reducing anthropogenic MMHg contamination in fish would result in even greater neurodevelopmental benefits from fish consumption.
In summary, the state of knowledge on MMHg toxicity and health effects has been making good progress over the last decade. National and international efforts in weighing the benefits and risks of fish consumption on child development are advocating an important paradigm shift: the benefits are higher in most cases. More research is also needed on the potential cardiovascular effects of MMHg exposure to adults, and on the confounding between beneficial PUFA and toxic MMHg observations in epidemiological studies (Stern and Korn, 2011). Finally, the risk/benefit analysis of fish consumption needs to be expanded to contaminants other than MMHg, and benefits other than PUFA. While fish advisories may require a re-think, MMHg neurotoxicity is not put into question and drastic Hg emission reductions are the only absolute means to lower the MMHg exposure risk for generations to come.
3 Atmospheric mercury cycling
3.1 Mercury oxidation – From ozone to bromine
GEM is the major component (more than 98%) of the atmospheric Hg burden. Due to its low reactivity, GEM is well mixed at a hemispheric scale. The GEM atmospheric lifetime is driven by gas phase oxidation processes. It was first established that ozone efficiently removed GEM from the gas phase (P’yankov, 1949); however there are still some uncertainties in the rate constants (Ariya et al., 2008). Hg oxidation by OH radicals is also an important pathway for GEM removal (Sommar et al., 2001). An atmospheric lifetime of less than a year for GEM can be derived from the combined OH/ozone oxidation reactions. In 1998, Canadian researchers found that the GEM lifetime could drop to a few hours during springtime in the high Arctic (Schroeder et al., 1998). These atmospheric mercury depletion events (AMDEs) were closely related to ozone depletion events and initiated a tremendous effort on both Arctic Hg research and more broadly on atmospheric Hg chemistry research. The AMDEs highlighted the role of halogen radicals as alternative oxidants for GEM and reactions with atomic (Cl, Br, I), molecular (Cl2, Br2, I2) or BrO/ClO were studied (Ariya et al., 2009). It is currently suggested that the oxidation of GEM by Br is likely involved in the fast removal process of atmospheric Hg. Under Arctic springtime conditions found during AMDEs, a lifetime of 6 h to 2.5 days for GEM can be derived (Donohoue et al., 2006). The discovery of AMDEs led to a reexamination of the role of Br on Hg removal processes on a global scale. Br atoms are today considered as a major oxidant for GEM in the marine boundary layer, in the upper troposphere and during polar sunrise (Brooks et al., 2006; Laurier et al., 2004; Lyman and Jaffe, 2012; Obrist et al., 2011; Steffen et al., 2008). Modeling studies even suggest that Hg oxidation by Br can consistently simulate GEM lifetime and distribution on a global scale (Holmes et al., 2010; Seigneur and Lohman, 2008). Parallel observations of atmospheric Hg and Br speciation are scarce, however, and continued research efforts are required. Finally, while Hg oxidation pathways have been examined in the gas phase, a recent review illustrates that heterogeneous pathways that occur in clouds, on snow grains, or on particle surfaces are poorly known (Subir et al., 2011). Environmental surfaces may therefore play a more important role than recognized in the oxidation and removal of GEM from the atmosphere.
3.2 Atmospheric mercury speciation measurements
There are still multiple uncertainties associated with atmospheric Hg dynamics due to experimental difficulties and field observation limitations. The development of the first automated instruments for the concurrent monitoring of GEM, GOM, and PHg species significantly improved our knowledge of atmospheric Hg speciation (Landis et al., 2002). GOM and PHg concentrations are low, typically around 5 pg/m3, and are operationally defined. GOM probably includes gaseous HgCl2, HgBr2 and/or HgO compounds, but there is a lack of knowledge on which and how much of the Hg(II) species are analyzed by different GOM methods and under different atmospheric conditions. There is a clear challenge to develop new methods that may accurately identify and quantify the different forms of GOM.
3.3 Polar mercury dynamics
Observation of Arctic AMDEs stimulated investigations in the Southern Hemisphere and a dramatic reactivity of GEM was subsequently observed in coastal Antarctica (for a review, see Dommergue et al., 2010). At first sight, modern Antarctic GEM depletion events looked similar to Arctic observations and it was initially thought that the Br oxidation pathway was once more operative. While Arctic AMDEs showed a positive correlation between GEM and ozone mixing ratio, some of the Antarctic GEM depletion events were not associated with ozone removal. A negative correlation between ozone and GEM was even observed in Antarctic summer. Investigations made on the vast and cold Antarctic Plateau (> 2000 m) revealed that major GEM depletion events could be observed far from the coast (Brooks et al., 2008). Recently, a study on the Antarctic plateau at Dome Concordia showed that daily GEM cycling involves atmospheric oxidation, deposition, and re-emission from snow via photochemical reactions (Dommergue et al., 2012). In this study, a negative correlation with ozone and GEM was measured. Both the source and the nature of the GEM oxidant remain unclear. Recent research also shows elevated levels of oxidants such as ozone and OH radicals in continental Antarctic air masses (Preunkert et al., 2012). Similarly, at Antarctic coastal locations, atmospheric iodine has been proposed as a significant oxidant for GEM (Saiz-Lopez et al., 2008). Clearly extreme Antarctic environments are unique locations to understand the role of a range of potential GEM oxidants.
4 The Arctic mercury cycle
In contrast to Antarctica, Arctic mercury has become a high-profile environmental case due to the excessive exposure of Arctic indigenous communities to seafood MMHg. The discovery that AMDEs deposit large amounts of Hg to Arctic sea-ice and the Arctic Ocean (Schroeder et al., 1998) has fueled the paradigm that mid-latitude anthropogenic Hg emissions are transported and deposited to Arctic ecosystems (AMAP, 2005). A decade of Arctic Hg research indicates, however, that approximately 80% of AMDE deposited Hg is photochemically re-emitted into the atmosphere (Douglas et al., 2012). Despite decreasing trends in Northern Hemisphere atmospheric Hg emissions and concentrations, multi-decade trends in Arctic biota Hg levels indicate continued increases in the western Arctic and stable or decreasing levels in the eastern Arctic (AMAP, 2006). A simple linear response between mid-latitude Hg emissions and Arctic MMHg exposure therefore does not appear to exist.
Modeling of the Arctic Hg cycle using an advanced Hg chemistry and transport model recently provided some new insights (Fisher et al., 2012). Multi-year monitoring of Arctic atmospheric Hg speciation shows that a spring minimum in atmospheric GEM concentrations is followed by a pronounced summer maximum in GEM (Steffen et al., 2005). The model is unable to capture these observed atmospheric GEM dynamics, unless a significant source of marine Hg to the Arctic atmosphere is included. The authors suggest that the missing Hg source consists of riverine and coastal erosion Hg inputs into the Arctic Ocean, that subsequently drive the summer GEM maximum through evasion. The riverine Hg inputs are suggested to be of the same order of magnitude, if not larger, than atmospheric deposition (Fig. 5). This strongly contrasts with the classical paradigm that Hg in the Arctic Ocean (and in biota) comes primarily from long-range atmospheric transport (AMAP, 2005). In addition, Sonke and Heimbürger (2012) emphasize that the model Arctic atmosphere exports Hg to, rather than imports Hg from, mid-latitudes on an annual basis (Fig. 5). This model observation would not imply that the Arctic region is not a sink for anthropogenic Hg emissions. Boreal soils could function as a temporary reservoir in the trafficking of anthropogenic Hg, deposited at high latitudes, to the Arctic Ocean through rivers. If the modeled river and erosional input and surface to deep ocean export can be confirmed by observations, then this would imply a major turnaround in understanding the inorganic Arctic Hg cycle.
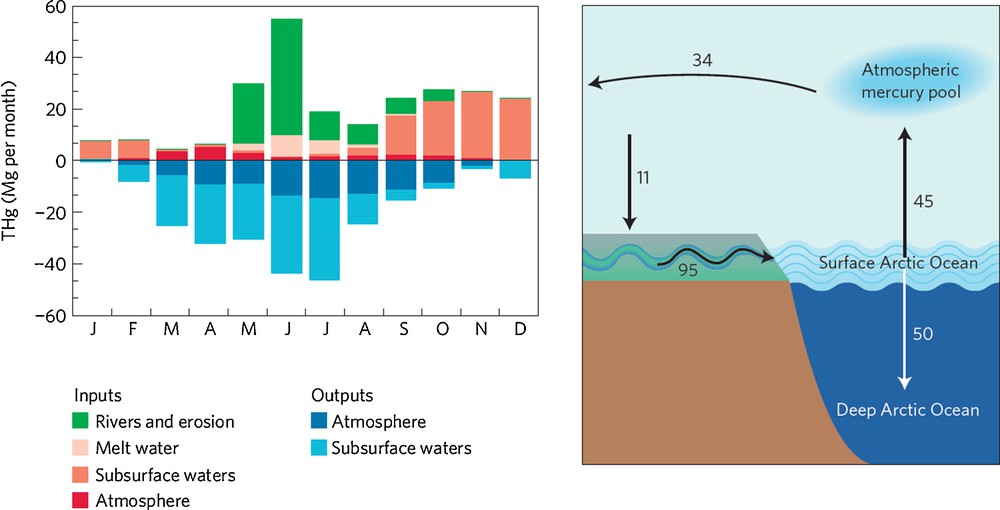
Mercury cycling between Arctic reservoirs. Conventional wisdom assumes that most mercury enters the Arctic region through the atmosphere. However, recent research suggests that boreal rivers deliver large quantities of mercury to the Arctic Ocean – left panel: seasonal budget of mercury in the Arctic surface ocean (Fisher et al., 2012). According to their revised mercury budget for the region, the Arctic atmosphere is a net source of mercury to lower latitudes on an annual basis – right panel (Sonke and Heimbürger, 2012). Annual fluxes (Mg/year) represent net transfers. Figure reproduced with permission.
5 Mercury methylation
5.1 Microbial methylation
The principal source of MMHg in the aqueous environment was early on attributed to biotic methylation of Hg by sulfate-reducing bacteria in anoxic sediments (Compeau and Bartha, 1985; Jensen and Jernelov, 1969). As shown more recently, iron-reducing bacteria (Fleming et al., 2006; Kerin et al., 2006) and methanogens (Hamelin et al., 2011) can also methylate Hg in anoxic environments. Hg methylation has been related to the production of acetyl-CoA and methylcobalamin (B12) (Choi et al., 1994) or to a methyltransferase pathway (Siciliano and Lean, 2002). Although Hg methylation by microorganisms is known for more than 40 years, the exact mechanisms remain elusive. A major breakthrough has been made recently with the discovery of two genes, hgcA and hgcB, that control anoxygenic Hg methylation in sulfate-reducing bacteria (Parks et al., 2013). Fifty-two bacteria and archaea for which genomes have been sequenced possess the hgcAB cluster, and include a psychrophile, thermophile, and a human intestinal methanogen.
While biotic methylation is mainly demonstrated to occur in anoxic environments, a growing body of field evidence suggests that oxic environments (such as arctic snow and seawater) may also host biotic methylation (Constant et al., 2007; Lehnherr et al., 2011; Monperrus et al., 2007). Genomic approaches that identify the presence of the hgcAB methylating gene cluster in bacteria present in seawater and snow may help understand Hg methylation in these oxic environments.
5.2 Marine methylmercury
It is well documented that microbial Hg methylation in estuarine and shelf sediments is an important source of MMHg to overlying waters and local food webs (Fitzgerald et al., 2007). The origin of MMHg in offshore marine fish remains however somewhat elusive. It has long been suggested that open ocean food webs accumulate MMHg via physical advection and bioadvection mechanisms (Chen et al., 2008). The potential for an open ocean MMHg source has been pointed out in the work of Mason and Fitzgerald (1990). In addition to MMHg, another form of methylated Hg exists in the oceans, dimethylmercury (DMHg), and most analysts measure the sum of MMHg + DMHg, hereafter indicated by ‘MeHg’. Renewed interest in open ocean water column Hg methylation stems from observations that:
- • inorganic Hg methylation takes place even in oxygenated surface waters (Heimbürger et al., 2010; Hammerschmidt and Bowman, 2012; Lehnherr et al., 2011; Monperrus et al., 2007);
- • MeHg profiles are closely related to apparent oxygen utilization and plankton dynamics (Cossa et al., 2009, 2011; Hammerschmidt and Bowman, 2012; Heimbürger et al., 2010; Sunderland et al., 2009; Wang et al., 2012).
These studies suggest that the remineralization of sinking particulate organic matter controls in situ methylation of Hg by providing the inorganic Hg as the substratum, and by stimulating the activity of methylating bacteria.
Little is known about the stability of MeHg in the oceanic water column, illustrated by a relatively wide range of biotic and abiotic demethylation rate constants (Mason et al., 2012). In the surface ocean, photochemical degradation of MeHg and evasion of volatile DMHg to the atmosphere decrease concentrations of the MeHg pool. Biotic decomposition of MMHg has been shown to be of the same order of magnitude in surface waters as at depth (Lehnherr et al., 2011). More observations are needed to affirm demethylation rates. This is important as it determines the residence times of MeHg in the deep ocean reservoir and as it may also help answer the question on the importance of advected vs. in situ produced MeHg.
Observed marine MeHg profiles are the result of a dynamic interplay between methylation and demethylation and appear largely governed by local features. For example, high MeHg concentrations have been observed in the oxygen minimum zones of upwelling regions (Conaway et al., 2009; Weiss-Penzias et al., 2012). Record MeHg concentrations up to 0.9 pM were observed in the remote upwelling zone close to Antarctica (Cossa et al., 2011). Here, the upwelling zone supports primary production, which together with large inputs of inorganic Hg substrate from sea-ice and atmospheric deposition can explain the high MeHg concentrations far from anthropogenic emission sources. Another example of local biogeochemical control is a time-series in the Mediterranean Sea, which shows that MeHg concentrations vary seasonally throughout the water column (Heimbürger et al., 2010). Both examples illustrate how MeHg dynamics depend more strongly on the organic carbon cycle than on variations in substrate inorganic Hg.
5.3 Atmospheric mono-methylmercury
MMHg constitutes up to about 5% of total Hg in rainwater (Bloom and Watras, 1989; Hall et al., 2005; Hultberg et al., 1995) or deposited snow (Marusczak et al., 2011), and thus up to 5% of wet atmospheric deposition of Hg (Conaway et al., 2010a). In atmospheric waters, MMHg levels are typically around 0.05 to 0.5 pM. In addition, atmospherically deposited inorganic Hg may be more available to methylation than strongly complexed inorganic substrate Hg already within an ecosystem (Conaway et al., 2010a; Harris et al., 2007; Rolfhus et al., 2003). Thus, atmospheric deposition of Hg may represent a significant source of substrate for MMHg production in aquatic environments. Abiotic methylation of atmospheric Hg(II) requires the presence of suitable methyl donors such as small organic molecules (CH3I, DMS), organometallic complexes (methylcobalamin, methyllead or methyltin compounds) or larger organic components of dissolved organic matter (Celo et al., 2006; Gardfeldt et al., 2003; Hammerschmidt et al., 2007; Pongratz and Heumann, 1999). Atmospheric methylation is considered to be abiotic, despite the fact that many of these methyl donors are of biological origin (Craig, 1986).
5.4 Dimethylmercury
Another hypothesis for the presence of MMHg in aqueous phases is the decomposition of DMHg that has been released to the atmosphere by evasion from upwelling oceanic water (Black et al., 2009; Mason et al., 1997). Relatively high concentrations of MMHg have been observed in rainwater in areas of coastal upwelling (Bloom and Watras, 1989), suggesting that the evasion of DMHg from the ocean and its subsequent degradation in the atmosphere may be a source of MMHg in precipitation in some areas. Although the existence of atmospheric DMHg is controversial, its importance as a precursor for atmospheric MMHg should be further examined for coastal and oceanic precipitation, especially in areas where coastal upwelling occurs and where DMHg is known to be seasonally present in marine surface waters (Conaway et al., 2010b).
5.5 Mercury bioavailability
Hg methylation and demethylation processes are affected by the physicochemical characteristics of the media (redox, pH, temperature, chemical composition) (Celo et al., 2006; Ullrich et al., 2001) and their influence on microbial communities and Hg speciation. In the case of microbial Hg methylation, the availability of Hg (or bioavailability) is an important factor that characterizes the ability of Hg to cross cell membranes of methylating microbes. Benoit et al. (1999, 2003) showed that sulfide chemistry exerts an important control on Hg uptake by determining the amount of bioavailable neutral HgS complexes. Uptake of Hg is also influenced by dissolved organic carbon (Gorski et al., 2008), chloride concentrations (Zhong and Wang, 2009), or the presence of divalent cations (Daguené et al., 2012). Recent studies pointed out that some Hg complexes (cysteine and other thiols) promote the uptake and methylation of Hg by anaerobic methylating microorganisms (Schaefer and Morel, 2009; Schaefer et al., 2011). Direct measurement of bioavailable Hg has been developed (Barkay et al., 1998), using genetically-modified cell biosensors and may be relevant proxies for evaluating the potential for bio-uptake of Hg. Finally, the bioavailability of MMHg and DMHg to plankton in marine systems is an overlooked but key factor in controlling the first step of Hg bioconcentration in marine food webs.
6 Novel tools
6.1 Modeling
The development of regional and global Hg chemistry and transport models (CTMs) has been instrumental in rendering complex Hg biogeochemical cycling accessible to scientists and policy makers. Global CTMs, which are rooted in early plume models for estimating pollutant dispersion around smoke stacks (Seigneur et al., 1997), have been elegantly used to illustrate to what extent the Hg issue is a global issue. Hg CTMs are 3D coupled ocean–atmosphere–land models that include up-to-date natural and anthropogenic Hg emission inventories, global meteorology fields, and detailed Hg transformation chemical kinetics. Typical global CTMs have a grid scale of 4 ° latitude and longitude, 50+ vertical atmospheric levels and 2D (2-level) soils and oceans. These models predict the fate and transport of various Hg species that correspond to their measured counterparts in the field. While complex in detail, model output in the form of simple visualizations is a very effective communication tool between scientists and policymakers. Fig. 6 illustrates how a global Hg CTMs generates straightforward maps of Hg deposition, and the contribution of different Hg emission sources to deposition over a particular region [North America (Selin and Jacob, 2008)]. Providing predictions of future Hg exposure based on different policy scenarios, i.e. Hg emission scenarios, can only be made by comprehensive models. Current CTMs are limited by the lack of knowledge on atmospheric Hg chemical kinetics, the lack of oceanic observations, a lack of high altitude atmospheric observations, etc. Some of the largest fluxes in the global Hg cycle, such as export to the deep ocean, are constrained by only a handful of observations on marine Hg-particle partition coefficients (Soerensen et al., 2010). A continuous dialogue is needed between modelers, observationalists and experimentalists to address the most crucial uncertainties in global Hg models.
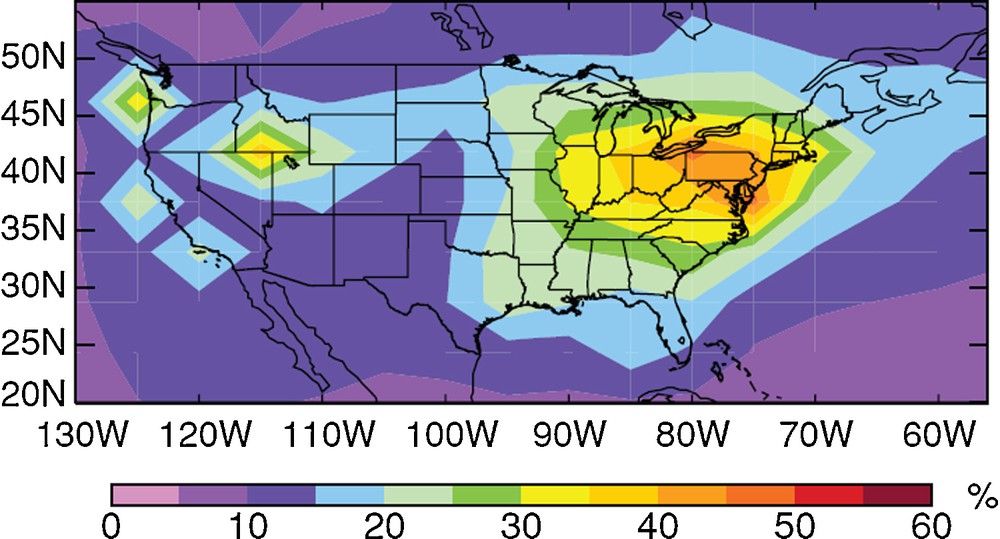
Simulated contribution from North American primary anthropogenic sources to total annual mercury deposition by the GEOS-Chem global mercury model for 2004–2005. The model suggests that on average only 20% of North American deposition results from North American Hg emission, and illustrates to what extent the Hg issue is a global issue.
Reproduced with permission from Selin and Jacob (2008).
6.2 Mercury stable isotopes
Following the development of trace-clean Hg sampling and analysis, and operational and molecular Hg speciation techniques, a more recent analytical advancement provides access to information embedded at the isotopic level. Hg has seven stable isotopes whose abundances can be precisely (0.01%) measured by mass spectrometry (Blum and Bergquist, 2007; Epov et al., 2011). A decade of research on the natural variations in Hg isotope abundances has shown large (1%) variations across biogeochemical reservoirs (Sonke and Blum, 2012). These variations result from the gradual separation of heavy/light or even/odd Hg isotopes during the numerous physicochemical processes that shuttle Hg across the Earth's surface. As a result, a Hg isotopic measurement gives rise to four useful isotope signatures (δ202Hg, Δ199Hg, Δ200Hg, Δ201Hg) that may characterize its source, or code for the transformations that Hg has undergone in the recent, but also geological past.
Case studies on tracing Hg emissions, deposition or discharge at the local or regional scale have confirmed the potential of Hg isotope signatures to discriminate between pollutant sources and background Hg (see reviews by: Bergquist and Blum, 2009; Blum, 2011; Sonke and Blum, 2012). Moreover, Hg mass-independent isotope fractionation (MIF) signatures, Δ199Hg and Δ201Hg, appear consistently unaffected by biochemical reactions within organisms (Kritee et al., 2012; Kwon et al., 2012). As such they are ideal for tracing Hg exposure sources to humans and wildlife. For example, Hg MIF in human hair, a biomarker for fish MMHg exposure, shows markedly different Δ199Hg and Δ201Hg for populations consuming fresh water or marine fish (Laffont et al., 2009, 2011). Beyond source tracing, Hg MIF in biological tissues ultimately reflects the photochemical transformations that affected MMHg before it was bioaccumulated up the food chain (Bergquist and Blum, 2007). Hg MIF in biomonitor tissues therefore provides a direct window on the aquatic photochemistry of MMHg, an aspect that is difficult to study using classical methods. For example, Hg MIF in marine seabird eggs from the Alaskan Arctic revealed a pronounced latitudinal gradient in Δ199Hg and Δ201Hg that was suggested to reflect the influence of sea-ice on surface ocean MMHg photodecomposition (Fig. 7; Point et al., 2011). Translating field observations on Hg isotope variations into meaningful numbers on Hg fluxes and cycling requires thorough understanding of the underlying isotope fractionation mechanisms. While there is a continued growth in field observations, there is a corresponding lack in laboratory studies to calibrate Hg isotope fractionation factors. More research is needed in this direction.
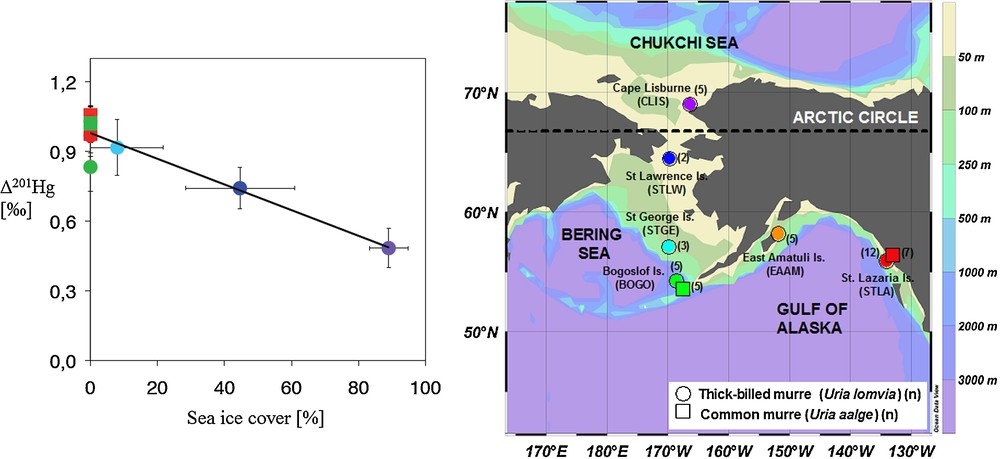
Mass-independent mercury stable isotope fractionation (MIF) is caused by aqueous photochemical breakdown of mono-methylmercury (MMHg) (amongst others). The figure shows how mercury MIF signatures (Δ201Hg shown) in an Alaskan seabird biomonitor decrease with increasing the sea-ice cover in their foraging area (Point et al., 2011). The authors used mercury MIF to quantify how Arctic climate change may potentially alter Arctic MMHg cycling. Although the disappearance of summer sea-ice will likely stimulate photochemical MMHg breakdown, other factors influencing the Arctic mercury cycle need to be understood before a net balance can be established.
7 Conclusions
Hg research has a long history and much is known about its biogeochemical cycle. Nevertheless, the low concentrations and the complexity that characterize Hg cycling and Hg toxicity continue to impose major paradigm shifts. In this review, we have drawn attention to some of the major outstanding questions on Hg cycling and some of the newest tools to address these. The 2013 UNEP Minamata Convention on Mercury is a clear step forward in preventing emissions and release of Hg to the environment. However, it does not specify concrete Hg emission reduction goals for the largest industrial sectors, in particular the energy sector. In the prospect of a ‘business as usual’ emission scenario, anticipating and mitigating the impact of Hg on human health demands continued basic research efforts in epidemiology, toxicology and biogeochemistry.
Global change, i.e. planetary-size change in Earth systems, in the 21st century is driven by human activities. As illustrated in the introduction and on Figs. 1 and 2, global change in the form of anthropogenic Hg emissions has severely modified the global Hg cycle. In addition, all physicochemical transformations of Hg compounds are not only temperature and thus climate dependent, but also depend on global change-related factors such as nutrient cycles, atmospheric composition (oxidants, aerosols) or biota (food web structure, foraging, microbiology). Other than Hg emissions, multiple aspects of the global Hg cycle can therefore be expected to vary with recent and ongoing trends in climate and global change (Stern et al., 2012).
Acknowledgments
J.E.S. acknowledges funding from the French Agence nationale de la recherche (ANR-09-JCJC-0035-01) and the European Research Council (ERC-2010-StG-20091028). A.D. is supported by the Fonds France Canada pour la recherche and the Rhône-Alpes Region. A.D. acknowledges the Institut universitaire de France. Daniel Cossa, Laurie Chan and Marc Chaussidon are thanked for constructive comments.